Pathways of Gastric Carcinogenesis, Helicobacter pylori Virulence and Interactions with Antioxidant Systems, Vitamin C and Phytochemicals


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Gastric Carcinogenesis
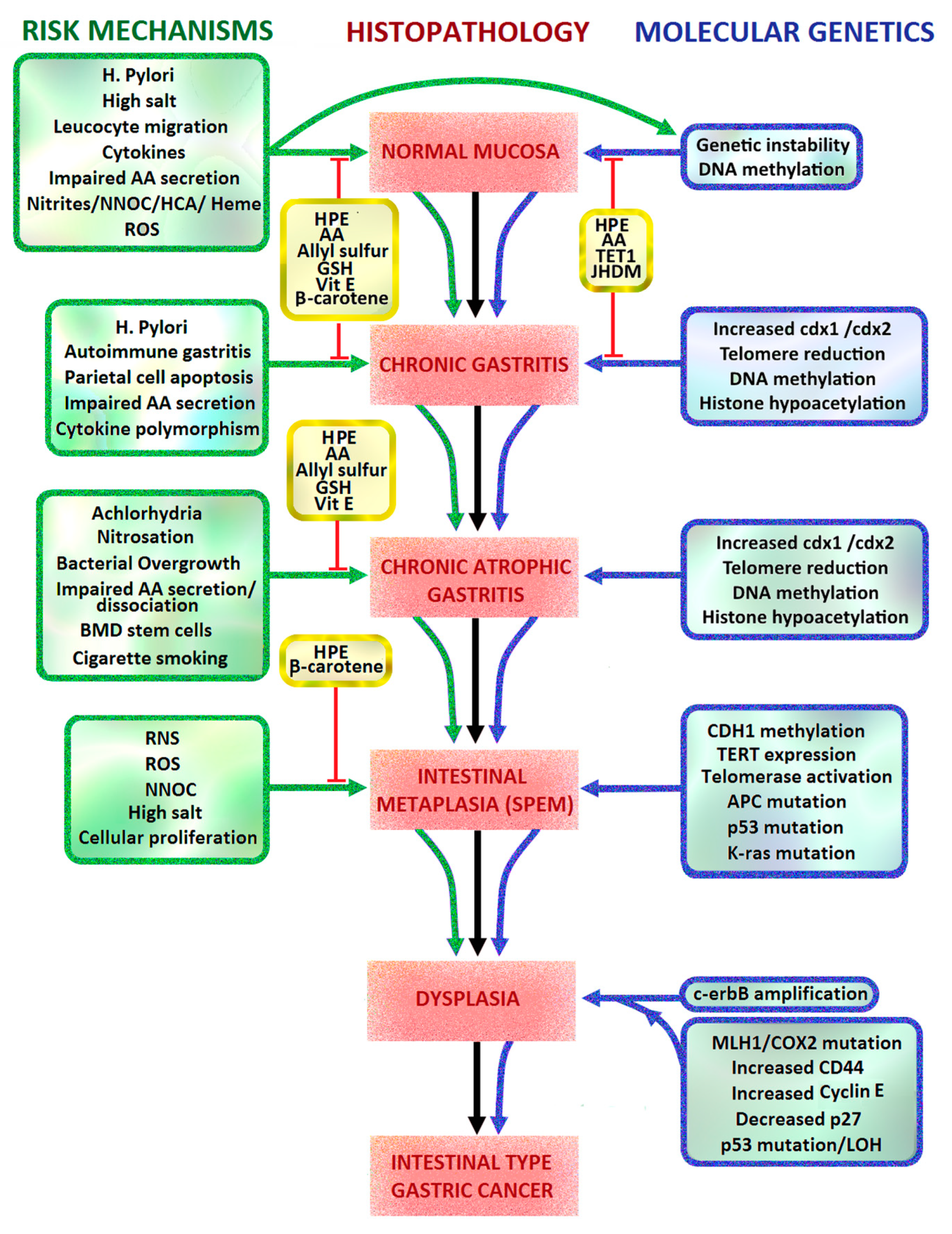
2.1. Correa Pathway and Sydney Classification of Intestinal Metaplasia
2.2. Helicobacter Pylori
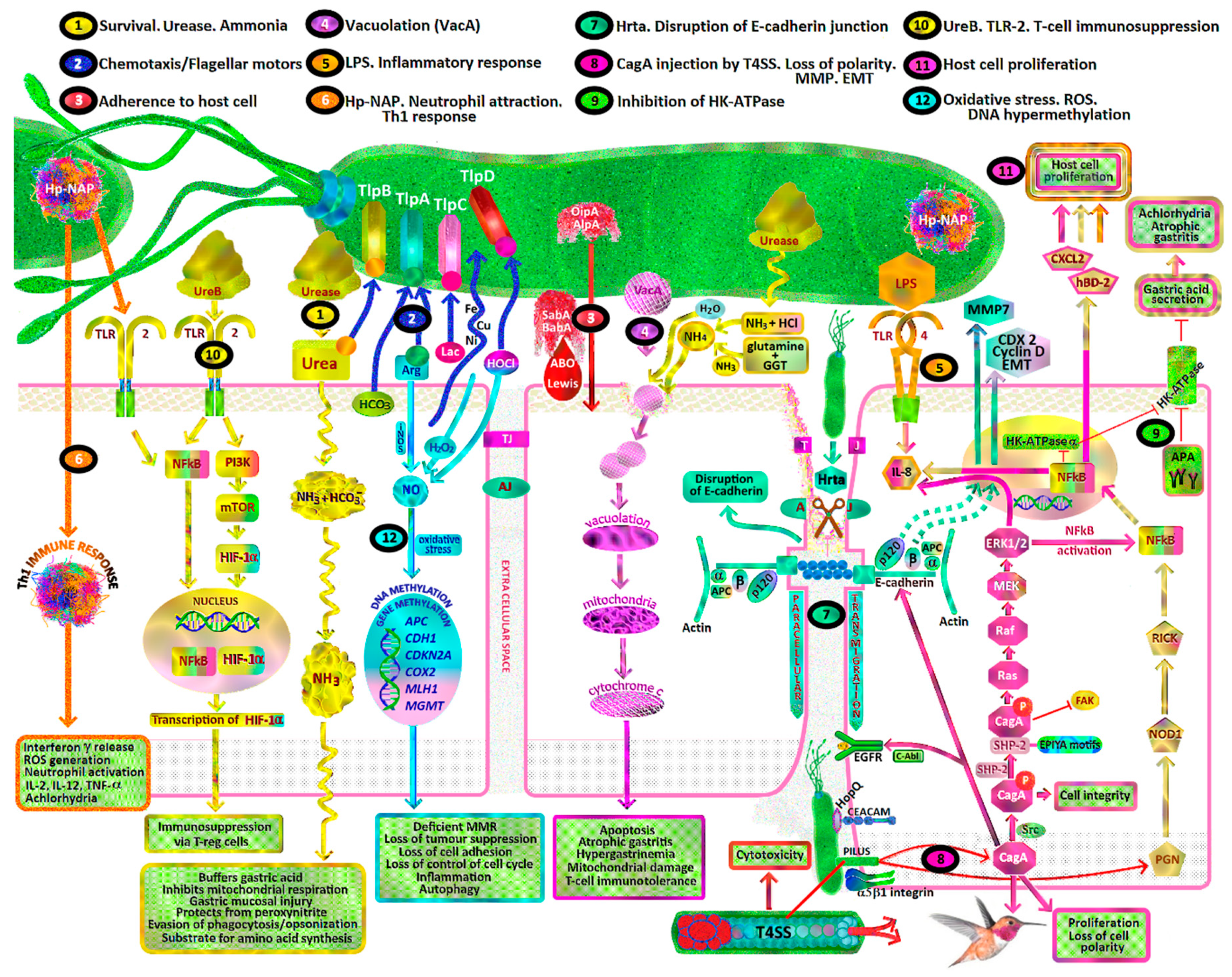
- H. pylori toxins which damage gastric mucosal epithelial cells leading to chronic atrophic gastritis, decreased gastric mucosal secretion of ascorbic acid, parietal cell apoptosis, achlorhydria, hypergastrinaemia, gastric dysbiosis, intestinal metaplasia, dysplasia and intestinal type gastric cancer (Figure 1, pathways 1, 4, 8, 9).
- Recruitment of inflammatory cells resulting in acute and/or chronic inflammation, activation of reactive oxygen species (ROS) pathways including neutrophil myeloperoxidase- hypochlorite (HOCl)-hydrogen peroxide (H2O2), macrophage nitric oxide (NO) and epithelial cell hydrogen peroxide production (Figure 1, pathway 6, 12).
- Oxidative stress due to reactive nitrogen species (RNS), ROS, lipid peroxidation and MDA/free radical formation overwhelming gastric antioxidant protection.
- Promotion of gastric epithelial proliferation, oncogenes and DNA damage via various mechanisms, including H. pylori CagA, VacA, BabA, SabA, Hp-NAP, ROS, RNS, urease, DNA hypermethylation and cellular tyrosine kinases (Figure 1, pathways 1, 3, 4, 7, 8, 10, 11, 12).
- Dysregulation of tyrosine kinase oncogenic pathways and loss of tumour suppressors (p53, CDH1/E-cadherin, APC, MGMT, MLH1, CDKN2A), leading to failure of apoptosis and Epithelial Mesenchymal Transition (EMT) (Figure 1, pathways 7, 8).
- Synergy with ingested carcinogens (nitrosamines/heterocyclic amines/nitrites/dietary salt/alcohol/tobacco smoke) and complex interactions with antioxidants resulting in decreased protective effects and promotion of carcinogenesis (Figure 1).
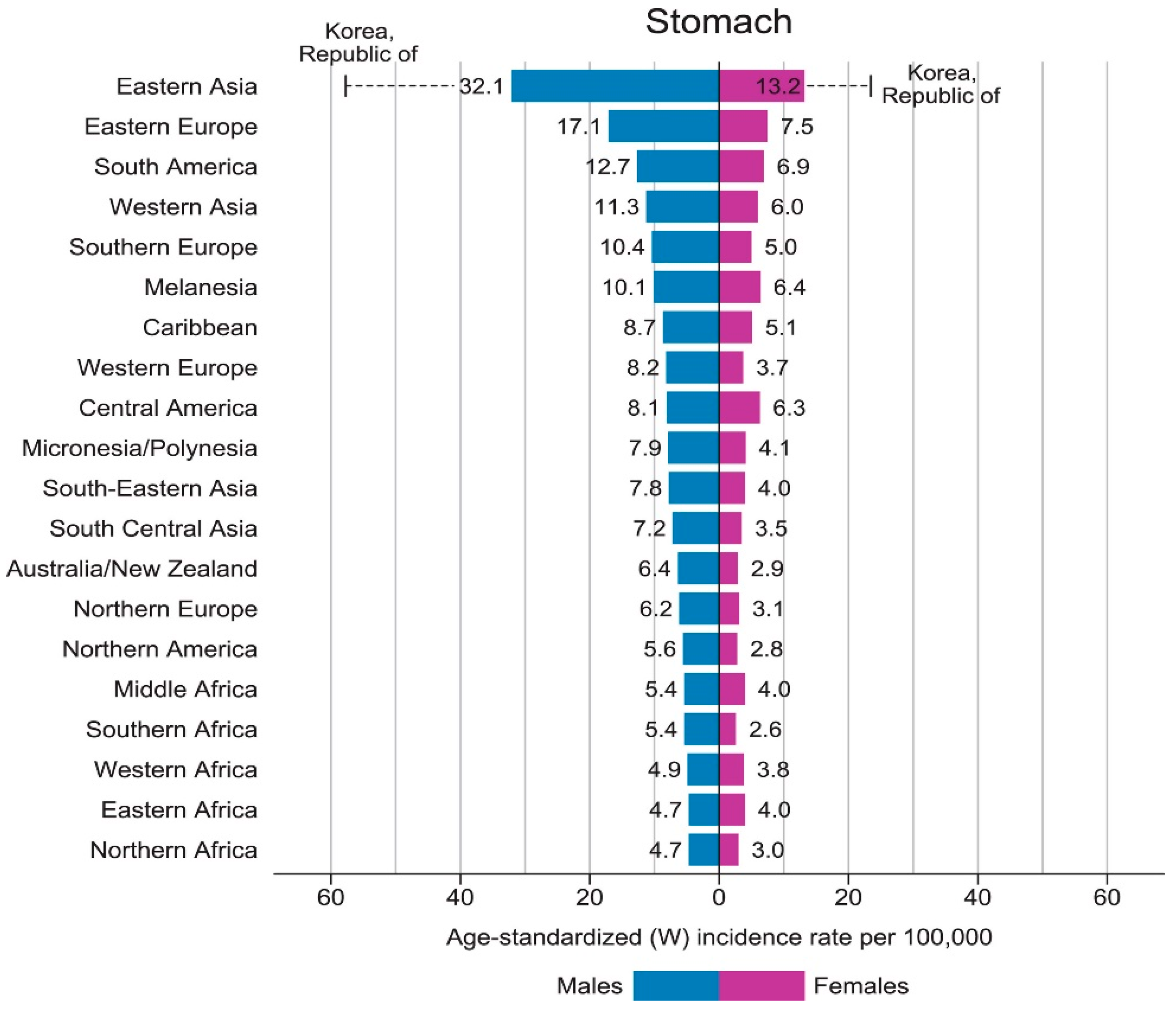
2.3. Epidemiology of Gastric Cancer
2.4. Lauren Classification
2.5. Gastric Cancer Molecular Subtypes
- Epstein Barr virus associated (EBV, 8.8%) with hypermethylation of DNA promoters. EBV gastric cancer is characteristically found in the proximal stomach. It is a poorly differentiated adenocarcinoma with lymphocytic infiltration on histology, PD-L1 and PD-L2 overexpression and CDKN2A silencing [22].
- Microsatellite instability (MSI, 21.7%) due mainly to mutations of the hMLH1 gene promoter, leading to deficient mismatch repair of DNA (dMMR). MSI is associated with Lynch syndrome, distal gastric cancers and Lauren intestinal subtype on histology.
- Chromosomal instability (CIN, 49.8%) with intestinal type cancer and cytosine and guanine (CpG) island methylator phenotype (CIMP). CIN gastric cancers arise more often in the gastro-oesophageal junction and gastric cardia (65%).
- Genomically stable (GS, 19.7%) with DGC [23].
- MSI-high (23%),
- Microsatellite stable/epithelial mesenchymal transition (MSS/EMT, 15%),
- Microsatellite stable/TP53 intact (MSS/TP53+, p53 active, 26%)
- Microsatellite stable/TP53 loss (MSS/TP53−, p53 inactive, 36%).
- Each subtype was associated with distinct treatment options and prognostic outcomes [27].
2.6. Cardia vs. Non-Cardia Gastric Cancer
2.7. Gastric Oxidative Stress
2.8. Gastric Cytoprotection
- Enzymatic antioxidants such as superoxide dismutase (SOD), catalase (CAT), thioredoxin reductase and glutathione peroxidase (GPX).
- Small molecule antioxidants such as α-tocopherol (vitamin E), ascorbic acid (vitamin C), beta-carotene, bilirubin, glutathione (GSH) and uric acid.
- Metal ion chelators including metallothionein, haptoglobulin, albumin, transferrin and ceruloplasmin (Figure 3).
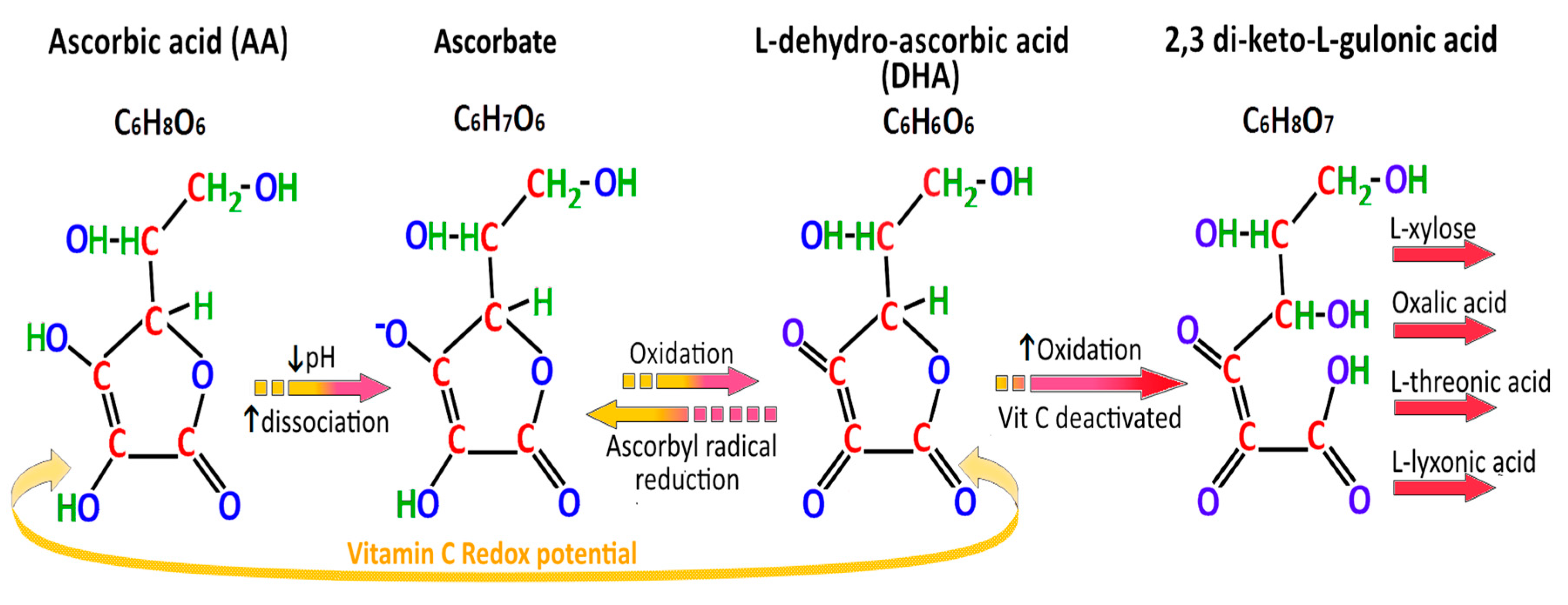
2.9. Absorption and Secretion of Ascorbic Acid
3. Helicobacter pylori-Virulence Factors and Pathways to Gastric Cancer
3.1. Helicobacter pylori
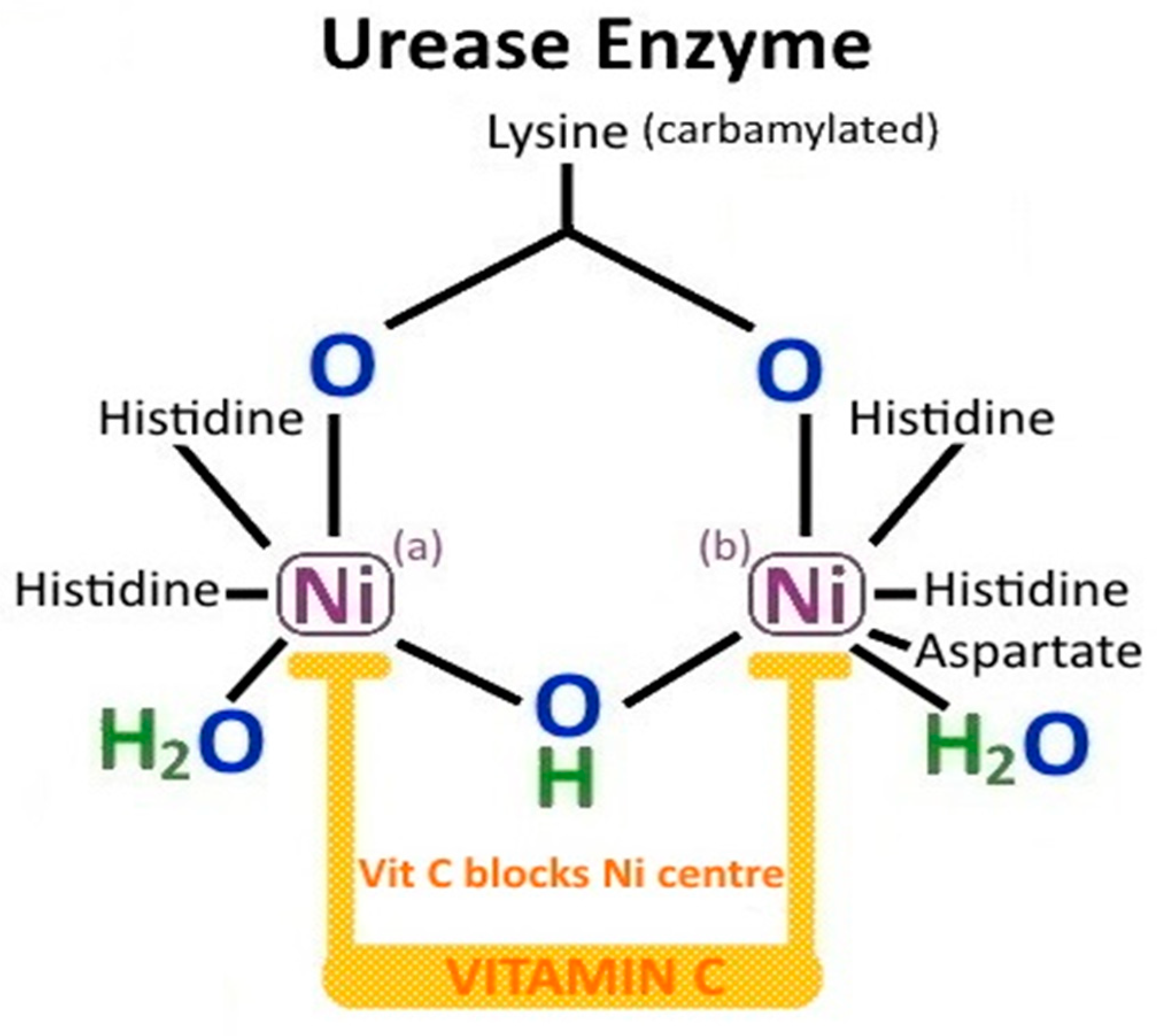
3.2. H. pylori Urease
3.3. H. pylori-Derived Ammonia
- buffer gastric acid
- inhibit gastric epithelial mitochondrial and isolated cellular respiration
- induce cytotoxicity in gastric epithelial cells
- contribute to gastric mucosal injury
- enable H. pylori to evade host phagocytosis and opsonisation
- provide protection to H. pylori from host generated peroxynitrite
3.4. H. pylori and Hypochlorhydria
- Increased T helper type 1 (Th1) cell secretion of Interleukin-1β (from mucosal neutrophils and monocytes), which inhibits gastric proton pump (H+/K+-ATPase) activity (Figure 1, pathway 6).
- Release of H. pylori fatty acids (tetradecanoic acid and cis 9,10-methyleneoctadecanoic acid), which inhibit proton pump H+/K+-ATPase and dissipate proton (H+) transport in parietal cell secretory vesicles [54].
- Suppression of acid secretion by neural inhibition of enterochromaffin cell histamine secretion and antral G-cell gastrin secretion [56].
- H. pylori VacA disrupts the incorporation of tubulovesicles (which contain H+/K+-ATPase) into the gastric parietal cell apical membrane [54].
3.5. H. pylori and Chemotaxis
- TlpA is able to sense gastric mucosal arginine/amino acids, acidic pH and bicarbonate
- TlpB senses urea and bacterial quorum (via auto-inducer (AI-2))
- TlpC senses lactate
- TlpD senses ROS, alkaline pH, neutrophil derived HOCl and inhibitors of electron transport (Figure 1, pathway 2).
3.6. Non-Enzymatic Effects of H. pylori Urease
- release of IL-8, a chemokine for inflammatory cells including monocytes and neutrophils
- activation of NF-κB pro-inflammatory pathways
- activation of primary mucosal macrophages
- release of gastric epithelial cell Th1 cytokines (IL-1β, IL-6, TNF-α, IL-8)
- disruption of gastric epithelial tight junctions
- release of IL-4 and antibody production by splenic lymphocytes
- platelet aggregation [53].
3.7. H. pylori Induction of HIF
3.8. H. pylori and Correa Pathway
- 21 (4.7%, p < 0.001) of the 445 patients with non-ulcer dyspepsia,
- 10 (3.4%, p = 0.002) of the 297 with gastric ulcers,
- 5 (2.2%, p = 0.02) of the 229 with gastric hyperplastic polyps, and
- 0 of the 275 with duodenal ulcers.
3.9. H. pylori-Induced Inflammatory Response
3.10. H. pylori and ROS
3.11. H. pylori and Evasion of Immunosurveillance
3.12. H. pylori and DNA Damage
3.13. H. pylori, iNOS, ROS and DNA Hypermethylation
- DNA repair [O-6-methylguanine DNA methyltransferase enzyme (MGMT)],
- DNA mismatch repair [MutL Homologue 1 (MLH1)],
- Cell cycle [Cyclin Dependent Kinase Inhibitor 2A (CDKN2A)],
- Inflammation [Trefoil factor 2 (TFF-2)],
- Transcription factors [Forkhead Box D3 (FOXD3), upstream stimulatory factors (USF1 and USF2), GATA4],
- Autophagy (ATG16L1),
3.14. H. pylori, NF-κB, STAT3, TNF-α, Vitamin C and β-carotene
4. Helicobacter CagA+/VacA+
4.1. CagA+/VacA+
4.2. CagA and EPIYA Carriage
4.3. CagA and Type IV Bacterial Secretion System (T4SS)
4.4. CagA and Src
4.5. CagA, Vitamin C and Epigenetic Programming
4.6. CagA, E-Cadherin and EMT
4.7. CagA and Inflammation
4.8. VacA
- Receptor-like protein tyrosine phosphatase α (RPTPα),
- Receptor-like protein tyrosine phosphatase β (RPTPβ),
- Epidermal Growth Factor Receptor (EGFR),
- Lipid raft/glycosylphosphatidylinositol-anchored proteins (GPI-AP),
- Sphingomyelin (SM),
- Fibronectin (FN),
- Heparin (H) and heparan sulfate (HS),
- Low-density Lipoprotein Receptor-related Protein-1 (LRP1),
- and one T lymphocyte receptor: CD18 [119].
4.9. VacA and Vacuolation
4.10. VacA and CagA
5. Helicobacter Adhesins, Blood Group and Vitamin C
6. Vitamin C and H. pylori
6.1. Chronic Atrophic Gastritis, H. pylori and Vitamin C
6.2. H. pylori and Gastric Ascorbic Acid Secretion
6.3. H. pylori Eradication and Vitamin C
6.4. Phytochemicals, CagA and Prevention of Gastric Cancer
7. Probiotics and Helicobacter Eradication
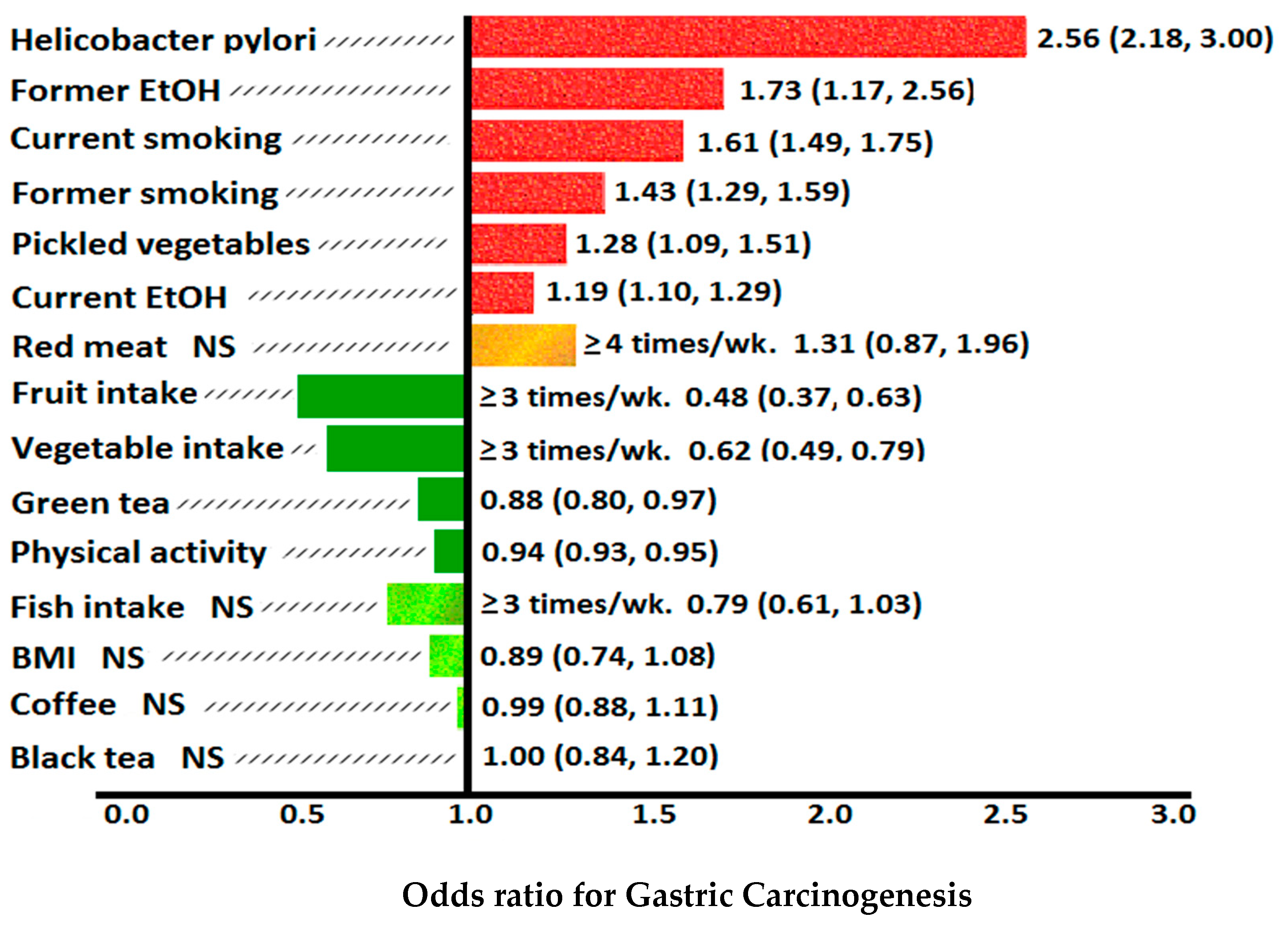
8. Dietary and Environmental Risk Factors for Gastric Cancer
8.1. Dietary Nitrosamines
8.2. Vitamin C, Allium Vegetables and Nitrosamines
8.3. Heterocyclic Amines
8.4. Salt and H. pylori
8.5. Tobacco Smoking, Vitamin C and H. pylori
- non-smokers/H. pylori negative: mean gastric vit C = 17.1 mcg/mL,
- non-smokers/H. pylori positive: mean gastric vit C = 12.6 mcg/mL,
- smokers/H. pylori negative: mean gastric vit C = 5.8 mcg/mL,
- smokers/H. pylori positive: mean gastric vit C = 3.9 mcg/mL.
- Consumption of ascorbic acid to DHA by ROS and oxidative stress in the stomach and other tissues exposed to cigarette smoke/tar.
- Vitamin C is used for regeneration of vitamin E, β-carotene or glutathione which have been directly oxidized during scavenging of free radicals and ROS from cigarette smoke.
- Smokers have lower dietary consumption of vitamin C containing foods.
- Cigarette smoke induced oxidative damage of proteins and peroxidation of lipids are accompanied by a marked drop in tissue ascorbate levels.
- Increased H. pylori infection in smokers vs. non-smokers.
- Failure of regeneration of ascorbate from DHA by GSH-dependent reductases.
8.6. Alcohol
9. Family History and Genetic Mutations
- Hereditary Diffuse Gastric Cancer (CDH1 germline mutation)
- Breast cancer syndrome (BRCA2)
- Hereditary non-polyposis colorectal cancer (HNPCC, Lynch II) syndrome (MSH2/MLH1/MSH6 mutation),
- Li Fraumeni syndrome (TP53 mutation)
- Familial adenomatous polyposis (FAP) syndrome (APC mutation).
9.1. Sodium Dependent Ascorbic Acid Transporters
9.2. Glutathione and Gastric Cancer
10. Gastric MALT Lymphoma and H. pylori Eradication
11. Prevention of Gastric Cancer by H. pylori Eradication
11.1. Helicobacter pylori Eradication and Gastric Cancer
11.2. H. pylori Eradication, Intestinal Metaplasia and Gastric Cancer
11.3. Shandong Intervention Trial
12. Conclusions
- Scavenging of superoxide anion radical (O2−·), singlet oxygen (1O2), hydroxyl radical (OH·)
- Regeneration of vitamin E and glutathione
- Inhibition of lipid peroxidation in conjunction with catechins
- Enhanced activity of HIF-α Prolyl hydroxylase
- Enhanced activity of α-KGDD TET DNA hydroxylases
- Enhanced activity of JHDMs
- Decreased DNA CpG hypermethylation (CIMP)
- Upregulation of transmembrane protein with epidermal growth factor (EGF)-like and two follistatin motifs 2 (TMEFF2)
- Inhibition of endogenous gastric N-nitrosation
- Reduced toxicity of ingested heterocyclic amines/NDMA
- Inhibition of H. pylori urease
- Inhibition of H. pylori colonization
- Improved H. pylori eradication rates
- Inhibition of H. pylori mediated activation of NF-κB and STAT3
- Increased synthesis of PGE2 and gastric mucus production.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-FU | 5-fluorouracil |
| α-KGDDs | α-ketoglutarate-dependent dioxygenases |
| AG | Atrophic gastritis |
| AGE | Advanced glycation end product |
| AGS | Human gastric adenocarcinoma hyperdiploid cell line |
| ALDH2 | Aldehyde Dehydrogenase 2 Family Member |
| ALE | Advanced lipid peroxidation end product |
| AlpA | Adherence associated lipoprotein A |
| Akt | Protein kinase B |
| APA | Anti-parietal cell antibodies |
| APC | Adenomatous polyposis coli protein |
| ATP | Adenosine triphosphate |
| BabA | Blood group antigen binding adhesin |
| BCL-2 | B cell lymphoma-2 |
| BRCA | Breast cancer susceptibility gene |
| CagA | Cytotoxin associated gene A |
| CAT | Catalase |
| CD | Cluster of differentiation |
| CDH1 | E-cadherin gene |
| CDH1 | E-cadherin protein |
| CDKN2A | Cyclin dependent Kinase Inhibitor 2A |
| CDX | Caudal type homeobox |
| CEACAM | Carcinoembryonic antigen-related cell adhesion molecule |
| CIMP | Cytosine and guanine (CpG) island methylator phenotype |
| CIN | Chromosome instability |
| COX | Cyclo-oxygenase |
| Csk | C-terminal Src kinase |
| CTGF | Connective tissue growth factor or CCN2 |
| CXCL2 | C-X-C chemokine ligand 2 or macrophage inflammatory protein 2-alpha (MIP) |
| CXCR4 | C-X-C chemokine receptor type 4 |
| DGC | Diffuse gastric cancer |
| DHA | Dehydroascorbic acid |
| dMMR | Deficient mismatch repair |
| EBV | Epstein–Barr virus |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial mesenchymal transition |
| ENOC | Endogenous nitrosamine |
| EPIYA | Glutamate-proline isoleucinetyrosine-alanine |
| EPO | Erythropoietin |
| erbB2 | Human epidermal growth factor receptor gene |
| ERK | Extracellular signal-regulated kinase |
| etv4 ets | (Erythroblast Transformation Specific) variant transcription factor 4 |
| FAK | Focal adhesion kinase |
| FAP | Familial adenomatous polyposis |
| FGFR4 | Fibroblast growth factor receptor gene |
| GGT | γ-glutamyl transpeptidase |
| GLUT | Membrane glucose transporter |
| GPX | Glutathione peroxidase |
| GSH | Glutathione |
| GSTM1 | Glutathione S-transferase Mu 1 |
| GSTP1 | Glutathione S-transferase Pi 1 |
| GSTT1 | Glutathione S-transferase Theta 1 |
| HBD | Human β-defensin |
| HCA | Heterocyclic amine |
| HER2 | Human epidermal growth factor |
| HGF-R | Hepatocyte growth factor receptor gene, c-MET |
| HIF-1α | Hypoxia inducible factor-1 alpha |
| HK | Hexokinase |
| HKalpha | Alpha subunit of the parietal cell proton pump |
| HMOX1 | Heme oxygenase-1 |
| hMLH1 | Human MutL Homologue 1 |
| HNO2 | Nitrous acid |
| HNPCC | Hereditary non polyposis colorectal cancer |
| HOCl | Hypochlorous acid |
| HopQ | Helicobacter pylori outer protein Q |
| Hp NAP | Helicobacter pylori neutrophil-activating protein |
| HRE | Hypoxia response element |
| H2O2 | Hydrogen peroxide |
| Htra | Serine protease high temperature requirement A |
| HSP | Heat shock protein |
| IFN | Interferon |
| IGFBP3 | Insulin-like growth factor-binding protein 3 |
| IκBα | Nuclear factor kappa light chain enhancer of activated B cells inhibitor, alpha |
| iNOS | Inducible nitric oxide synthase |
| IL | Interleukin |
| ILK | Integrin linked kinase |
| IM | Intestinal metaplasia |
| JHDM | Jumonji-C domain-containing histone demethylases |
| JNK | c-Jun N-terminal kinase |
| KRAS | Kirsten rat sarcoma virus oncogene |
| LDH | Lactate dehydrogenase |
| LOX | lysyl oxidase |
| L-GULO | L-gulono-gamma-lactone-oxidase |
| LPS | lipopolysaccharide |
| MALT | Mucosa associated lymphoid tissue |
| MAP3K | Mitogen-activating protein kinase |
| MDA | Malondialdehyde |
| MGMT | O-6-methylguanine DNA methyltransferase enzyme |
| MHC | Major histocompatibility complex |
| MMP | Matrix metalloprotease |
| MSI | Microsatellite instability |
| MSS | Microsatellite stable |
| mTOR | Mammalian target of rapamycin |
| NAC | N-acetyl cysteine |
| NDMA | Nitrosodimethylamine |
| NF-κB | Nuclear factor kappa light chain enhancer of activated B cells |
| NO | Nitric oxide |
| NOX | NADPH oxidase |
| OipA | Outer inflammatory protein A |
| PAMP | Pathogen associated molecular pattern |
| PDK1 | Pyruvate dehydrogenase kinase 1 |
| PDGFβ | Platelet derived growth factor beta |
| PD-L1 | Programmed death-ligand 1 |
| PGE2 | Prostaglandin E2 |
| PGK | Phosphoglycerate kinase |
| PGN | Peptidoglycan |
| PHD | Prolyl hydroxylase |
| PKM2 | Pyruvate kinase muscle isoenzyme 2 |
| PPI | Proton pump inhibitor |
| PTEN | Phosphatase and tensin homolog |
| PUFA | Polyunsaturated fatty acid |
| pVHL | Von-Hippel–Lindau protein |
| RAGE | Receptor for advanced glycation end product |
| RhoA | Ras homolog family member A |
| RICK | Receptor-interacting protein serine-threonine kinase 2 |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| SabA | Sialic acid binding adhesin |
| SFK | Src family kinase |
| SH2 | Src Homology 2 |
| SHIP2 | Src Homology 2 inositol phosphatase |
| SIRT3 | [sirtuin (silent mating type information regulation 2 homolog) 3] |
| SLC23A1 | Solute carrier family 23 member 1 |
| SLUG | Zinc finger protein SNAI2 |
| SOD | Superoxide dismutase |
| SOX2 | Sex determining region Y-box 2 |
| SNAIL | Zinc finger protein SNAI1 |
| SPEM | Spasmolytic polypeptide expressing metaplasia |
| Src | Non receptor associated sarcoma tyrosine kinase |
| STAT3 | Signal transducer and activator of transcription 3 |
| TET | Ten eleven translocation |
| TET1 | TET methylcytosine dioxygenase 1 |
| TF | Thompsen-Friedenreich antigen |
| T4SS | Type IV bacterial secretion system |
| TGF-β | Transforming growth factor beta gene |
| Th1 | T helper type 1 cell |
| TLR | Toll-like receptor |
| Tlp | Transducer like protein |
| TNF-α | Tumor necrosis factor-alpha |
| TRAF | TNF Receptor associated factor |
| TRAIL | Tumour necrosis factor apoptosis inducing ligand |
| Tregs | Regulatory T cells |
| TWIST1 | Twist basic helix loop helix transcription factor 1 |
| vacA | Vacuolating cytotoxin gene A |
| VEGF | Vascular endothelial growth factor |
| VM | Vimentin |
| ZEB | Zinc finger E-box-binding homeobox 1 |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics, 2002. CA A Cancer J. Clin. 2005, 55, 74–108. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA A Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Y.; Lee, Y.C.; Graham, D.Y. The eradication of Helicobacter pylori to prevent gastric cancer: A critical appraisal. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Machlowska, J.; Baj, J.; Sitarz, M.; Maciejewski, R.; Sitarz, R. Gastric Cancer: Epidemiology, Risk Factors, Classification, Genomic Characteristics and Treatment Strategies. Int. J. Mol. Sci. 2020, 21, 4012. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.H.; Kim, N. Review of atrophic gastritis and intestinal metaplasia as a premalignant lesion of gastric cancer. J. Cancer Prev. 2015, 20, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Schistosomes, Liver Flukes and Helicobacter Pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7–14 June 1994. Int. Agency Res. Cancer 1994, 61, 1–241. [Google Scholar]
- Correa, P. Human gastric carcinogenesis: A multistep and multifactorial process--First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992, 52, 6735–6740. [Google Scholar]
- Jencks, D.S.; Adam, J.D.; Borum, M.L.; Koh, J.M.; Stephen, S.; Doman, D.B. Overview of Current Concepts in Gastric Intestinal Metaplasia and Gastric Cancer. Gastroenterol. Hepatol. (N. Y.) 2018, 14, 92–101. [Google Scholar]
- Kinoshita, H.; Hayakawa, Y.; Koike, K. Metaplasia in the Stomach-Precursor of Gastric Cancer? Int. J. Mol. Sci. 2017, 18, 2063. [Google Scholar] [CrossRef]
- Banks, M.; Graham, D.; Jansen, M.; Gotoda, T.; Coda, S.; di Pietro, M.; Uedo, N.; Bhandari, P.; Pritchard, D.M.; Kuipers, E.J.; et al. British Society of Gastroenterology guidelines on the diagnosis and management of patients at risk of gastric adenocarcinoma. Gut 2019, 68, 1545–1575. [Google Scholar] [CrossRef] [PubMed]
- Ansari, S.; Gantuya, B.; Tuan, V.P.; Yamaoka, Y. Diffuse Gastric Cancer: A Summary of Analogous Contributing Factors for Its Molecular Pathogenicity. Int. J. Mol. Sci. 2018, 19, 2424. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.E.; Smyrk, T.C.; Zhang, L. Histologic and immunohistochemical differences between hereditary and sporadic diffuse gastric carcinoma. Hum. Pathol. 2018, 74, 64–72. [Google Scholar] [CrossRef]
- Compare, D.; Rocco, A.; Nardone, G. Risk factors in gastric cancer. Eur. Rev. Med. Pharmacol. Sci. 2010, 14, 302–308. [Google Scholar]
- Rawla, P.; Barsouk, A. Epidemiology of gastric cancer: Global trends, risk factors and prevention. Prz. Gastroenterol. 2019, 14, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Reed, P.I. Vitamin C Helicobacter pylori infection and gastric carcinogenesis. Int. J. Vitam. Nutr. Res. 1999, 69, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Nouraie, M.; Pietinen, P.; Kamangar, F.; Dawsey, S.M.; Abnet, C.C.; Albanes, D.; Virtamo, J.; Taylor, P.R. Fruits, vegetables, and antioxidants and risk of gastric cancer among male smokers. Cancer Epidemiol. Biomark. Prev. 2005, 14, 2087–2092. [Google Scholar] [CrossRef]
- Hoang, B.V.; Lee, J.; Choi, I.J.; Kim, Y.W.; Ryu, K.W.; Kim, J. Effect of dietary vitamin C on gastric cancer risk in the Korean population. World J. Gastroenterol. WJG 2016, 22, 6257–6267. [Google Scholar] [CrossRef]
- Moss, S.F. The Clinical Evidence Linking Helicobacter pylori to Gastric Cancer. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 183–191. [Google Scholar] [CrossRef]
- Forman, D. Gastric cancer and Helicobacter pylori: A combined analysis of 12 case control studies nested within prospective cohorts. Gut 2001, 49, 347–353. [Google Scholar]
- Ma, J.; Shen, H.; Kapesa, L.; Zeng, S. Lauren classification and individualized chemotherapy in gastric cancer. Oncol. Lett. 2016, 11, 2959–2964. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, J.; Iizasa, H.; Yoshiyama, H.; Shimokuri, K.; Kobayashi, Y.; Sasaki, S.; Nakamura, M.; Yanai, H.; Sakai, K.; Suehiro, Y.; et al. Clinical Importance of Epstein–Barr Virus-Associated Gastric Cancer. Cancers 2018, 10, 167. [Google Scholar] [CrossRef] [PubMed]
- Tavera, G.; Morgan, D.R.; Williams, S.M. Tipping the Scale toward Gastric Disease: A Host-Pathogen Genomic Mismatch? Curr. Genet. Med. Rep. 2018, 6, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.W.T.; Tan, P. Dissection of gastric cancer heterogeneity for precision oncology. Cancer Sci. 2019, 110, 3405–3414. [Google Scholar] [CrossRef]
- Lott, P.C.; Carvajal-Carmona, L.G. Resolving gastric cancer aetiology: An update in genetic predisposition. Lancet Gastroenterol. Hepatol. 2018, 3, 874–883. [Google Scholar] [CrossRef]
- Nagini, S. Carcinoma of the stomach: A review of epidemiology, pathogenesis, molecular genetics and chemoprevention. World J. Gastrointest. Oncol. 2012, 4, 156–169. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, G.; Hu, C. Molecular Classification of Gastric Adenocarcinoma. Gastroenterol. Res. 2019, 12, 275–282. [Google Scholar] [CrossRef]
- Avila, F.; Theoduloz, C.; Lopez-Alarcon, C.; Dorta, E.; Schmeda-Hirschmann, G. Cytoprotective Mechanisms Mediated by Polyphenols from Chilean Native Berries against Free Radical-Induced Damage on AGS Cells. Oxidative Med. Cell. Longev. 2017, 2017, 9808520. [Google Scholar] [CrossRef]
- Kanner, J.; Selhub, J.; Shpaizer, A.; Rabkin, B.; Shacham, I.; Tirosh, O. Redox homeostasis in stomach medium by foods: The Postprandial Oxidative Stress Index (POSI) for balancing nutrition and human health. Redox Biol. 2017, 12, 929–936. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef]
- Everett, S.M.; Singh, R.; Leuratti, C.; White, K.L.; Neville, P.; Greenwood, D.; Marnett, L.J.; Schorah, C.J.; Forman, D.; Shuker, D.; et al. Levels of malondialdehyde-deoxyguanosine in the gastric mucosa: Relationship with lipid peroxidation, ascorbic acid, and Helicobacter pylori. Cancer Epidemiol. Biomark. Prev. 2001, 10, 369–376. [Google Scholar]
- Deng, R.; Mo, F.; Chang, B.; Zhang, Q.; Ran, H.; Yang, S.; Zhu, Z.; Hu, L.; Su, Q. Glucose-derived AGEs enhance human gastric cancer metastasis through RAGE/ERK/Sp1/MMP2 cascade. Oncotarget 2017, 8, 104216–104226. [Google Scholar] [CrossRef] [PubMed]
- Zoidis, E.; Seremelis, I.; Kontopoulos, N.; Danezis, G.P. Selenium-Dependent Antioxidant Enzymes: Actions and Properties of Selenoproteins. Antioxidants 2018, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Pasupathi, P.; Saravanan, G.; Chinnaswamy, P.; Bakthavathsalam, G. Effect of chronic smoking on lipid peroxidation and antioxidant status in gastric carcinoma patients. Indian J. Gastroenterol. Off. J. Indian Soc. Gastroenterol. 2009, 28, 65–67. [Google Scholar] [CrossRef]
- Abotaleb, M.; Liskova, A.; Kubatka, P.; Büsselberg, D. Therapeutic Potential of Plant Phenolic Acids in the Treatment of Cancer. Biomolecules 2020, 10, 221. [Google Scholar] [CrossRef]
- Carr, A.C.; Vissers, M.C. Synthetic or food-derived vitamin C—Are they equally bioavailable? Nutrients 2013, 5, 4284–4304. [Google Scholar] [CrossRef]
- Kamiya, Y.; Ohta, Y.; Imai, Y.; Arisawa, T.; Nakano, H. A critical role of gastric mucosal ascorbic acid in the progression of acute gastric mucosal lesions induced by compound 48/80 in rats. World J. Gastroenterol. WJG 2005, 11, 1324–1332. [Google Scholar] [CrossRef]
- Blaszczak, W.; Barczak, W.; Masternak, J.; Kopczyński, P.; Zhitkovich, A.; Rubiś, B. Vitamin C as a Modulator of the Response to Cancer Therapy. Molecules 2019, 24, 453. [Google Scholar] [CrossRef]
- Carr, A.; Frei, B. Does vitamin C act as a pro-oxidant under physiological conditions? FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1999, 13, 1007–1024. [Google Scholar] [CrossRef]
- Cui, J.; Pan, Y.H.; Zhang, Y.; Jones, G.; Zhang, S. Progressive pseudogenization: Vitamin C synthesis and its loss in bats. Mol. Biol. Evol. 2011, 28, 1025–1031. [Google Scholar] [CrossRef]
- Aditi, A.; Graham, D.Y. Vitamin C, gastritis, and gastric disease: A historical review and update. Dig. Dis. Sci. 2012, 57, 2504–2515. [Google Scholar] [CrossRef] [PubMed]
- Tuo, B.G.; Yan, Y.H.; Ge, Z.L.; Ou, G.W.; Zhao, K. Ascorbic acid secretion in the human stomach and the effect of gastrin. World J. Gastroenterol. WJG 2000, 6, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Wohlrab, C.; Phillips, E.; Dachs, G.U. Vitamin C Transporters in Cancer: Current Understanding and Gaps in Knowledge. Front. Oncol. 2017, 7, 74. [Google Scholar] [CrossRef] [PubMed]
- Rood, J.C.; Ruiz, B.; Fontham, E.T.; Malcom, G.T.; Hunter, F.M.; Sobhan, M.; Johnson, W.D.; Correa, P. Helicobacter pylori-associated gastritis and the ascorbic acid concentration in gastric juice. Nutr. Cancer 1994, 22, 65–72. [Google Scholar] [CrossRef]
- Grollman, A.P.; Lehninger, A.L. Enzymic synthesis of L-ascorbic acid in different animal species. Arch. Biochem. Biophys. 1957, 69, 458–467. [Google Scholar] [CrossRef]
- Drouin, G.; Godin, J.R.; Page, B. The genetics of vitamin C loss in vertebrates. Curr. Genom. 2011, 12, 371–378. [Google Scholar] [CrossRef]
- Tsujii, M.; Kawano, S.; Tsuji, S.; Fusamoto, H.; Kamada, T.; Sato, N. Mechanism of gastric mucosal damage induced by ammonia. Gastroenterology 1992, 102, 1881–1888. [Google Scholar] [CrossRef]
- Valenzuela-Valderrama, M.; Cerda-Opazo, P.; Backert, S.; González, M.F.; Carrasco-Véliz, N.; Jorquera-Cordero, C.; Wehinger, S.; Canales, J.; Bravo, D.; Quest, A.F.G. The Helicobacter pylori Urease Virulence Factor Is Required for the Induction of Hypoxia-Induced Factor-1α in Gastric Cells. Cancers 2019, 11, 799. [Google Scholar] [CrossRef]
- Wen, Y.; Scott, D.R.; Vagin, O.; Tokhtaeva, E.; Marcus, E.A.; Sachs, G. Measurement of Internal pH in Helicobacter pylori by Using Green Fluorescent Protein Fluorimetry. J. Bacteriol. 2018, 200. [Google Scholar] [CrossRef]
- Scott, D.R.; Marcus, E.A.; Wen, Y.; Singh, S.; Feng, J.; Sachs, G. Cytoplasmic histidine kinase (HP0244)-regulated assembly of urease with UreI, a channel for urea and its metabolites, CO2, NH3, and NH4(+), is necessary for acid survival of Helicobacter pylori. J. Bacteriol. 2010, 192, 94–103. [Google Scholar] [CrossRef]
- Stingl, K.; Uhlemann, E.M.; Schmid, R.; Altendorf, K.; Bakker, E.P. Energetics of Helicobacter pylori and its implications for the mechanism of urease-dependent acid tolerance at pH 1. J. Bacteriol. 2002, 184, 3053–3060. [Google Scholar] [CrossRef] [PubMed][Green Version]
- McGee, D.J.; Zabaleta, J.; Viator, R.J.; Testerman, T.L.; Ochoa, A.C.; Mendz, G.L. Purification and characterization of Helicobacter pylori arginase, RocF: Unique features among the arginase superfamily. Eur. J. Biochem. 2004, 271, 1952–1962. [Google Scholar] [CrossRef] [PubMed]
- Schoep, T.D.; Fulurija, A.; Good, F.; Lu, W.; Himbeck, R.P.; Schwan, C.; Choi, S.S.; Berg, D.E.; Mittl, P.R.; Benghezal, M.; et al. Surface properties of Helicobacter pylori urease complex are essential for persistence. PLoS ONE 2010, 5, e15042. [Google Scholar] [CrossRef] [PubMed]
- Beil, W.; Sewing, K.F.; Busche, R.; Wagner, S. Helicobacter pylori augments the acid inhibitory effect of omeprazole on parietal cells and gastric H(+)/K(+)-ATPase. Gut 2001, 48, 157–162. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Waldum, H.L.; Kleveland, P.M.; Sørdal, Ø.F. Helicobacter pylori and gastric acid: An intimate and reciprocal relationship. Ther. Adv. Gastroenterol. 2016, 9, 836–844. [Google Scholar] [CrossRef]
- Yao, X.; Smolka, A.J. Gastric Parietal Cell Physiology and Helicobacter pylori-Induced Disease. Gastroenterology 2019, 156, 2158–2173. [Google Scholar] [CrossRef]
- Bockerstett, K.A.; Osaki, L.H.; Petersen, C.P.; Cai, C.W.; Wong, C.F.; Nguyen, T.M.; Ford, E.L.; Hoft, D.F.; Mills, J.C.; Goldenring, J.R.; et al. Interleukin-17A Promotes Parietal Cell Atrophy by Inducing Apoptosis. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 678–690.e1. [Google Scholar] [CrossRef]
- Ling, S.S.; Khoo, L.H.; Hwang, L.A.; Yeoh, K.G.; Ho, B. Instrumental Role of Helicobacter pylori gamma-Glutamyl Transpeptidase in VacA-Dependent Vacuolation in Gastric Epithelial Cells. PLoS ONE 2015, 10, e0131460. [Google Scholar] [CrossRef]
- Keilberg, D.; Ottemann, K.M. How Helicobacter pylori senses, targets and interacts with the gastric epithelium. Environ. Microbiol. 2016, 18, 791–806. [Google Scholar] [CrossRef]
- Haley, K.P.; Gaddy, J.A. Nutrition and Helicobacter pylori: Host Diet and Nutritional Immunity Influence Bacterial Virulence and Disease Outcome. Gastroenterol. Res. Pract. 2016, 2016, 3019362. [Google Scholar] [CrossRef]
- Machuca, M.A.; Johnson, K.S.; Liu, Y.C.; Steer, D.L.; Ottemann, K.M.; Roujeinikova, A. Helicobacter pylori chemoreceptor TlpC mediates chemotaxis to lactate. Sci. Rep. 2017, 7, 14089. [Google Scholar] [CrossRef] [PubMed]
- Perkins, A.; Tudorica, D.A.; Amieva, M.R.; Remington, S.J.; Guillemin, K. Helicobacter pylori senses bleach (HOCl) as a chemoattractant using a cytosolic chemoreceptor. PLoS Biol. 2019, 17, e3000395. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.Y.; Goers Sweeney, E.; Guillemin, K.; Amieva, M.R. Multiple Acid Sensors Control Helicobacter pylori Colonization of the Stomach. PLoS Pathog. 2017, 13, e1006118. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.; Konradt, M.; Groll, C.; Scheid, P.; Hanauer, G.; Werling, H.O.; Josenhans, C.; Suerbaum, S. The spatial orientation of Helicobacter pylori in the gastric mucus. Proc. Natl. Acad. Sci. USA 2004, 101, 5024–5029. [Google Scholar] [CrossRef]
- Wilson, R.B. Hypoxia, cytokines and stromal recruitment: Parallels between pathophysiology of encapsulating peritoneal sclerosis, endometriosis and peritoneal metastasis. Pleura Peritoneum. 2018, 3, 20180103. [Google Scholar] [CrossRef]
- Marshall, B.J. One Hundred Years of Discovery and Rediscovery of Helicobacter pylori and Its Association with Peptic Ulcer Disease. In Helicobacter Pylori: Physiology and Genetics; Mobley, H.L.T., Mendz, G.L., Hazell, S.L., Eds.; ASM Press: Washington, DC, USA, 2001. [Google Scholar]
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef]
- Furuta, T.; El-Omar, E.M.; Xiao, F.; Shirai, N.; Takashima, M.; Sugimura, H. Interleukin 1β polymorphisms increase risk of hypochlorhydria and atrophic gastritis and reduce risk of duodenal ulcer recurrence in Japan. Gastroenterology 2002, 123, 92–105. [Google Scholar] [CrossRef]
- Bang, C.S.; Lee, J.J.; Baik, G.H. Prediction of Chronic Atrophic Gastritis and Gastric Neoplasms by Serum Pepsinogen Assay: A Systematic Review and Meta-Analysis of Diagnostic Test Accuracy. J. Clin. Med. 2019, 8, 657. [Google Scholar] [CrossRef]
- Eslick, G.D. Helicobacter pylori infection causes gastric cancer? A review of the epidemiological, meta-analytic, and experimental evidence. World J. Gastroenterol. WJG 2006, 12, 2991–2999. [Google Scholar] [CrossRef]
- Gonzalez, C.A.; Megraud, F.; Buissonniere, A.; Lujan Barroso, L.; Agudo, A.; Duell, E.J.; Boutron-Ruault, M.C.; Clavel-Chapelon, F.; Palli, D.; Krogh, V.; et al. Helicobacter pylori infection assessed by ELISA and by immunoblot and noncardia gastric cancer risk in a prospective study: The Eurgast-EPIC project. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. ESMO 2012, 23, 1320–1324. [Google Scholar] [CrossRef]
- Kim, S.S.; Ruiz, V.E.; Carroll, J.D.; Moss, S.F. Helicobacter pylori in the pathogenesis of gastric cancer and gastric lymphoma. Cancer Lett. 2011, 305, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Kaczanowska, S.; Davila, E. IL-1 Receptor-Associated Kinase Signaling and Its Role in Inflammation, Cancer Progression, and Therapy Resistance. Front. Immunol. 2014, 5, 553. [Google Scholar] [CrossRef]
- Peleteiro, B.; Lunet, N. Gastritis and Gastric Cancer-New Insights into Gastroprotection, Diagnosis and Treatment; Tonino, P., Ed.; InTech: London, UK, 2011; Available online: https://www.researchgate.net/publication/259391426_Gastritis_and_Gastric_Cancer_-_New_Insights_in_Gastroprotection_Diagnosis_and_Treatments_Edited_by_Paola_Tonino_ISBN_978-953-307-375-0_296_pages_Publisher_InTech_Chapters_published_September_15_2011_u (accessed on 8 February 2020).
- Youn, H.S.; Ko, G.H.; Chung, M.H.; Lee, W.K.; Cho, M.J.; Rhee, K.H. Pathogenesis and prevention of stomach cancer. J. Korean Med. Sci. 1996, 11, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Knaapen, A.M.; Güngör, N.; Schins, R.P.; Borm, P.J.; Van Schooten, F.J. Neutrophils and respiratory tract DNA damage and mutagenesis: A review. Mutagenesis 2006, 21, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Marcus, E.A.; Sachs, G.; Scott, D.R. Acid-regulated gene expression of Helicobacter pylori: Insight into acid protection and gastric colonization. Helicobacter 2018, 23, e12490. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, T.; Liao, T.; Debowski, A.W.; Nilsson, H.O.; Fulurija, A.; Haslam, S.M.; Mulloy, B.; Dell, A.; Stubbs, K.A.; et al. The redefinition of Helicobacter pylori lipopolysaccharide O-antigen and core-oligosaccharide domains. PLoS Pathog. 2017, 13, e1006280. [Google Scholar] [CrossRef]
- Allison, C.C.; Ferrero, R.L. Role of virulence factors and host cell signaling in the recognition of Helicobacter pylori and the generation of immune responses. Future Microbiol. 2010, 5, 1233–1255. [Google Scholar] [CrossRef]
- Yao, Y.; Tao, H.; Park, D.I.; Sepulveda, J.L.; Sepulveda, A.R. Demonstration and characterization of mutations induced by Helicobacter pylori organisms in gastric epithelial cells. Helicobacter 2006, 11, 272–286. [Google Scholar] [CrossRef]
- Wang, Y.K.; Chiang, W.C.; Kuo, F.C.; Wu, M.C.; Shih, H.Y.; Wang, S.S.W.; Liu, C.J.; Chen, Y.H.; Wu, D.C.; Su, W.W.; et al. Levels of malondialdehyde in the gastric juice: Its association with Helicobacter pylori infection and stomach diseases. Helicobacter 2018, 23, e12460. [Google Scholar] [CrossRef]
- Ding, S.Z.; Goldberg, J.B.; Hatakeyama, M. Helicobacter pylori infection, oncogenic pathways and epigenetic mechanisms in gastric carcinogenesis. Future Oncol. 2010, 6, 851–862. [Google Scholar] [CrossRef]
- Muhammad, J.S.; Eladl, M.A.; Khoder, G. Helicobacter pylori-induced DNA Methylation as an Epigenetic Modulator of Gastric Cancer: Recent Outcomes and Future Direction. Pathogens 2019, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Xu, J.; Ji, W.; Wang, L.; Wang, A. Upregulation of TMEFF2 is involved in the antiproliferative effects of vitamin C and tyrphostin AG490 on GES-1 and AGS cells. Oncol. Lett. 2019, 17, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Lee, H.; Lim, J.W.; Kim, H. Inhibitory Effect of β-Carotene on Helicobacter pylori-Induced TRAF Expression and Hyper-Proliferation in Gastric Epithelial Cells. Antioxidants 2019, 8, 637. [Google Scholar] [CrossRef] [PubMed]
- da Costa, D.M.; Pereira Edos, S.; Rabenhorst, S.H. What exists beyond cagA and vacA? Helicobacter pylori genes in gastric diseases. World J. Gastroenterol. WJG 2015, 21, 10563–10572. [Google Scholar] [CrossRef]
- Chang, C.C.; Kuo, W.S.; Chen, Y.C.; Perng, C.L.; Lin, H.J.; Ou, Y.H. Fragmentation of CagA Reduces Hummingbird Phenotype Induction by Helicobactor pylori. PLoS ONE 2016, 11, e0150061. [Google Scholar] [CrossRef]
- Vaziri, F.; Peerayeh, S.N.; Alebouyeh, M.; Maghsoudi, N.; Azimzadeh, P.; Siadat, S.D.; Zali, M.R. Novel effects of Helicobacter pylori CagA on key genes of gastric cancer signal transduction: A comparative transfection study. Pathog. Dis. 2015, 73. [Google Scholar] [CrossRef]
- Roebuck, K.A. Regulation of interleukin-8 gene expression. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 1999, 19, 429–438. [Google Scholar] [CrossRef]
- Tegtmeyer, N.; Wessler, S.; Necchi, V.; Rohde, M.; Harrer, A.; Rau, T.T.; Asche, C.I.; Boehm, M.; Loessner, H.; Figueiredo, C.; et al. Helicobacter pylori Employs a Unique Basolateral Type IV Secretion Mechanism for CagA Delivery. Cell Host. Microbe 2017, 22, 552–560.e5. [Google Scholar] [CrossRef]
- Pero, R.; Coretti, L.; Nigro, E.; Lembo, F.; Laneri, S.; Lombardo, B.; Daniele, A.; Scudiero, O. β-Defensins in the Fight against Helicobacter pylori. Molecules 2017, 22, 424. [Google Scholar] [CrossRef]
- Vergara, D.; Simeone, P.; Damato, M.; Maffia, M.; Lanuti, P.; Trerotola, M. The Cancer Microbiota: EMT and Inflammation as Shared Molecular Mechanisms Associated with Plasticity and Progression. J. Oncol. 2019, 2019, 1253727. [Google Scholar] [CrossRef]
- Schneider, B.G.; Peng, D.F.; Camargo, M.C.; Piazuelo, M.B.; Sicinschi, L.A.; Mera, R.; Romero-Gallo, J.; Delgado, A.G.; Bravo, L.E.; Wilson, K.T.; et al. Promoter DNA hypermethylation in gastric biopsies from subjects at high and low risk for gastric cancer. Int. J. Cancer 2010, 127, 2588–2597. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.O.; Peng, J.Z.; Lam, S.K.; Lai, K.C.; Yuen, M.F.; Cheung, H.K.; Kwong, Y.L.; Rashid, A.; Chan, C.K.; Wong, B.C. Eradication of Helicobacter pylori infection reverses E-cadherin promoter hypermethylation. Gut 2006, 55, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhang, X.; Jin, M.; Hu, L.; Zang, M.; Qiu, W.; Wang, S.; Liu, B.; Liu, S.; Guo, D. CagA increases DNA methylation and decreases PTEN expression in human gastric cancer. Mol. Med. Rep. 2019, 19, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Starczak, M.; Zarakowska, E.; Modrzejewska, M.; Dziaman, T.; Szpila, A.; Linowiecka, K.; Guz, J.; Szpotan, J.; Gawronski, M.; Labejszo, A.; et al. In vivo evidence of ascorbate involvement in the generation of epigenetic DNA modifications in leukocytes from patients with colorectal carcinoma, benign adenoma and inflammatory bowel disease. J. Transl. Med. 2018, 16, 204. [Google Scholar] [CrossRef]
- Pawlowska, E.; Szczepanska, J.; Blasiak, J. Pro- and Antioxidant Effects of Vitamin C in Cancer in correspondence to Its Dietary and Pharmacological Concentrations. Oxidative Med. Cell. Longev. 2019, 2019, 7286737. [Google Scholar] [CrossRef]
- Lee Chong, T.; Ahearn, E.L.; Cimmino, L. Reprogramming the Epigenome With Vitamin C. Front. Cell Dev. Biol. 2019, 7, 128. [Google Scholar] [CrossRef]
- Zeng, X.Q.; Wang, J.; Chen, S.Y. Methylation modification in gastric cancer and approaches to targeted epigenetic therapy (Review). Int. J. Oncol. 2017, 50, 1921–1933. [Google Scholar] [CrossRef]
- Chichirau, B.E.; Diechler, S.; Posselt, G.; Wessler, S. Tyrosine Kinases in Helicobacter pylori Infections and Gastric Cancer. Toxins 2019, 11, 591. [Google Scholar] [CrossRef]
- Takahashi-Kanemitsu, A.; Knight, C.T.; Hatakeyama, M. Molecular anatomy and pathogenic actions of Helicobacter pylori CagA that underpin gastric carcinogenesis. Cell. Mol. Immunol. 2020, 17, 50–63. [Google Scholar] [CrossRef]
- Shi, J.; Qu, Y.P.; Hou, P. Pathogenetic mechanisms in gastric cancer. World J. Gastroenterol. WJG 2014, 20, 13804–13819. [Google Scholar] [CrossRef]
- Baj, J.; Korona-Głowniak, I.; Forma, A.; Maani, A.; Sitarz, E.; Rahnama-Hezavah, M.; Radzikowska, E.; Portincasa, P. Mechanisms of the Epithelial-Mesenchymal Transition and Tumor Microenvironment in Helicobacter pylori-Induced Gastric Cancer. Cells 2020, 9, 1055. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.I.; Yoon, C.; Park, M.R.; Lee, D.; Kook, M.C.; Lin, J.X.; Kang, J.H.; Ashktorab, H.; Smoot, D.T.; Yoon, S.S.; et al. CDX1 Expression Induced by CagA-Expressing Helicobacter pylori Promotes Gastric Tumorigenesis. Mol. Cancer Res. 2019, 17, 2169–2183. [Google Scholar] [CrossRef]
- Murata-Kamiya, N.; Kurashima, Y.; Teishikata, Y.; Yamahashi, Y.; Saito, Y.; Higashi, H.; Aburatani, H.; Akiyama, T.; Peek, R.M., Jr.; Azuma, T.; et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the beta-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene 2007, 26, 4617–4626. [Google Scholar] [CrossRef] [PubMed]
- Buda, A.; Pignatelli, M. E-cadherin and the cytoskeletal network in colorectal cancer development and metastasis. Cell Commun. Adhes 2011, 18, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Devaux, C.A.; Mezouar, S.; Mege, J.L. The E-Cadherin Cleavage Associated to Pathogenic Bacteria Infections Can Favor Bacterial Invasion and Transmigration, Dysregulation of the Immune Response and Cancer Induction in Humans. Front Microbiol. 2019, 10, 2598. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Zhang, P.Y.; Aboul-Soud, M.A. From inflammation to gastric cancer: Role of Helicobacter pylori. Oncol. Lett. 2017, 13, 543–548. [Google Scholar] [CrossRef]
- Molina Castro, S.E.; Tiffon, C.; Giraud, J.; Boeuf, H.; Sifre, E.; Giese, A.; Belleannee, G.; Lehours, P.; Bessede, E.; Megraud, F.; et al. The Hippo Kinase LATS2 Controls Helicobacter pylori-Induced Epithelial-Mesenchymal Transition and Intestinal Metaplasia in Gastric Mucosa. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 257–276. [Google Scholar] [CrossRef]
- Wada, Y.; Takemura, K.; Tummala, P.; Uchida, K.; Kitagaki, K.; Furukawa, A.; Ishige, Y.; Ito, T.; Hara, Y.; Suzuki, T.; et al. Helicobacter pylori induces somatic mutations in TP53 via overexpression of CHAC1 in infected gastric epithelial cells. FEBS Open Bio 2018, 8, 671–679. [Google Scholar] [CrossRef]
- Ruggiero, P. Helicobacter pylori infection: What’s new. Curr. Opin. Infect. Dis. 2012, 25, 337–344. [Google Scholar] [CrossRef]
- Lee, D.Y.; Jung, D.E.; Yu, S.S.; Lee, Y.S.; Choi, B.K.; Lee, Y.C. Regulation of SIRT3 signal related metabolic reprogramming in gastric cancer by Helicobacter pylori oncoprotein CagA. Oncotarget 2017, 8, 78365–78378. [Google Scholar] [CrossRef]
- Kido, M.; Watanabe, N.; Aoki, N.; Iwamoto, S.; Nishiura, H.; Maruoka, R.; Ikeda, A.; Azuma, T.; Chiba, T. Dual roles of CagA protein in Helicobacterpylori-induced chronic gastritis in mice. Biochem. Biophys. Res. Commun. 2011, 412, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.W.; Patchett, S.E.; Perrett, D.; Katelaris, P.H.; Domizio, P.; Farthing, M.J. The relation between gastric vitamin C concentrations, mucosal histology, and CagA seropositivity in the human stomach. Gut 1998, 43, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Meliț, L.E.; Mărginean, C.O.; Mărginean, C.D.; Mărginean, M.O. The Relationship between Toll-like Receptors and Helicobacter pylori-Related Gastropathies: Still a Controversial Topic. J. Immunol. Res. 2019, 2019, 8197048. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Cong, X.; Gao, H.; Lan, X.; Li, Z.; Wang, W.; Song, S.; Wang, Y.; Li, C.; Zhang, H.; et al. Tumor-associated neutrophils induce EMT by IL-17a to promote migration and invasion in gastric cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, L.; Guo, Z.; Liu, L.; Ji, J.; Zhang, J.; Chen, X.; Liu, B.; Zhang, J.; Ding, Q.; et al. A unique feature of iron loss via close adhesion of Helicobacter pylori to host erythrocytes. PLoS ONE 2012, 7, e50314. [Google Scholar] [CrossRef]
- Poplawski, T.; Chojnacki, C.; Czubatka, A.; Klupinska, G.; Chojnacki, J.; Blasiak, J. Helicobacter pylori infection and antioxidants can modulate the genotoxic effects of heterocyclic amines in gastric mucosa cells. Mol. Biol. Rep. 2013, 40, 5205–5212. [Google Scholar] [CrossRef]
- Fahimi, F.; Tohidkia, M.R.; Fouladi, M.; Aghabeygi, R.; Samadi, N.; Omidi, Y. Pleiotropic cytotoxicity of VacA toxin in host cells and its impact on immunotherapy. Bioimpacts 2017, 7, 59–71. [Google Scholar] [CrossRef]
- Bridge, D.R.; Merrell, D.S. Polymorphism in the Helicobacter pylori CagA and VacA toxins and disease. Gut Microbes 2013, 4, 101–117. [Google Scholar] [CrossRef]
- Palframan, S.L.; Kwok, T.; Gabriel, K. Vacuolating cytotoxin A (VacA), a key toxin for Helicobacter pylori pathogenesis. Front. Cell. Infect. Microbiol. 2012, 2, 92. [Google Scholar] [CrossRef]
- Mahdavi, J.; Sonden, B.; Hurtig, M.; Olfat, F.O.; Forsberg, L.; Roche, N.; Angstrom, J.; Larsson, T.; Teneberg, S.; Karlsson, K.A.; et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 2002, 297, 573–578. [Google Scholar] [CrossRef]
- Su, Y.L.; Huang, H.L.; Huang, B.S.; Chen, P.C.; Chen, C.S.; Wang, H.L.; Lin, P.H.; Chieh, M.S.; Wu, J.J.; Yang, J.C.; et al. Combination of OipA, BabA, and SabA as candidate biomarkers for predicting Helicobacter pylori-related gastric cancer. Sci. Rep. 2016, 6, 36442. [Google Scholar] [CrossRef] [PubMed]
- Wiener, A.S. Blood-groups and disease. A critical review. Lancet 1962, 1, 813–816. [Google Scholar] [CrossRef]
- Horwich, L.; Evans, D.A.; McConnell, R.B.; Donohoe, W.T. ABO blood groups in gastric bleeding. Gut 1966, 7, 680–685. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Edgren, G.; Hjalgrim, H.; Rostgaard, K.; Norda, R.; Wikman, A.; Melbye, M.; Nyren, O. Risk of gastric cancer and peptic ulcers in relation to ABO blood type: A cohort study. Am. J. Epidemiol. 2010, 172, 1280–1285. [Google Scholar] [CrossRef] [PubMed]
- Alkout, A.M.; Blackwell, C.C.; Weir, D.M. Increased inflammatory responses of persons of blood group O to Helicobacter pylori. J. Infect. Dis. 2000, 181, 1364–1369. [Google Scholar] [CrossRef]
- Nakao, M.; Matsuo, K.; Ito, H.; Shitara, K.; Hosono, S.; Watanabe, M.; Ito, S.; Sawaki, A.; Iida, S.; Sato, S.; et al. ABO genotype and the risk of gastric cancer, atrophic gastritis, and Helicobacter pylori infection. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1665–1672. [Google Scholar] [CrossRef]
- Kurtenkov, O.; Klaamas, K. Hidden IgG Antibodies to the Tumor-Associated Thomsen-Friedenreich Antigen in Gastric Cancer Patients: Lectin Reactivity, Avidity, and Clinical Relevance. Biomed. Res. Int. 2017, 2017, 6097647. [Google Scholar] [CrossRef]
- Jaff, M.S. Relation between ABO blood groups and Helicobacter pylori infection in symptomatic patients. Clin. Exp. Gastroenterol. 2011, 4, 221–226. [Google Scholar] [CrossRef]
- Sievers, M.L. Hereditary aspects of gastric secretory function; race and ABO blood groups in relationship to acid and pepsin production. Am. J. Med. 1959, 27, 246–255. [Google Scholar] [CrossRef]
- Akatsuka, S.; Yamashita, Y.; Ohara, H.; Liu, Y.T.; Izumiya, M.; Abe, K.; Ochiai, M.; Jiang, L.; Nagai, H.; Okazaki, Y.; et al. Fenton reaction induced cancer in wild type rats recapitulates genomic alterations observed in human cancer. PLoS ONE 2012, 7, e43403. [Google Scholar] [CrossRef]
- Conteduca, V.; Sansonno, D.; Lauletta, G.; Russi, S.; Ingravallo, G.; Dammacco, F. H. pylori infection and gastric cancer: State of the art (review). Int. J. Oncol. 2013, 42, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Correa, P.; Houghton, J. Carcinogenesis of Helicobacter pylori. Gastroenterology 2007, 133, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Minalyan, A.; Benhammou, J.N.; Artashesyan, A.; Lewis, M.S.; Pisegna, J.R. Autoimmune atrophic gastritis: Current perspectives. Clin. Exp. Gastroenterol. 2017, 10, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Cavalcoli, F.; Zilli, A.; Conte, D.; Massironi, S. Micronutrient deficiencies in patients with chronic atrophic autoimmune gastritis: A review. World J. Gastroenterol. WJG 2017, 23, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Tabrez, E.; Peela, J.; Honnavar, P.D.; Tabrez, S.S.M. Vitamin C: A Preventative, Therapeutic Agent Against Helicobacter pylori. Cureus 2018, 10, e3062. [Google Scholar] [CrossRef]
- Feiz, H.R.; Mobarhan, S. Does vitamin C intake slow the progression of gastric cancer in Helicobacter pylori-infected populations? Nutr. Rev. 2002, 60, 34–36. [Google Scholar] [PubMed]
- Waring, A.J.; Drake, I.M.; Schorah, C.J.; White, K.L.; Lynch, D.A.; Axon, A.T.; Dixon, M.F. Ascorbic acid and total vitamin C concentrations in plasma, gastric juice, and gastrointestinal mucosa: Effects of gastritis and oral supplementation. Gut 1996, 38, 171–176. [Google Scholar] [CrossRef]
- Rokkas, T.; Papatheodorou, G.; Karameris, A.; Mavrogeorgis, A.; Kalogeropoulos, N.; Giannikos, N. Helicobacter pylori infection and gastric juice vitamin C levels. Impact of eradication. Dig. Dis. Sci. 1995, 40, 615–621. [Google Scholar] [CrossRef]
- Banerjee, S.; Hawksby, C.; Miller, S.; Dahill, S.; Beattie, A.D.; McColl, K.E. Effect of Helicobacter pylori and its eradication on gastric juice ascorbic acid. Gut 1994, 35, 317–322. [Google Scholar] [CrossRef]
- Sobala, G.M.; Schorah, C.J.; Shires, S.; Lynch, D.A.; Gallacher, B.; Dixon, M.F.; Axon, A.T. Effect of eradication of Helicobacter pylori on gastric juice ascorbic acid concentrations. Gut 1993, 34, 1038–1041. [Google Scholar] [CrossRef]
- Annibale, B.; Capurso, G.; Lahner, E.; Passi, S.; Ricci, R.; Maggio, F.; Delle Fave, G. Concomitant alterations in intragastric pH and ascorbic acid concentration in patients with Helicobacter pylori gastritis and associated iron deficiency anaemia. Gut 2003, 52, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.J.; Yu, L.Z.; Bai, J.F.; Peng, Y.S.; Sun, G.; Zhao, H.L.; Miu, K.; Lu, X.Z.; Zhang, X.Y.; Zhao, Z.Q. Multiple genetic alterations and behavior of cellular biology in gastric cancer and other gastric mucosal lesions:H. pylori infection, histological types and staging. World J. Gastroenterol. WJG 2000, 6, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.Q.; Xu, X.Y.; Shang, A.; Gan, R.Y.; Wu, D.T.; Atanasov, A.G.; Li, H.B. Phytochemicals for the Prevention and Treatment of Gastric Cancer: Effects and Mechanisms. Int. J. Mol. Sci. 2020, 21, 570. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Wei, J.; He, X.; An, P.; Wang, H.; Jiang, L.; Shao, D.; Liang, H.; Li, Y.; Wang, F.; et al. Landscape of dietary factors associated with risk of gastric cancer: A systematic review and dose-response meta-analysis of prospective cohort studies. Eur. J. Cancer 2015, 51, 2820–2832. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Cai, Q.; Geng, Q.; Wang, J.; Lan, Y.; Zhan, Y.; Xu, D. Vitamin intake reduce the risk of gastric cancer: Meta-analysis and systematic review of randomized and observational studies. PLoS ONE 2014, 9, e116060. [Google Scholar] [CrossRef]
- Sasazuki, S.; Sasaki, S.; Tsubono, Y.; Okubo, S.; Hayashi, M.; Kakizoe, T.; Tsugane, S. The effect of 5-year vitamin C supplementation on serum pepsinogen level and Helicobacter pylori infection. Cancer Sci. 2003, 94, 378–382. [Google Scholar] [CrossRef]
- Mera, R.; Fontham, E.T.; Bravo, L.E.; Bravo, J.C.; Piazuelo, M.B.; Camargo, M.C.; Correa, P. Long term follow up of patients treated for Helicobacter pylori infection. Gut 2005, 54, 1536–1540. [Google Scholar] [CrossRef]
- Bjelakovic, G.; Nikolova, D.; Simonetti, R.G.; Gluud, C. Antioxidant supplements for prevention of gastrointestinal cancers: A systematic review and meta-analysis. Lancet 2004, 364, 1219–1228. [Google Scholar] [CrossRef]
- López-Carrillo, L.; López-Cervantes, M.; Ward, M.H.; Bravo-Alvarado, J.; Ramírez-Espitia, A. Nutrient intake and gastric cancer in Mexico. Int. J. Cancer 1999, 83, 601–605. [Google Scholar] [CrossRef]
- Pelucchi, C.; Tramacere, I.; Bertuccio, P.; Tavani, A.; Negri, E.; La Vecchia, C. Dietary intake of selected micronutrients and gastric cancer risk: An Italian case-control study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. ESMO 2009, 20, 160–165. [Google Scholar] [CrossRef]
- Epplein, M.; Shu, X.O.; Xiang, Y.B.; Chow, W.H.; Yang, G.; Li, H.L.; Ji, B.T.; Cai, H.; Gao, Y.T.; Zheng, W. Fruit and vegetable consumption and risk of distal gastric cancer in the Shanghai Women’s and Men’s Health studies. Am. J. Epidemiol. 2010, 172, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cai, H.; Sasazuki, S.; Tsugane, S.; Zheng, W.; Cho, E.R.; Jee, S.H.; Michel, A.; Pawlita, M.; Xiang, Y.B.; et al. Fruit and vegetable consumption, Helicobacter pylori antibodies, and gastric cancer risk: A pooled analysis of prospective studies in China, Japan, and Korea. Int. J. Cancer 2017, 140, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, S.; Yonezawa, H.; Osaki, T. Role of Probiotics in Eradication Therapy for Helicobacter pylori Infection. Adv. Exp. Med. Biol. 2019, 1149, 243–255. [Google Scholar] [PubMed]
- Eslami, M.; Yousefi, B.; Kokhaei, P.; Jazayeri Moghadas, A.; Sadighi Moghadam, B.; Arabkari, V.; Niazi, Z. Are probiotics useful for therapy of Helicobacter pylori diseases? Comp. Immunol. Microbiol. Infect. Dis. 2019, 64, 99–108. [Google Scholar] [CrossRef]
- Song, H.; Zhou, L.; Liu, D.; Ge, L.; Li, Y. Probiotic effect on Helicobacter pylori attachment and inhibition of inflammation in human gastric epithelial cells. Exp. Med. 2019, 18, 1551–1562. [Google Scholar] [CrossRef]
- Ji, J.; Yang, H. Using Probiotics as Supplementation for Helicobacter pylori Antibiotic Therapy. Int. J. Mol. Sci. 2020, 21, 1136. [Google Scholar] [CrossRef]
- Elangovan, A.; Fischer, M. When to use probiotics in luminal gastrointestinal disorders? Curr. Opin. Clin. Nutr. Metab. Care 2020, 23, 336–343. [Google Scholar] [CrossRef]
- Russo, F.; Linsalata, M.; Orlando, A. Probiotics against neoplastic transformation of gastric mucosa: Effects on cell proliferation and polyamine metabolism. World J. Gastroenterol. WJG 2014, 20, 13258–13272. [Google Scholar] [CrossRef]
- Lijinsky, W. N-Nitroso compounds in the diet. Mutat. Res. 1999, 443, 129–138. [Google Scholar] [CrossRef]
- Ohgaki, H.; Takayama, S.; Sugimura, T. Carcinogenicities of heterocyclic amines in cooked food. Mutat. Res. 1991, 259, 399–410. [Google Scholar] [CrossRef]
- Tsugane, S. Salt, salted food intake, and risk of gastric cancer: Epidemiologic evidence. Cancer Sci. 2005, 96, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Poorolajal, J.; Moradi, L.; Mohammadi, Y.; Cheraghi, Z.; Gohari-Ensaf, F. Risk factors for stomach cancer: A systematic review and meta-analysis. Epidemiol. Health 2020, 42, e2020004. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Bergkvist, L.; Wolk, A. Processed meat consumption, dietary nitrosamines and stomach cancer risk in a cohort of Swedish women. Int. J. Cancer 2006, 119, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Wynder, E.L.; Bross, I.J. A study of etiological factors in cancer of the esophagus. Cancer 1961, 14, 389–413. [Google Scholar] [CrossRef]
- Stillwell, W.G.; Glogowski, J.; Xu, H.X.; Wishnok, J.S.; Zavala, D.; Montes, G.; Correa, P.; Tannenbaum, S.R. Urinary excretion of nitrate, N-nitrosoproline, 3-methyladenine, and 7-methylguanine in a Colombian population at high risk for stomach cancer. Cancer Res. 1991, 51, 190–194. [Google Scholar] [PubMed]
- Bouvard, V.; Loomis, D.; Guyton, K.Z.; Grosse, Y.; Ghissassi, F.E.; Benbrahim-Tallaa, L.; Guha, N.; Mattock, H.; Straif, K. Carcinogenicity of consumption of red and processed meat. Lancet Oncol. 2015, 16, 1599–1600. [Google Scholar] [CrossRef]
- Mard, S.A.; Khadem Haghighian, H.; Sebghatulahi, V.; Ahmadi, B. Dietary Factors in Relation to Helicobacter pylori Infection. Gastroenterol. Res. Pract. 2014, 2014, 826910. [Google Scholar] [CrossRef]
- Jakszyn, P.; Bingham, S.; Pera, G.; Agudo, A.; Luben, R.; Welch, A.; Boeing, H.; Del Giudice, G.; Palli, D.; Saieva, C.; et al. Endogenous versus exogenous exposure to N-nitroso compounds and gastric cancer risk in the European Prospective Investigation into Cancer and Nutrition (EPIC-EURGAST) study. Carcinogenesis 2006, 27, 1497–1501. [Google Scholar] [CrossRef]
- Tannenbaum, S.R.; Sinskey, A.J.; Weisman, M.; Bishop, W. Nitrite in human saliva. Its possible relationship to nitrosamine formation. J. Natl. Cancer Inst. 1974, 53, 79–84. [Google Scholar] [CrossRef]
- Tannenbaum, S.R.; Archer, M.C.; Wishnok, J.S.; Bishop, W.W. Nitrosamine formation in human saliva. J. Natl. Cancer Inst. 1978, 60, 251–253. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, Y.; Gerhard, M.; Gao, J.J.; Mejias-Luque, R.; Zhang, L.; Vieth, M.; Ma, J.L.; Bajbouj, M.; Suchanek, S.; et al. Effect of Helicobacter pylori on gastrointestinal microbiota: A population-based study in Linqu, a high-risk area of gastric cancer. Gut 2019. [Google Scholar] [CrossRef]
- Nicastro, H.L.; Ross, S.A.; Milner, J.A. Garlic and onions: Their cancer prevention properties. Cancer Prev. Res. (Phila) 2015, 8, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Kyrtopoulos, S.A.; Pignatelli, B.; Karkanias, G.; Golematis, B.; Esteve, J. Studies in gastric carcinogenesis. V. The effects of ascorbic acid on N-nitroso compound formation in human gastric juice in vivo and in vitro. Carcinogenesis 1991, 12, 1371–1376. [Google Scholar] [CrossRef] [PubMed]
- D’Elia, L.; Rossi, G.; Ippolito, R.; Cappuccio, F.P.; Strazzullo, P. Habitual salt intake and risk of gastric cancer: A meta-analysis of prospective studies. Clin. Nutr. 2012, 31, 489–498. [Google Scholar] [CrossRef]
- Fox, J.G.; Wang, T.C. Dietary factors modulate Helicobacter-associated gastric cancer in rodent models. Toxicol. Pathol. 2014, 42, 162–181. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.F. Helicobacter pylori associated Asian enigma: Does diet deserve distinction? World J. Gastrointest. Oncol. 2016, 8, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Shikata, K.; Kiyohara, Y.; Kubo, M.; Yonemoto, K.; Ninomiya, T.; Shirota, T.; Tanizaki, Y.; Doi, Y.; Tanaka, K.; Oishi, Y.; et al. A prospective study of dietary salt intake and gastric cancer incidence in a defined Japanese population: The Hisayama study. Int. J. Cancer 2006, 119, 196–201. [Google Scholar] [CrossRef]
- Loh, J.T.; Beckett, A.C.; Scholz, M.B.; Cover, T.L. High-Salt Conditions Alter Transcription of Helicobacter pylori Genes Encoding Outer Membrane Proteins. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef]
- Gonzalez, C.A.; Pera, G.; Agudo, A.; Palli, D.; Krogh, V.; Vineis, P.; Tumino, R.; Panico, S.; Berglund, G.; Siman, H.; et al. Smoking and the risk of gastric cancer in the European Prospective Investigation Into Cancer and Nutrition (EPIC). Int. J. Cancer 2003, 107, 629–634. [Google Scholar] [CrossRef]
- Zaridze, D.; Borisova, E.; Maximovitch, D.; Chkhikvadze, V. Alcohol consumption, smoking and risk of gastric cancer: Case-control study from Moscow, Russia. Cancer Causes Control CCC 2000, 11, 363–371. [Google Scholar] [CrossRef]
- Koizumi, Y.; Tsubono, Y.; Nakaya, N.; Kuriyama, S.; Shibuya, D.; Matsuoka, H.; Tsuji, I. Cigarette smoking and the risk of gastric cancer: A pooled analysis of two prospective studies in Japan. Int. J. Cancer 2004, 112, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, K. Tobacco smoke: Involvement of reactive oxygen species and stable free radicals in mechanisms of oxidative damage, carcinogenesis and synergistic effects with other respirable particles. Int. J. Environ. Res. Public Health 2009, 6, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Bohn, S.K.; Myhrstad, M.C.; Thoresen, M.; Holden, M.; Karlsen, A.; Tunheim, S.H.; Erlund, I.; Svendsen, M.; Seljeflot, I.; Moskaug, J.O.; et al. Blood cell gene expression associated with cellular stress defense is modulated by antioxidant-rich food in a randomised controlled clinical trial of male smokers. BMC Med. 2010, 8, 54. [Google Scholar] [CrossRef] [PubMed]
- McCready, D.R.; Clark, L.; Cohen, M.M. Cigarette smoking reduces human gastric luminal prostaglandin E2. Gut 1985, 26, 1192–1196. [Google Scholar] [CrossRef]
- Jarosz, M.; Dzieniszewski, J.; Dabrowska-Ufniarz, E.; Wartanowicz, M.; Ziemlanski, S. Tobacco smoking and vitamin C concentration in gastric juice in healthy subjects and patients with Helicobacter pylori infection. Eur. J. Cancer Prev. 2000, 9, 423–428. [Google Scholar] [CrossRef]
- Gonzalez, C.A.; Lopez-Carrillo, L. Helicobacter pylori, nutrition and smoking interactions: Their impact in gastric carcinogenesis. Scand. J. Gastroenterol. 2010, 45, 6–14. [Google Scholar] [CrossRef]
- Wang, H.; Ma, L.; Li, Y.; Cho, C.H. Exposure to cigarette smoke increases apoptosis in the rat gastric mucosa through a reactive oxygen species-mediated and p53-independent pathway. Free Radic. Biol. Med. 2000, 28, 1125–1131. [Google Scholar] [CrossRef]
- Kneller, R.W.; You, W.C.; Chang, Y.S.; Liu, W.D.; Zhang, L.; Zhao, L.; Xu, G.W.; Fraumeni, J.F., Jr.; Blot, W.J. Cigarette smoking and other risk factors for progression of precancerous stomach lesions. J. Natl. Cancer Inst. 1992, 84, 1261–1266. [Google Scholar] [CrossRef]
- Butt, J.; Varga, M.G.; Wang, T.; Tsugane, S.; Shimazu, T.; Zheng, W.; Abnet, C.C.; Yoo, K.Y.; Park, S.K.; Kim, J.; et al. Smoking, Helicobacter Pylori Serology, and Gastric Cancer Risk in Prospective Studies from China, Japan, and Korea. Cancer Prev. Res. (Phila) 2019, 12, 667–674. [Google Scholar] [CrossRef]
- Han, X.; Xiao, L.; Yu, Y.; Chen, Y.; Shu, H.H. Alcohol consumption and gastric cancer risk: A meta-analysis of prospective cohort studies. Oncotarget 2017, 8, 83237–83245. [Google Scholar] [CrossRef]
- Ma, S.H.; Jung, W.; Weiderpass, E.; Jang, J.; Hwang, Y.; Ahn, C.; Ko, K.P.; Chang, S.H.; Shin, H.R.; Yoo, K.Y.; et al. Impact of alcohol drinking on gastric cancer development according to Helicobacter pylori infection status. Br. J. Cancer 2015, 113, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Moy, K.A.; Fan, Y.; Wang, R.; Gao, Y.T.; Yu, M.C.; Yuan, J.M. Alcohol and tobacco use in relation to gastric cancer: A prospective study of men in Shanghai, China. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2287–2297. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, H.; Nair, J. Accumulation of lipid peroxidation-derived DNA lesions: Potential lead markers for chemoprevention of inflammation-driven malignancies. Mutat. Res. 2005, 591, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Sjodahl, K.; Lu, Y.; Nilsen, T.I.; Ye, W.; Hveem, K.; Vatten, L.; Lagergren, J. Smoking and alcohol drinking in relation to risk of gastric cancer: A population-based, prospective cohort study. Int. J. Cancer 2007, 120, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T. Total number of genome alterations in sporadic gastrointestinal cancer inferred from pooled analyses in the literature. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2008, 29, 343–350. [Google Scholar] [CrossRef]
- Gonzalez, C.A.; Sala, N.; Rokkas, T. Gastric cancer: Epidemiologic aspects. Helicobacter 2013, 18, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.E.; Andreotti, G.; Lissowska, J.; Yeager, M.; Zatonski, W.; Chanock, S.J.; Chow, W.H.; Hou, L. Genetic variation in sodium-dependent ascorbic acid transporters and risk of gastric cancer in Poland. Eur. J. Cancer 2009, 45, 1824–1830. [Google Scholar] [CrossRef]
- Duell, E.J.; Lujan-Barroso, L.; Llivina, C.; Munoz, X.; Jenab, M.; Boutron-Ruault, M.C.; Clavel-Chapelon, F.; Racine, A.; Boeing, H.; Buijsse, B.; et al. Vitamin C transporter gene (SLC23A1 and SLC23A2) polymorphisms, plasma vitamin C levels, and gastric cancer risk in the EPIC cohort. Genes Nutr. 2013, 8, 549–560. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, L.P.; Xing, C.Z.; Xu, Q.; He, C.Y.; Li, P.; Gong, Y.H.; Liu, Y.P.; Yuan, Y. Interaction between GSTP1 Val allele and H. pylori infection, smoking and alcohol consumption and risk of gastric cancer among the Chinese population. PLoS ONE 2012, 7, e47178. [Google Scholar] [CrossRef]
- Kim, J.; Lee, H.S.; Bae, S.I.; Lee, Y.M.; Kim, W.H. Silencing and CpG island methylation of GSTP1 is rare in ordinary gastric carcinomas but common in Epstein-Barr virus-associated gastric carcinomas. Anticancer Res. 2005, 25, 4013–4019. [Google Scholar]
- Lao, X.; Peng, Q.; Lu, Y.; Li, S.; Qin, X.; Chen, Z.; Chen, J. Glutathione S-transferase gene GSTM1, gene-gene interaction, and gastric cancer susceptibility: Evidence from an updated meta-analysis. Cancer Cell Int. 2014, 14, 127. [Google Scholar] [CrossRef] [PubMed]
- Gong, E.J.; Ahn, J.Y.; Jung, H.Y.; Park, H.; Ko, Y.B.; Na, H.K.; Jung, K.W.; Kim, D.H.; Lee, J.H.; Choi, K.D.; et al. Helicobacter pylori Eradication Therapy Is Effective as the Initial Treatment for Patients with H. pylori-Negative and Disseminated Gastric Mucosa-Associated Lymphoid Tissue Lymphoma. Gut Liver 2016, 10, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Floch, P.; Mégraud, F.; Lehours, P. Helicobacter pylori Strains and Gastric MALT Lymphoma. Toxins 2017, 9, 132. [Google Scholar] [CrossRef] [PubMed]
- Wündisch, T.; Stolte, M. Current status of gastric MALT lymphoma. Curr. Gastroenterol. Rep. 2006, 8, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Santacroce, L.; Cagiano, R.; Del Prete, R.; Bottalico, L.; Sabatini, R.; Carlaio, R.G.; Prejbeanu, R.; Vermesan, H.; Dragulescu, S.I.; Vermesan, D.; et al. Helicobacter pylori infection and gastric MALTomas: An up-to-date and therapy highlight. Clin. Ter. 2008, 159, 457–462. [Google Scholar]
- Mazloom, A.; Rodriguez, A.; Ha, C.S.; Medeiros, L.J.; Wogan, C.; Shihadeh, F.; Allen, P.; Fowler, N.; Dabaja, B. Incidence of gastric involvement in patients with nongastrointestinal extranodal marginal zone lymphoma. Cancer 2011, 117, 2461–2466. [Google Scholar] [CrossRef]
- Pervez, S.; Ali, N.; Aaqil, H.; Mumtaz, K.; Siddiq Ullah, S.; Akhtar, N. Gastric MALT lymphoma: A rarity. J. Coll. Physicians Surg. Pak. 2011, 21, 171–172. [Google Scholar]
- Saito, Y.; Suzuki, H.; Tsugawa, H.; Imaeda, H.; Matsuzaki, J.; Hirata, K.; Hosoe, N.; Nakamura, M.; Mukai, M.; Saito, H.; et al. Overexpression of miR-142-5p and miR-155 in gastric mucosa-associated lymphoid tissue (MALT) lymphoma resistant to Helicobacter pylori eradication. PLoS ONE 2012, 7, e47396. [Google Scholar] [CrossRef]
- Zucca, E.; Copie-Bergman, C.; Ricardi, U.; Thieblemont, C.; Raderer, M.; Ladetto, M. Gastric marginal zone lymphoma of MALT type: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. ESMO 2013, 24, vi144–vi148. [Google Scholar] [CrossRef]
- Raderer, M.; Wöhrer, S.; Kiesewetter, B.; Dolak, W.; Lagler, H.; Wotherspoon, A.; Muellauer, L.; Chott, A. Antibiotic treatment as sole management of Helicobacter pylori-negative gastric MALT lymphoma: A single center experience with prolonged follow-up. Ann. Hematol. 2015, 94, 969–973. [Google Scholar] [CrossRef]
- Hatakeyama, M. Malignant Helicobacter pylori-Associated Diseases: Gastric Cancer and MALT Lymphoma. Adv. Exp. Med. Biol. 2019, 1149, 135–149. [Google Scholar] [PubMed]
- Lee, Y.C.; Chiang, T.H.; Chou, C.K.; Tu, Y.K.; Liao, W.C.; Wu, M.S.; Graham, D.Y. Association Between Helicobacter pylori Eradication and Gastric Cancer Incidence: A Systematic Review and Meta-analysis. Gastroenterology 2016, 150, 1113–1124.e5. [Google Scholar] [CrossRef] [PubMed]
- Seta, T.; Takahashi, Y.; Noguchi, Y.; Shikata, S.; Sakai, T.; Sakai, K.; Yamashita, Y.; Nakayama, T. Effectiveness of Helicobacter pylori eradication in the prevention of primary gastric cancer in healthy asymptomatic people: A systematic review and meta-analysis comparing risk ratio with risk difference. PLoS ONE 2017, 12, e0183321. [Google Scholar] [CrossRef] [PubMed]
- Sugano, K. Effect of Helicobacter pylori eradication on the incidence of gastric cancer: A systematic review and meta-analysis. Gastric Cancer 2019, 22, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Ford, A.C.; Yuan, Y.; Forman, D.; Hunt, R.; Moayyedi, P. Helicobacter pylori eradication for the prevention of gastric neoplasia. Cochrane Database Syst. Rev. 2020, 7, Cd005583. [Google Scholar]
- Kumar, S.; Metz, D.C.; Ellenberg, S.; Kaplan, D.E.; Goldberg, D.S. Risk Factors and Incidence of Gastric Cancer After Detection of Helicobacter pylori Infection: A Large Cohort Study. Gastroenterology 2020, 158, 527–536.e7. [Google Scholar] [CrossRef]
- Malfertheiner, P.; Megraud, F.; O’Morain, C.A.; Gisbert, J.P.; Kuipers, E.J.; Axon, A.T.; Bazzoli, F.; Gasbarrini, A.; Atherton, J.; Graham, D.Y.; et al. Management of Helicobacter pylori infection-the Maastricht V/Florence Consensus Report. Gut 2017, 66, 6–30. [Google Scholar] [CrossRef]
- Lee, Y.C.; Chen, T.H.; Chiu, H.M.; Shun, C.T.; Chiang, H.; Liu, T.Y.; Wu, M.S.; Lin, J.T. The benefit of mass eradication of Helicobacter pylori infection: A community-based study of gastric cancer prevention. Gut 2013, 62, 676–682. [Google Scholar] [CrossRef]
- Choi, I.J.; Kim, C.G.; Lee, J.Y.; Kim, Y.I.; Kook, M.C.; Park, B.; Joo, J. Family History of Gastric Cancer and Helicobacter pylori Treatment. N. Engl. J. Med. 2020, 382, 427–436. [Google Scholar] [CrossRef]
- Michigami, Y.; Watari, J.; Ito, C.; Nakai, K.; Yamasaki, T.; Kondo, T.; Kono, T.; Tozawa, K.; Tomita, T.; Oshima, T.; et al. Long-term effects of H. pylori eradication on epigenetic alterations related to gastric carcinogenesis. Sci. Rep. 2018, 8, 14369. [Google Scholar] [CrossRef]
- Li, W.Q.; Zhang, J.Y.; Ma, J.L.; Li, Z.X.; Zhang, L.; Zhang, Y.; Guo, Y.; Zhou, T.; Li, J.Y.; Shen, L.; et al. Effects of Helicobacter pylori treatment and vitamin and garlic supplementation on gastric cancer incidence and mortality: Follow-up of a randomized intervention trial. BMJ (Clin. Res. Ed.) 2019, 366, l5016. [Google Scholar] [CrossRef] [PubMed]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toh, J.W.T.; Wilson, R.B. Pathways of Gastric Carcinogenesis, Helicobacter pylori Virulence and Interactions with Antioxidant Systems, Vitamin C and Phytochemicals. Int. J. Mol. Sci. 2020, 21, 6451. https://doi.org/10.3390/ijms21176451
Toh JWT, Wilson RB. Pathways of Gastric Carcinogenesis, Helicobacter pylori Virulence and Interactions with Antioxidant Systems, Vitamin C and Phytochemicals. International Journal of Molecular Sciences. 2020; 21(17):6451. https://doi.org/10.3390/ijms21176451
Chicago/Turabian StyleToh, James W. T., and Robert B. Wilson. 2020. "Pathways of Gastric Carcinogenesis, Helicobacter pylori Virulence and Interactions with Antioxidant Systems, Vitamin C and Phytochemicals" International Journal of Molecular Sciences 21, no. 17: 6451. https://doi.org/10.3390/ijms21176451
APA StyleToh, J. W. T., & Wilson, R. B. (2020). Pathways of Gastric Carcinogenesis, Helicobacter pylori Virulence and Interactions with Antioxidant Systems, Vitamin C and Phytochemicals. International Journal of Molecular Sciences, 21(17), 6451. https://doi.org/10.3390/ijms21176451