Structural Features of 1,3,4-Thiadiazole-Derived Ligands and Their Zn(II) and Cu(II) Complexes Which Demonstrate Synergistic Antibacterial Effects with Kanamycin

 ,
,  ,
, 
 ,
, 
Abstract
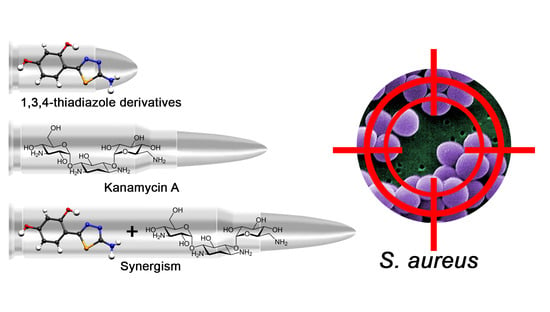
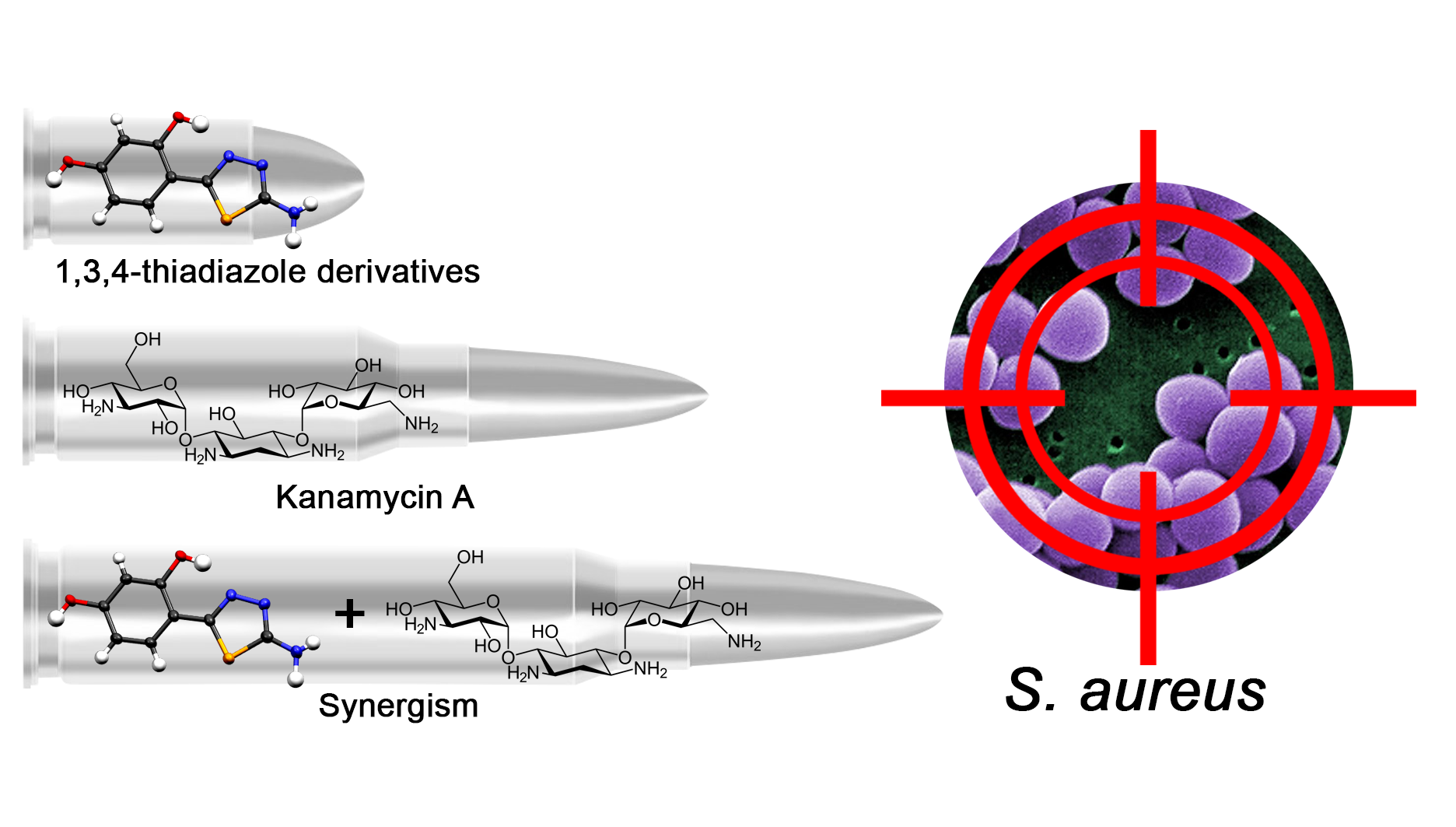
1. Introduction
2. Results
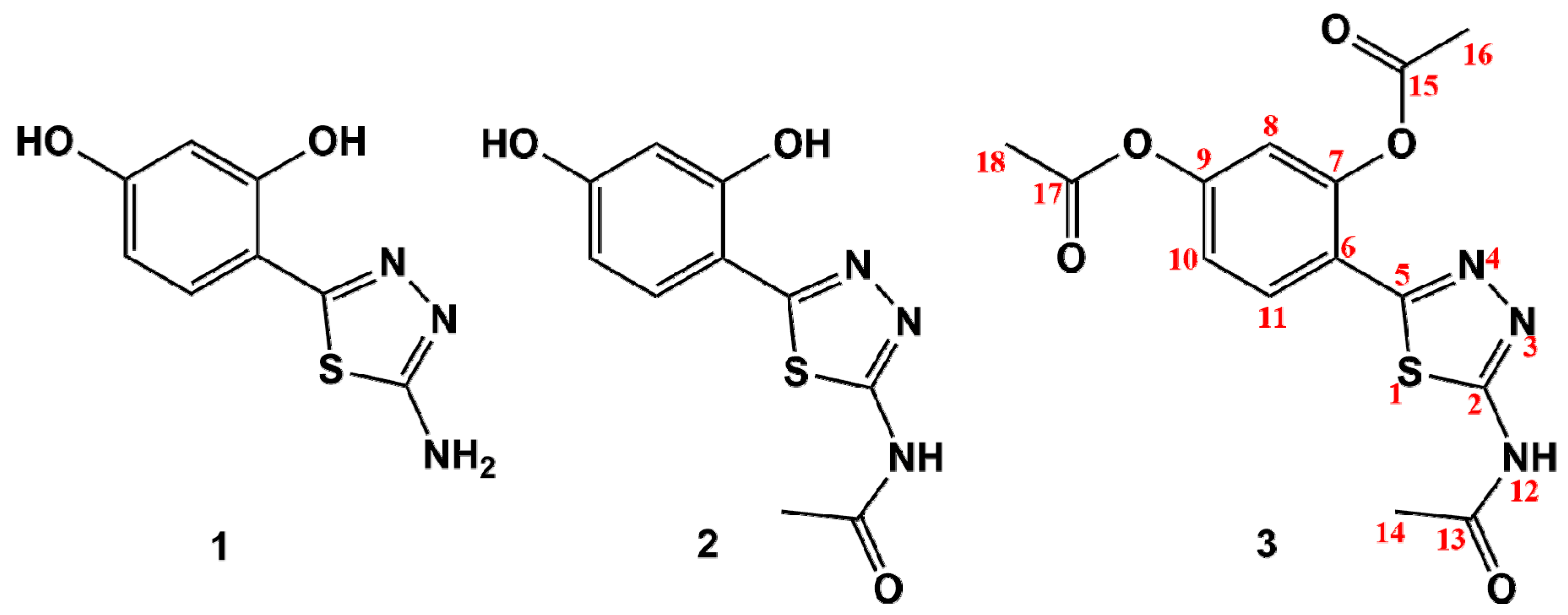
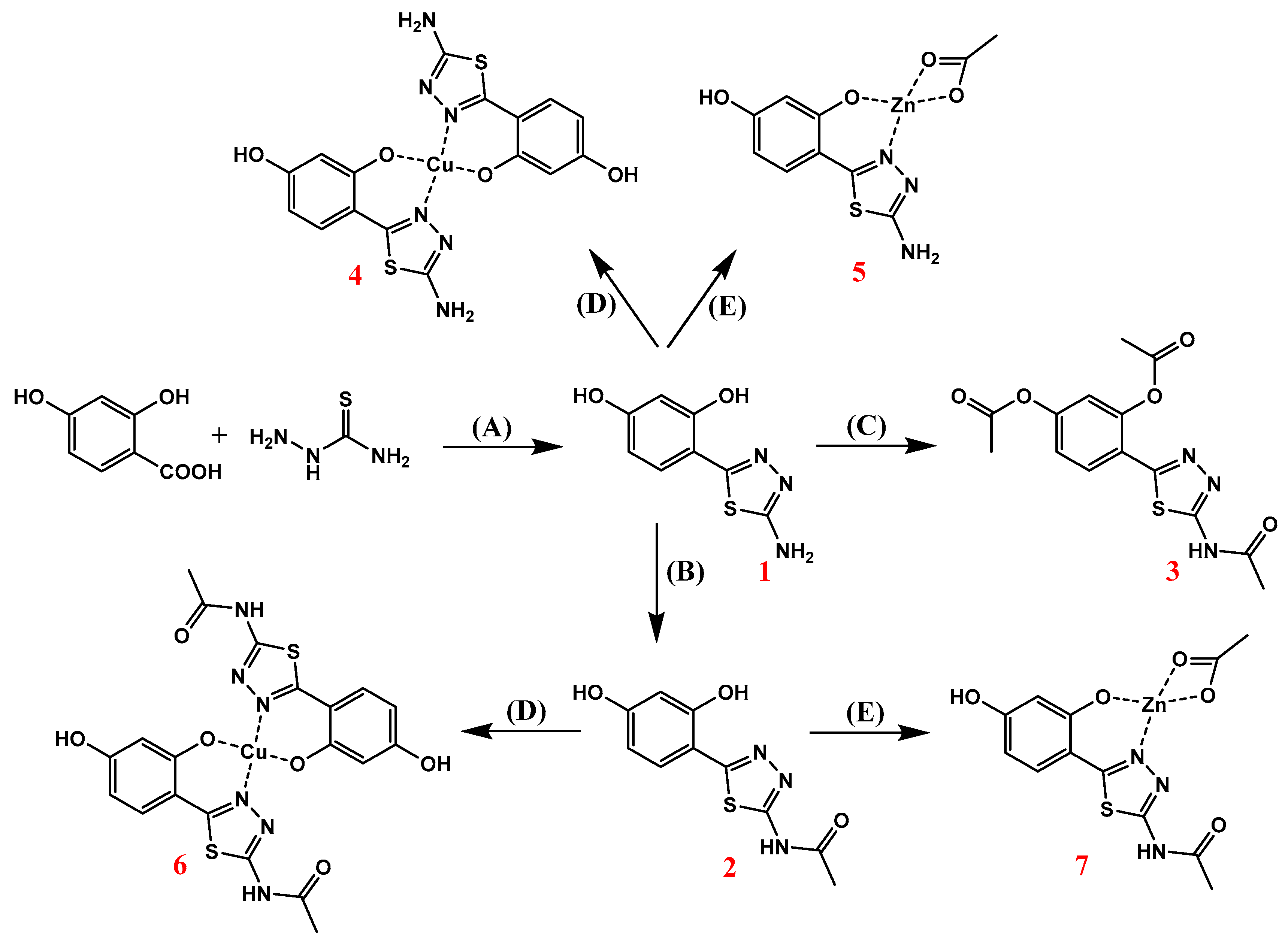
2.1. Synthesis of 1,3,4-Thiadiazole-Derived Ligands and Their Cu(II) and Zn(II) Complexes
2.2. H-NMR Spectroscopy
2.3. C-NMR Spectroscopy
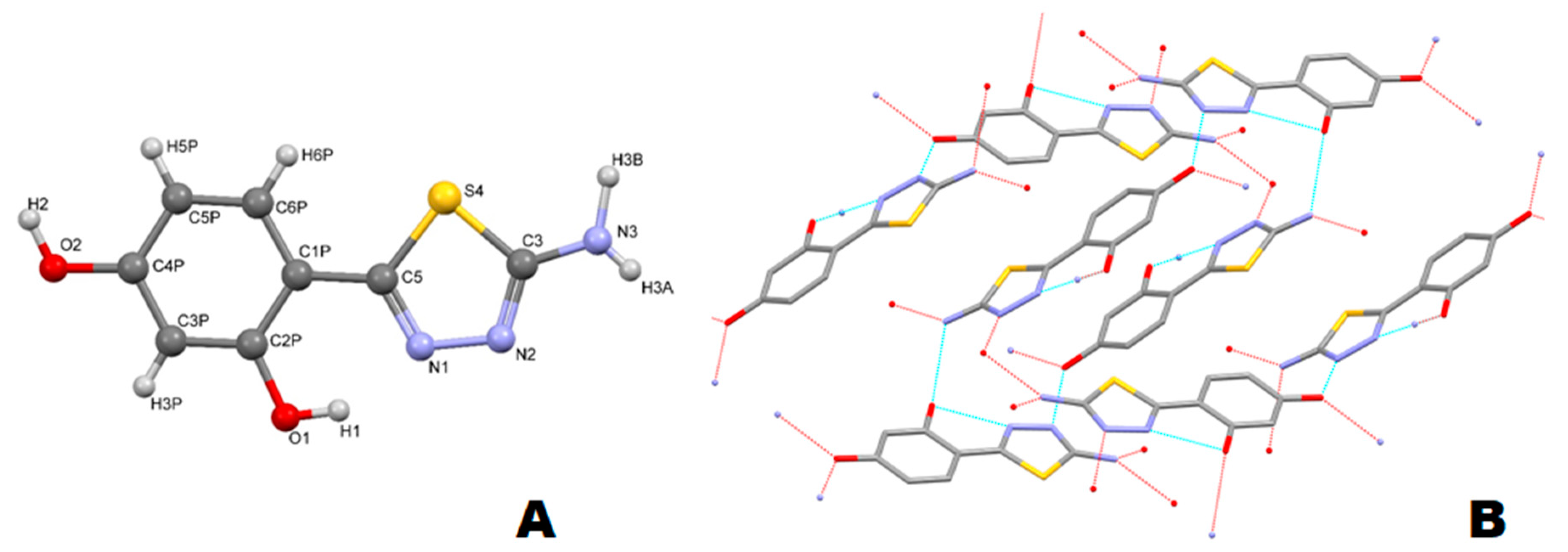
2.4. X-ray Diffraction
2.5. IR (ATR) Spectroscopy
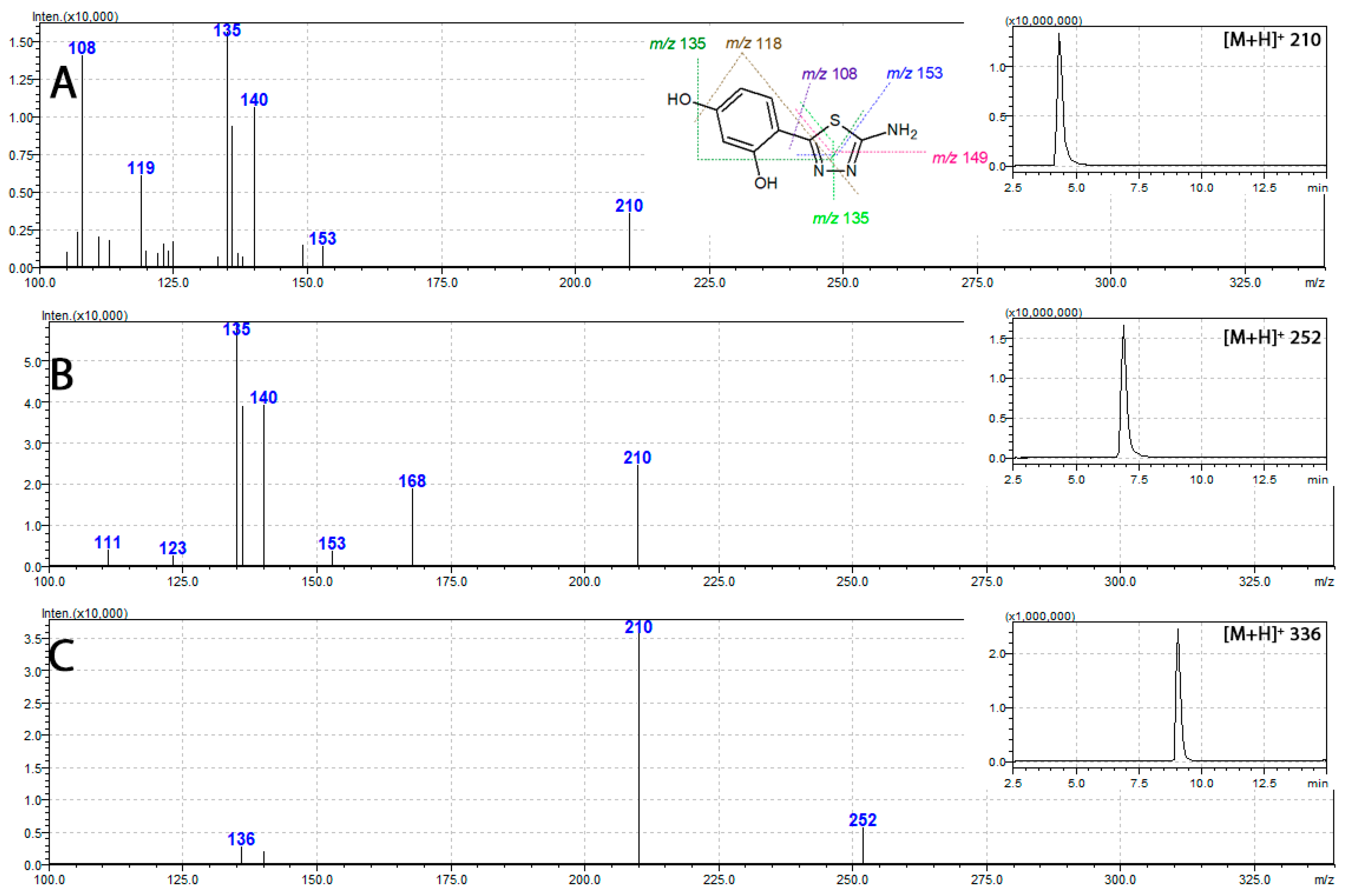
2.6. Mass Spectrometry
2.7. Elemental Analysis (C,H,N) and Atomic Absorption (AAS) Spectroscopy
2.8. UV–Vis Spectroscopy
2.9. Fluorescence Spectroscopy
2.10. Antioxidant Activity
2.11. Antibacterial Activity
3. Materials and Methods
| Synthesis of 2-amino-5-(2,4-dihydroksyphenyl)-1,3,4-thiadiazole | (1) |
| Synthesis of 2-acetamido-5-(2,4-dihydroksyphenyl)-1,3,4-thiadiazole | (2) |
| Synthesis of 2-acetamido-5-((phenyl-2,4-diacetate)-yl)-1,3,4-thiadiazole | (3) |
| Synthesis of Zn(II) and Cu(II) complexes | (4–7) |
3.1. Antioxidant Assay
3.2. Antibacterial Activity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Czernel, G.; Matwijczuk, A.; Karcz, D.; Górecki, A.; Niemczynowicz, A.; Szcześ, A.; Gładyszewski, G.; Matwijczuk, A.; Gładyszewska, B.; Niewiadomy, A. Spectroscopic Studies of Dual Fluorescence in 2-(4-Fluorophenylamino)-5-(2,4-dihydroxybenzeno)-1,3,4-thiadiazole: Effect of Molecular Aggregation in a Micellar System. Molecules 2018, 23, 2861. [Google Scholar] [CrossRef]
- Budziak, I.; Karcz, D.; Makowski, M.; Myśliwa-Kurdziel, B.; Kasprzak, K.; Matwijczuk, A.; Chruściel, E.; Oniszczuk, A.; Adwent, L.; Matwijczuk, A. Spectroscopic and theoretical investigation into substituent- and aggregation-related dual fluorescence effects in the selected 2-amino-1,3,4-thiadiazoles. J. Mol. Liq. 2019, 291, 111261. [Google Scholar] [CrossRef]
- Matwijczuk, A.; Kamiński, D.; Górecki, A.; Ludwiczuk, A.; Niewiadomy, A.; Maćkowski, S.; Gagos, M. Spectroscopic Studies of Dual Fluorescence in 2-((4-Fluorophenyl)amino)-5-(2,4-dihydroxybenzeno)-1,3,4-thiadiazole. J. Phys. Chem. A 2015, 119, 10791–10805. [Google Scholar] [CrossRef] [PubMed]
- Kamiński, D.; Hoser, A.A.; Gagoś, M.; Matwijczuk, A.; Arczewska, M.; Niewiadomy, A.; Woźniak, K. Solvatomorphism of 2-(4-Fluorophenylamino)-5-(2,4-dihydroxybenzeno)-1,3,4-thiadiazole Chloride. Cryst. Growth Des. 2010, 10, 3480–3488. [Google Scholar] [CrossRef]
- Matwijczuk, A.; Karcz, D.; Walkowiak, R.; Furso, J.; Gładyszewska, B.; Wybraniec, S.; Niewiadomy, A.; Karwasz, G.P.; Gagoś, M. Effect of Solvent Polarizability on the Keto/Enol Equilibrium of Selected Bioactive Molecules from the 1,3,4-Thiadiazole Group with a 2,4-Hydroxyphenyl Function. J. Phys. Chem. A 2017, 121, 1402–1411. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Verma, A.; Prajapati, S.; Sharma, U.K. Synthetic Methods, Chemistry, and the Anticonvulsant Activity of Thiadiazoles. Int. J. Med. Chem. 2013, 2013, 1–16. [Google Scholar] [CrossRef]
- Jain, A.K.; Sharma, S.; Vaidya, A.; Ravichandran, V.; Agrawal, R.K. 1,3,4-Thiadiazole and its Derivatives: A Review on Recent Progress in Biological Activities. Chem. Biol. Drug Des. 2013, 81, 557–576. [Google Scholar] [CrossRef]
- Hu, Y.; Li, C.-Y.; Wang, X.-M.; Yang, Y.-H.; Zhu, H.-L. 1,3,4-Thiadiazole: Synthesis, Reactions, and Applications in Medicinal, Agricultural, and Materials Chemistry. Chem. Rev. 2014, 114, 5572–5610. [Google Scholar] [CrossRef]
- Matysiak, J.; Maliński, Z. 2-(2,4-dihydroxyphenyl)-1,3,4-thiadiazole analogues: Antifungal activity in vitro against Candida species. Russ. J. Bioorganic Chem. 2007, 33, 594–601. [Google Scholar] [CrossRef]
- Matysiak, J.; Skrzypek, A.; Niewiadomy, A. Synthesis and antifungal activity of novel 5-substituted 4-(1,3,4-thiadiazol-2-yl)benzene-1,3-diols. Heteroat. Chem. 2010, 21, 533–540. [Google Scholar] [CrossRef]
- Luszczki, J.J.; Karpińska, M.; Matysiak, J.; Niewiadomy, A. Characterization and preliminary anticonvulsant assessment of some 1,3,4-thiadiazole derivatives. Pharmacol. Rep. 2015, 67, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Chudzik, B.; Bonio, K.; Dąbrowski, W.; Pietrzak, D.; Niewiadomy, A.; Olender, A.; Małodobry, K.; Gagoś, M. Synergistic antifungal interactions of amphotericin B with 4-(5-methyl-1,3,4-thiadiazole-2-yl) benzene-1,3-diol. Sci. Rep. 2019, 9, 12945. [Google Scholar] [CrossRef] [PubMed]
- Chudzik, B.; Bonio, K.; Dąbrowski, W.; Pietrzak, D.; Niewiadomy, A.; Olender, A.; Pawlikowska-Pawlęga, B.; Gagoś, M. Antifungal effects of a 1,3,4-thiadiazole derivative determined by cytochemical and vibrational spectroscopic studies. PLoS ONE 2019, 14, e0222775. [Google Scholar] [CrossRef] [PubMed]
- Skrzypek, A.; Matysiak, J.; Niewiadomy, A.; Bajda, M.; Szymański, P. Synthesis and biological evaluation of 1,3,4-thiadiazole analogues as novel AChE and BuChE inhibitors. Eur. J. Med. Chem. 2013, 62, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, H.; Janicka-Klos, A.; Brasun, J.; Gaggelli, E.; Valensin, D.; Valensin, G. Copper, iron, and zinc ions homeostasis and their role in neurodegenerative disorders (metal uptake, transport, distribution and regulation). Coord. Chem. Rev. 2009, 253, 2665–2685. [Google Scholar] [CrossRef]
- Kozlowski, H.; Luczkowski, M.; Remelli, M.; Valensin, D. Copper, zinc and iron in neurodegenerative diseases (Alzheimer’s, Parkinson’s and prion diseases). Coord. Chem. Rev. 2012, 256, 2129–2141. [Google Scholar] [CrossRef]
- Valensin, D.; Gabbiani, C.; Messori, L. Metal compounds as inhibitors of β-amyloid aggregation. Perspectives for an innovative metallotherapeutics on Alzheimer’s disease. Coord. Chem. Rev. 2012, 256, 2357–2366. [Google Scholar] [CrossRef]
- Hureau, C.; Dorlet, P. Coordination of redox active metal ions to the amyloid precursor protein and to amyloid-β peptides involved in Alzheimer disease. Part 2: Dependence of Cu(II) binding sites with Aβ sequences. Coord. Chem. Rev. 2012, 256, 2175–2187. [Google Scholar] [CrossRef]
- Gaeta, A.; Hider, R.C. The crucial role of metal ions in neurodegeneration: The basis for a promising therapeutic strategy. Br. J. Pharmacol. 2009, 146, 1041–1059. [Google Scholar] [CrossRef]
- Zatta, P.; Drago, D.; Bolognin, S.; Sensi, S.L. Alzheimer’s disease, metal ions and metal homeostatic therapy. Trends Pharmacol. Sci. 2009, 30, 346–355. [Google Scholar] [CrossRef]
- Karcz, D.; Matwijczuk, A.; Boroń, B.; Creaven, B.; Fiedor, L.; Niewiadomy, A.; Gagos, M. Isolation and spectroscopic characterization of Zn(II), Cu(II), and Pd(II) complexes of 1,3,4-thiadiazole-derived ligand. J. Mol. Struct. 2017, 1128, 44–50. [Google Scholar] [CrossRef]
- Chohan, Z.H.; Pervez, H.; Rauf, A.; Khan, K.M.; Supuran, C.T. Antibacterial cobalt (II), copper (II), nickel (II) and zinc (II) complexes of mercaptothiadiazole—Derived furanyl, thienyl, pyrrolyl, salicylyl and pyridinyl Schiff bases. J. Enzym. Inhib. Med. Chem. 2006, 21, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Obaleye, J.A.; Adediji, J.F.; Adebayo, M.A. Synthesis and Biological Activities on Metal Complexes of 2,5-Diamino-1,3,4-thiadiazole Derived from Semicarbazide Hydrochloride. Molecules 2011, 16, 5861–5874. [Google Scholar] [CrossRef] [PubMed]
- Serbest, K.; Kayi, H.; Er, M.; Sancak, K.; Değirmencioğlu, İ. Ni(II), Cu(II), and Zn(II) complexes of tetradentate schiff base containing two thiadiazoles units: Structural, spectroscopic, magnetic properties, and molecular modeling studies. Heteroat. Chem. 2008, 19, 700–712. [Google Scholar] [CrossRef]
- Dar, A.A.; Sharma, S.K.; Murugavel, R. Is Single-4-Ring the Most Basic but Elusive Secondary Building Unit That Transforms to Larger Structures in Zinc Phosphate Chemistry? Inorg. Chem. 2015, 54, 4882–4894. [Google Scholar] [CrossRef]
- Vishnoi, P.; Sharma, S.K.; Kaleeswaran, D.; Murugavel, R. Zinc Dialkylhydroxybenzoates with Unusual Structures: First Example of a Discrete Three-Blade Paddle-Wheel and a Solvent Engulfed Coordination Polymer. ChemistrySelect 2016, 1, 6658–6668. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Bassler, G.C. Spectrometric identification of organic compounds. J. Chem. Educ. 1962, 39, 546. [Google Scholar] [CrossRef]
- Turan, N.; Şekerci, M. Synthesis, characterization and thermal behavior of some Zn(II) complexes with ligands having 1,3,4-thiadiazole moieties. Heteroat. Chem. 2010, 21, 14–23. [Google Scholar] [CrossRef]
- Pihlaja, K.; Agirbas, H.; Ovcharenko, V.; Valtamo, P. Electron ionization induced fragmentation of some oxadiazole and thiadiazole derivatives. Rapid Commun. Mass Spectrom. 2004, 18, 760–764. [Google Scholar] [CrossRef]
- Karcz, D.; Starzak, K.; Matwijczuk, A.; Ciszkowicz, E.; Lecka-Szlachta, K.; Matwijczuk, A.; Ciupak, A.; Gładyszewska, B.; Niewiadomy, A. Synthesis, spectroscopy and biological activity of novel Cu(II) and Zn(II) complexes with 1,3,4-thiadiazole derivatives. Przemysł Chem. 2018, 97, 1095–1101. [Google Scholar] [CrossRef]
- Matwijczuk, A.; Karcz, D.; Pustuła, K.; Makowski, M.; Górecki, A.; Kluczyk, D.; Karpińska, M.M.; Niewiadomy, A.; Gagos, M. Spectroscopic and theoretical studies of fluorescence effects in bio-active: 4-(5-(methyl-1,3,4-thiadiazol-2-yl))benzene-1,3-diol and 4-(5-(methylamino-1,3,4-thiadiazol-2-yl))benzene-1,3-diol compounds: Effect of molecular aggregation and amino group position. J. Lumin. 2018, 201, 44–56. [Google Scholar] [CrossRef]
- Budziak, I.; Karcz, D.; Makowski, M.A.; Rachwał, K.; Starzak, K.; Matwijczuk; Myśliwa-Kurdziel, B.; Oniszczuk, A.; Combrzyński, M.; Podleśna, A.; et al. Non-Typical Fluorescence Effects and Biological Activity in Selected 1,3,4-thiadiazole Derivatives: Spectroscopic and Theoretical Studies on Substituent, Molecular Aggregation, and pH Effects. Int. J. Mol. Sci. 2019, 20, 5494. [Google Scholar] [CrossRef]
- Mereshchenko, A.S.; Pal, S.K.; Karabaeva, K.E.; El-Khoury, P.Z.; Tarnovsky, A.N. Photochemistry of Monochloro Complexes of Copper(II) in Methanol Probed by Ultrafast Transient Absorption Spectroscopy. J. Phys. Chem. A 2011, 116, 2791–2799. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Starzak, K.; Creaven, B.; Matwijczuk, A.; Matwijczuk, A.; Karcz, D. Anti-Hypochlorite and Catalytic Activity of Commercially Available Moringa oleifera Diet Supplement. Molecules 2019, 24, 3330. [Google Scholar] [CrossRef]
- Starzak, K.; Świergosz, T.; Matwijczuk, A.; Creaven, B.; Podleśny, J.; Karcz, D. Anti-Hypochlorite, Antioxidant, and Catalytic Activity of Three Polyphenol-Rich Super-Foods Investigated with the Use of Coumarin-Based Sensors. Biomolecules 2020, 10, 723. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Sofic, E.; Prior, R.L. Antioxidant and Prooxidant Behavior of Flavonoids: Structure-Activity Relationships. Free. Radic. Biol. Med. 1997, 22, 749–760. [Google Scholar] [CrossRef]
- Aquilano, K.; Baldelli, S.; Rotilio, G.; Ciriolo, M.R. Role of Nitric Oxide Synthases in Parkinson’s Disease: A Review on the Antioxidant and Anti-inflammatory Activity of Polyphenols. Neurochem. Res. 2008, 33, 2416–2426. [Google Scholar] [CrossRef]
- Heim, K.E.; Tagliaferro, A.R.; Bobilya, D.J. Flavonoid antioxidants: Chemistry, metabolism and structure-activity relationships. J. Nutr. Biochem. 2002, 13, 572–584. [Google Scholar] [CrossRef]
- Woźnicka, E.; Zapała, L.; Pieniążek, E.; Kosińska, M.; Ciszkowicz, E.; Lecka-Szlachta, K.; Pusz, J.; Maciołek, U.; Dronka, J. Synthesis, characterization and antibacterial studies of Tm(III), Yb(III) and Lu(III) complexes of morin. J. Coord. Chem. 2017, 70, 1–1463. [Google Scholar] [CrossRef]
- European Committee for Antimicrobial Susceptibility Testing (EUCAST) of the European Society of Clinical Microbiology; Infectious Diseases (ESCMID). Terminology relating to methods for the determination of susceptibility of bacteria to antimicrobial agents. Clin. Microbiol. Infect. 2000, 6, 503–508. [Google Scholar] [CrossRef]
- Andrews, J.M. Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 2001, 48, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Bonapace, C.R.; Bosso, J.A.; Friedrich, L.V.; White, R.L. Comparison of methods of interpretation of checkerboard synergy testing. Diagn. Microbiol. Infect. Dis. 2002, 44, 363–366. [Google Scholar] [CrossRef]
- Grzybowska, W.; Banaszczyk-Ruś, M.; Wójcik, A.; Tyski, S. [Comparison of checkerboard and time-kill methods for the analysis of two antibiotics combined]. Med. Doświadczalna I Mikrobiol. 2004, 56, 391–403. [Google Scholar]
- Lu, X.; Liu, X.; Wan, B.; Franzblau, S.G.; Chen, L.; Zhou, C.; You, Q. Synthesis and evaluation of anti-tubercular and antibacterial activities of new 4-(2,6-dichlorobenzyloxy)phenyl thiazole, oxazole and imidazole derivatives. Part 2. Eur. J. Med. Chem. 2012, 49, 164–171. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history ofSHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2007, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Thiadiazole Serivative | 1H-NMR Signals: δ (ppm) *, Multiplicity **, and J Value (Hz) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| H7 | H8 | H9 | H10 | H11 | H12 | H14 | H16 | H18 | |
| 1 | 10.91, s (-OH) | 6.38, d, J = 2.3 | 9.84, s, (-OH) | 6.36, dd, J1 = 8.5, J2 = 2.3 | 7.53, d, J = 8.5 | 7.15, s, (-NH2) | - | - | - |
| 2 | 10.90, s (-OH) | 6.45, d, J = 2.3 | 9.92, s, (-OH) | 6.40, dd, J1 = 8.7, J2 = 2.3 | 7.91, d, J = 8.7 | 12.32, s, (-NH-) | 2.18, s | - | - |
| 3 | - | 7.29, d, J = 2.3 | - | 7.26, dd, J1 = 8.5, J2 = 2.3 | 8.2, d, J = 8.5 | 12.68, s, (-NH-) | 2.22, s | 2.31, s | 2.38, s |
| Thiadiazole Derivative | 13C-NMR Signals (ppm) * | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C2 | C5 | C6 | C7 | C8 | C9 | C10 | C11 | C13 | C14 | C15 | C16 | C17 | C18 | |
| 1 | 167.84 | 156.03 | 108.98 | 156.43 | 103.00 | 160.22 | 108.35 | 129.20 | - | - | - | - | - | - |
| 2 | 168.79 | 158.87 | 109.15 | 156.35 | 102.88 | 160.94 | 108.62 | 129.03 | 158.89 | 22.88 | - | - | - | - |
| 3 | 169.24 | 155.92 | 121.36 | 147.89 | 118.21 | 152.53 | 121.02 | 129.72 | 160.03 | 22.87 | 168.96 | 21.31 | 169.33 | 21.68 |
| Parameter | Value |
|---|---|
| Molecular formula | C8H7N3O3S |
| Temperature (K) | 120(2) |
| Crystal system | monoclinic |
| Space group | I 2/a |
| a (Å) | 13.421(2) |
| b (Å) | 7.1590(10) |
| c (Å) | 18.454(2) |
| α (°) | 90 |
| β (°) | 100.79(2) |
| γ (°) | 90 |
| V (Å3) | 1741.73 |
| Z | 8 |
| Calculated density (g cm−3) | 1.596 |
| Absorption coefficient (mm−1) | 3.133 |
| F (000) | 864 |
| Completeness | 97% |
| θ range for data collection (°) | 4.88–76.15 |
| Index ranges | −13 ≤ h ≤ 16 −8 ≤ k ≤ 7 −22 ≤ l ≤ 15 |
| Reflections collected/unique | 6137/3789 (Rint = 0.0353) |
| Observed/restraints/parameters | 1761/0/151 |
| Goodness-of-fit on F2 | 1.217 |
| Final R indices (I > 2 sigma(I)) | R1 = 0.0551 wR2 = 0.1679 |
| R indices (all data) | R1 = 0.0709 wR2 = 0.2225 |
| Largest diff. peak and hole (e Å−3) | 0.5/−0.7 |
| CCDC number | 1845297 |
| Compound No | %C | %H | %N | %M * | Mw ** (g/mol) | Chemical Formula ** | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Calc | Found | Calc | Found | Calc | Found | Calc | Found | |||
| 1 | 45.93 | 42.96 | 3.37 | 3.30 | 20.08 | 17.84 | - | - | 209.22 | C8H7N3O2S |
| 2 | 47.80 | 46.34 | 3.61 | 3.41 | 16.72 | 16.21 | - | - | 251.26 | C10H9N3O3S |
| 3 | 50.15 | 49.02 | 3.91 | 3.66 | 12.53 | 12.50 | - | - | 335.33 | C14H13N3O5S |
| 4 | 37.24 | 26.85 | 3.13 | 2.34 | 16.29 | 10.69 | 12.31 | 24.67 | 516.01 | C16H16CuN6O6S2 |
| 5 | 32.58 | 30.98 | 3.55 | 2.74 | 11.40 | 12.02 | 17.73 | 16.93 | 368.67 | C10H13N3O6SZn |
| 6 | 40.03 | 37.67 | 3.36 | 2.95 | 14.01 | 12.90 | 10.59 | 12.04 | 600.08 | C20H20CuN6O8S2 |
| 7 | 35.09 | 37.01 | 3.68 | 3.02 | 10.23 | 11.55 | 15.92 | 13.21 | 470.71 | C12H15N3O7SZn |
| Compound | mM Antioxidants/mM TE | IC50 (mM) | ||
|---|---|---|---|---|
| 1 | 0.08 | ±0.00 | 0.60 | ±0.01 |
| 2 | 0.35 | ±0.03 | 0.13 | ±0.00 |
| 3 | 0.41 × 10−3 | ±0.02 × 10−3 | 126.78 | ±2.25 |
| TROLOX | 1.00 | ±0.01 | 0.05 | ±0.00 |
| Compound Bacteria | MIC (μg/mL) (±0.01) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 5 | 7 | Ampicillin | Tetracycline | Erythromycin | Kanamycin | |
| Staphylococcus aureus | 500 | - | - | 500 | 500 | 1.9 | 0.1 | 250 | 3.9 |
| Escherichia coli | 1000 | - | - | 1000 | 1000 | 31.2 | 0.5 | 62.5 | 7.8 |
| Pseudomonas aeruginosa | - | - | - | - | - | 62.5 | 500 | 250 | - |
| Compound | MIC a (µg/mL) (±0.01) | Combination | MIC b (µg/mL) (±0.01) | FIC c |
|---|---|---|---|---|
| 1 | 500 | 1/kanamycin | 125 | 0.375 |
| kanamycin | 3.9 | 0.5 | ||
| 5 | 500 | 5/kanamycin | 125 | 0.375 |
| kanamycin | 3.9 | 0.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karcz, D.; Matwijczuk, A.; Kamiński, D.; Creaven, B.; Ciszkowicz, E.; Lecka-Szlachta, K.; Starzak, K. Structural Features of 1,3,4-Thiadiazole-Derived Ligands and Their Zn(II) and Cu(II) Complexes Which Demonstrate Synergistic Antibacterial Effects with Kanamycin. Int. J. Mol. Sci. 2020, 21, 5735. https://doi.org/10.3390/ijms21165735
Karcz D, Matwijczuk A, Kamiński D, Creaven B, Ciszkowicz E, Lecka-Szlachta K, Starzak K. Structural Features of 1,3,4-Thiadiazole-Derived Ligands and Their Zn(II) and Cu(II) Complexes Which Demonstrate Synergistic Antibacterial Effects with Kanamycin. International Journal of Molecular Sciences. 2020; 21(16):5735. https://doi.org/10.3390/ijms21165735
Chicago/Turabian StyleKarcz, Dariusz, Arkadiusz Matwijczuk, Daniel Kamiński, Bernadette Creaven, Ewa Ciszkowicz, Katarzyna Lecka-Szlachta, and Karolina Starzak. 2020. "Structural Features of 1,3,4-Thiadiazole-Derived Ligands and Their Zn(II) and Cu(II) Complexes Which Demonstrate Synergistic Antibacterial Effects with Kanamycin" International Journal of Molecular Sciences 21, no. 16: 5735. https://doi.org/10.3390/ijms21165735
APA StyleKarcz, D., Matwijczuk, A., Kamiński, D., Creaven, B., Ciszkowicz, E., Lecka-Szlachta, K., & Starzak, K. (2020). Structural Features of 1,3,4-Thiadiazole-Derived Ligands and Their Zn(II) and Cu(II) Complexes Which Demonstrate Synergistic Antibacterial Effects with Kanamycin. International Journal of Molecular Sciences, 21(16), 5735. https://doi.org/10.3390/ijms21165735