Lysosomal Storage Disease-Associated Neuropathy: Targeting Stable Nucleic Acid Lipid Particle (SNALP)-Formulated siRNAs to the Brain as a Therapeutic Approach

 ,
, 

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Lysosomal Storage Disorders
The Unmet Need—Defeating Central Nervous System Pathology
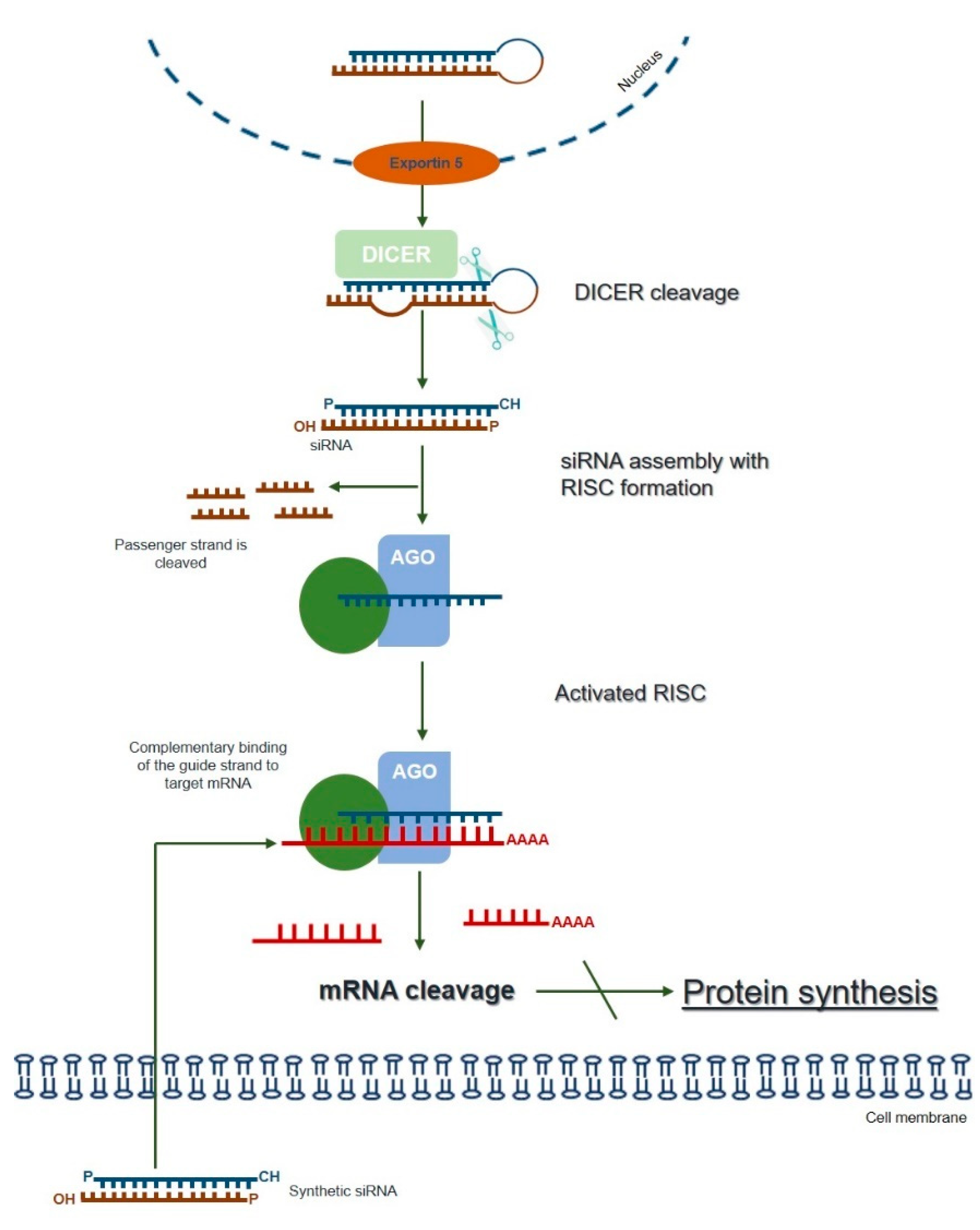
3. RNA Interference (RNAi)—An Overview of the Silencing Machinery
4. RNAi as a Potential Therapeutic Approach for LSDs
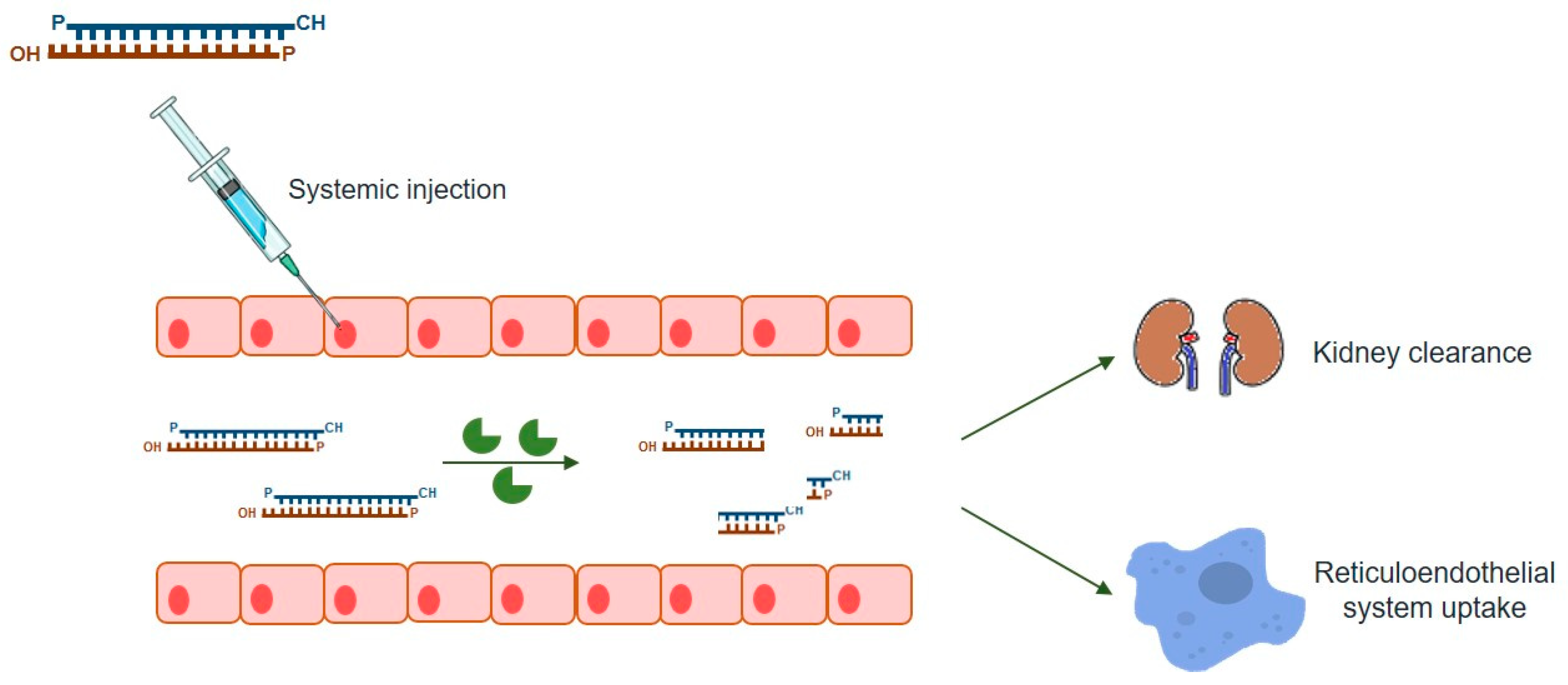
5. RNAi-Based Therapy Challenges
5.1. Some Examples of Small Interfering RNAs (siRNAs) Delivery Systems
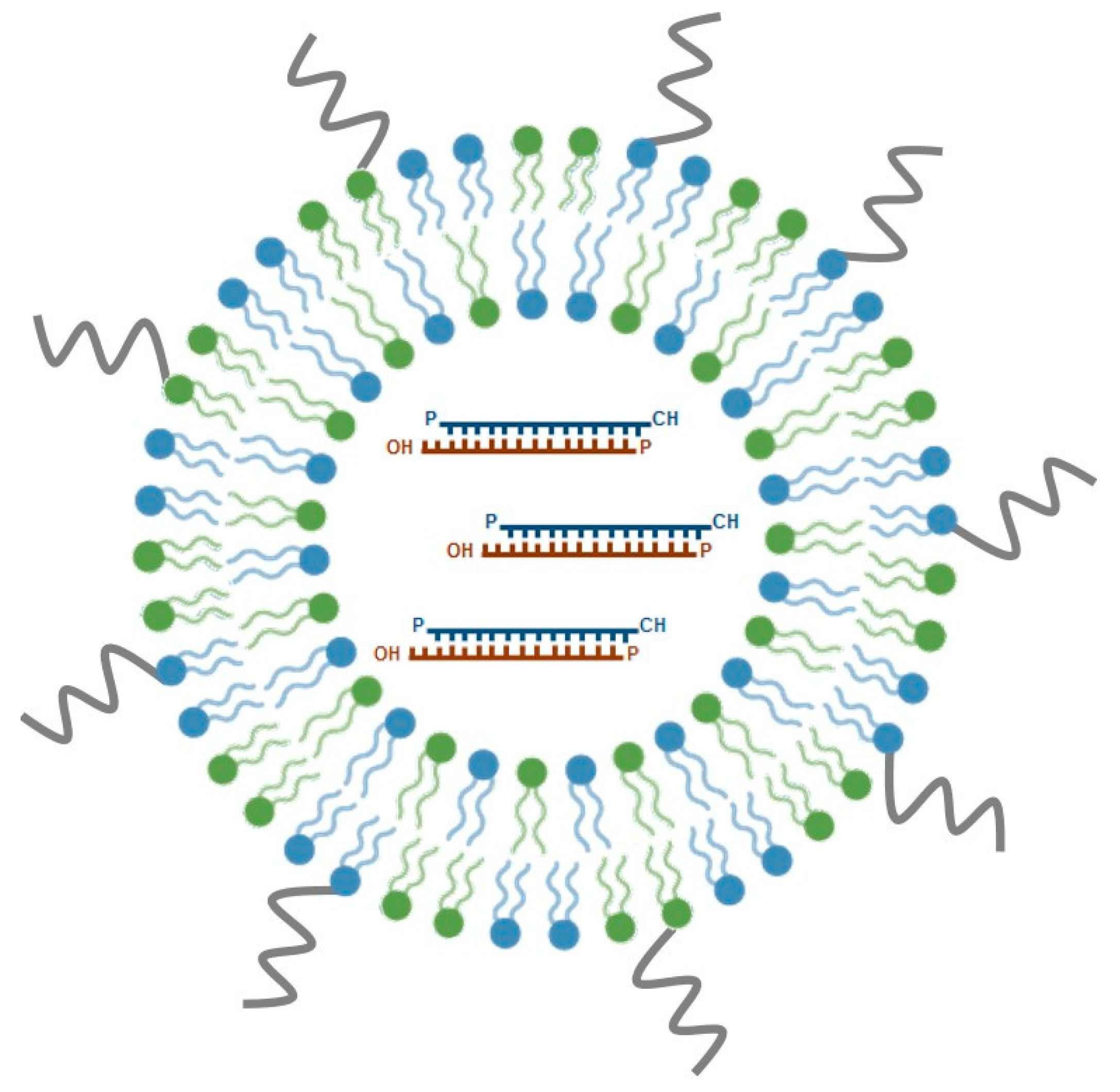
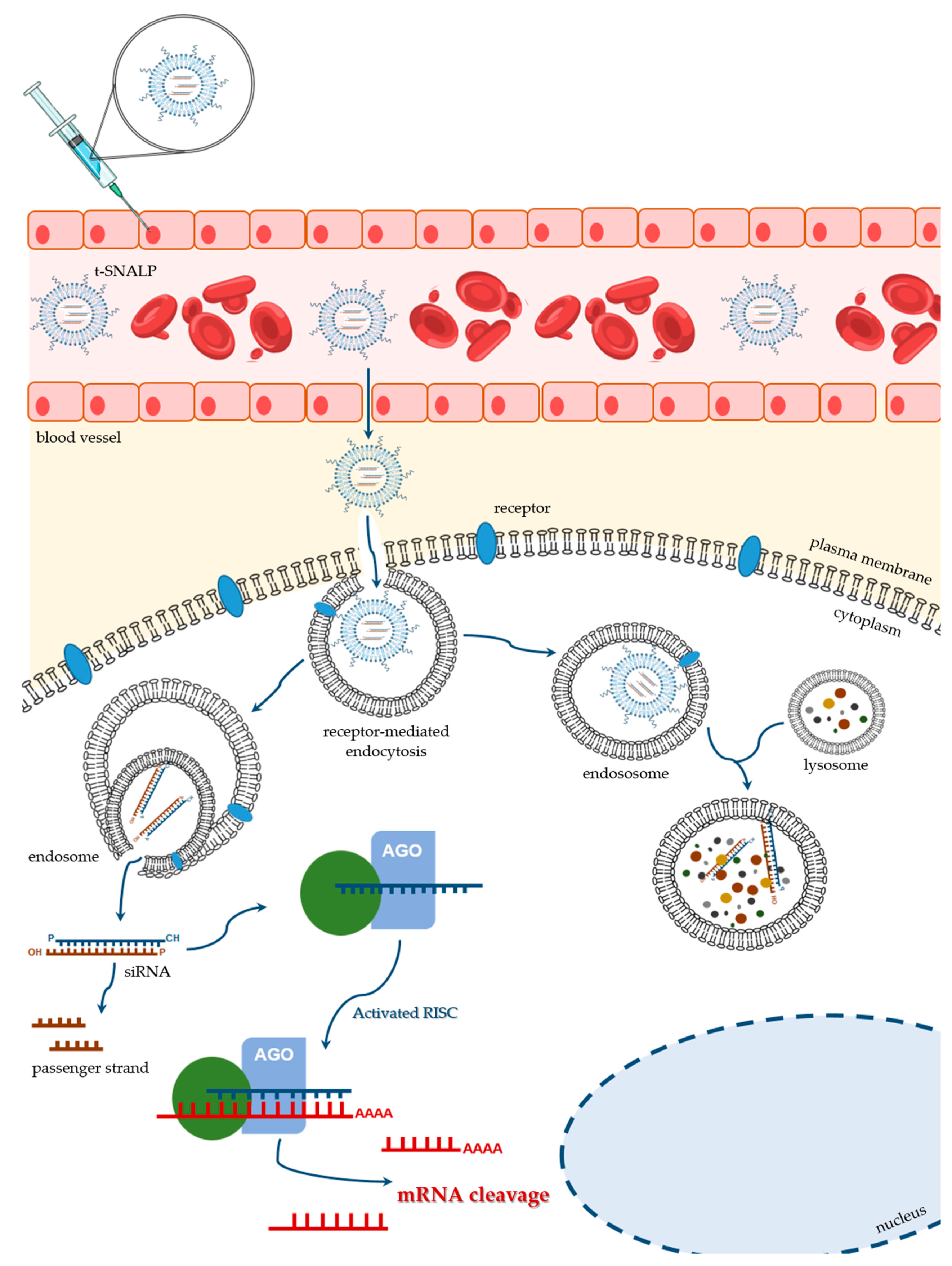
5.2. Stable Nucleic Acid Lipid Particles (SNALPs)
5.3. Ligand-Mediated SNALP Targeting
6. General Considerations on the Pre-Clinical Development of RNAi-Based Substrate Reduction Approaches for LSDs
7. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AGO2 | Argonaute 2 |
| ApoB | Apolipoprotein B |
| ASOs | Antisense oligonucleotides |
| BBB | Blood-brain barrier |
| C6-Cer | C6-ceramide |
| CML | Chronic myeloid leukemia |
| CNS | Central nervous system |
| CSCs | Cancer stem cells |
| CSF | Cerebrospinal fluid |
| CTX | Chlorotoxin |
| DAGs | Diacylglycerols |
| DSPC | 1,2-distearoyl-sn-glycero-3-phosphocholine |
| dsRNA | Double-stranded RNA |
| DXR | Doxorubicin |
| EBOV | Ebola virus |
| ERT | Enzyme replacement therapy |
| FD | Fabry disease |
| GAGs | Glycosaminoglycans |
| GD | Gaucher disease |
| GBM | Glioblastoma |
| gSRT | Genetic substrate reduction therapy |
| hATTR | Hereditary TTR-mediated amyloidosis |
| HBV | Hepatitis B virus |
| HF | Hemorrhagic fever |
| HIV | Human immunodeficiency virus |
| HMEC-1 | Angiogenic endothelial cells |
| HS | Heparan sulphate |
| ICV | Intracerebroventricular |
| iPSCs | Induced pluripotent stem cells |
| IT | Intrathecal |
| IV | Intravenous |
| KSP | Kinesin spindle protein |
| LNPs | Lipid nanoparticles |
| LDL | Low-density lipoprotein |
| LSDs | Lysosomal storage disorders |
| miRNA | microRNA |
| MLD | Metachromatic Leukodystrophy |
| MPSs | Mucopolysaccharidoses |
| mRNA | messenger RNA |
| NCAM | Neural cell adhesion molecule |
| NCLs | Neuronal Ceroid Lipofuscinoses |
| nACh | Nicotinic acetylcholine |
| nAChR | Nicotinic acetylcholine receptor |
| n-SCC | Non-stem cancer cells |
| PD | Parkinson’s disease |
| PLK1 | Polo-like kinase 1 |
| PEG | Polyethylene glycol |
| PEI | Polyethylenimine |
| PC3 | Prostate cancer cells |
| PKR | Protein kinase receptor |
| P75NTR | Neurotrophin receptor 75 |
| RDP | Rabies virus derived peptide |
| RVG | Rabies virus glycoprotein |
| RVG-9r | Rabies virus peptide derivative |
| RME | Receptor-mediated endocytosis |
| rhTPP1 | Recombinant human tripeptidyl peptidase 1 |
| RES | Reticuloendothelial system |
| RNAi | RNA interference |
| RISC | RNA-induced silencing complex |
| shRNA | Short hairpin RNA |
| siRNAs | small interfering RNAs |
| SNALP | Stable nucleic acid lipid particle |
| SRT | Substrate reduction therapy |
| TLRs | Toll-like receptors |
| Trf | Transferrin |
| TrfR | Transferrin receptor |
| TTR | Transthyretin |
| T1/2 | Elimination/circulation half-time |
| U.S. FDA | United States Food and drug administration |
| VLDL | Very-low-density lipoprotein |
| ZEBOV | Zaire species of EBOV |
| 3’-UTR | 3’ untranslated region |
References
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.J.; Baulcombe, D.C. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science 1999, 286, 950–952. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, S.M.; Lendeckel, W.; Tuschl, T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001, 15, 188–200. [Google Scholar] [CrossRef]
- Sandy, P.; Ventura, A.; Jacks, T. Mammalian RNAi: A practical guide. Biotechniques 2005, 39, 215–224. [Google Scholar] [CrossRef]
- Coutinho, M.F.; Matos, L.; Santos, J.I.; Alves, S. RNA Therapeutics: How Far Have We Gone? Adv. Exp. Med. Biol. 2019, 1157, 133–177. [Google Scholar] [CrossRef]
- Deng, Y.; Wang, C.C.; Choy, K.W.; Du, Q.; Chen, J.; Wang, Q.; Li, L.; Chung, T.K.; Tang, T. Therapeutic potentials of gene silencing by RNA interference: Principles, challenges, and new strategies. Gene 2014, 538, 217–227. [Google Scholar] [CrossRef]
- Kelly, J.M.; Bradbury, A.; Martin, D.R.; Byrne, M.E. Emerging therapies for neuropathic lysosomal storage disorders. Prog. Neurobiol. 2017, 152, 166–180. [Google Scholar] [CrossRef]
- Wraith, J.E. Lysosomal disorders. Paediatr. Child Health 2011, 21, 76–79. [Google Scholar] [CrossRef]
- Gritti, A. Gene therapy for lysosomal storage disorders. Expert Opin. Biol. 2011, 11, 1153–1167. [Google Scholar] [CrossRef]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Author Correction: Lysosomal storage diseases. Nat. Rev. Dis. Primers 2018, 4, 36. [Google Scholar] [CrossRef] [PubMed]
- Micsenyi, M.C.; Walkley, S.U. The lysosomal system: Physiology and pathology. In Lysosomal Storage Disorders—A Practial Guide, 1st ed.; Mehta, A., Winchester, B., Eds.; Wiley-Blackwell: West Sussex, UK, 2012; pp. 3–12. [Google Scholar]
- Raas-Rothschild, A.; Cormier-Daire, V.; Bao, M.; Genin, E.; Salomon, R.; Brewer, K.; Zeigler, M.; Mandel, H.; Toth, S.; Roe, B.; et al. Molecular basis of variant pseudo-hurler polydystrophy (mucolipidosis IIIC). J. Clin. Investig. 2000, 105, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Tiede, S.; Storch, S.; Lübke, T.; Henrissat, B.; Bargal, R.; Raas-Rothschild, A.; Braulke, T. Mucolipidosis II is caused by mutations in GNPTA encoding the alpha/beta GlcNAc-1-phosphotransferase. Nat. Med. 2005, 11, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Anikster, Y.; Lucero, C.; Guo, J.; Huizing, M.; Shotelersuk, V.; Bernardini, I.; McDowell, G.; Iwata, F.; Kaiser-Kupfer, M.I.; Jaffe, R.; et al. Ocular nonnephropathic cystinosis: Clinical, biochemical, and molecular correlations. Pediatr. Res. 2000, 47, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Winchester, B. Laboratory diagnosis of Lysosomal Storage Diseases. In Lysosomal Storage Disorders—A Practial Guide, 1st ed.; Metha, A., Winchester, B., Eds.; Wiley-Blackwell: West Sussex, UK, 2012; pp. 20–28. [Google Scholar]
- Lieberman, A.P.; Puertollano, R.; Raben, N.; Slaugenhaupt, S.; Walkley, S.U.; Ballabio, A. Autophagy in lysosomal storage disorders. Autophagy 2012, 8, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Walkley, S.U. Pathogenic cascades in lysosomal disease-Why so complex? J. Inherit. Metab. Dis. 2009, 32, 181–189. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Cox, T. Current Treatments. In Lysosomal Storage Disorders—A Practial Guide, 1st ed.; Mehta, A.B., Winchester, B., Eds.; Wiley-Blackwell: West Sussex, UK, 2012; pp. 153–165. [Google Scholar]
- Schultz, N.; Marenstein, D.R.; De Angelis, D.A.; Wang, W.Q.; Nelander, S.; Jacobsen, A.; Marks, D.S.; Massagué, J.; Sander, C. Off-target effects dominate a large-scale RNAi screen for modulators of the TGF-β pathway and reveal microRNA regulation of TGFBR2. Silence 2011, 2, 3. [Google Scholar] [CrossRef]
- Begley, D.; Scarpa, M. Central Nervous System Aspects, Neurodegeneration and the Blood-Brain Barrier. In Lysosomal Storage Disorders—A Practical Guide, 1st ed.; Mehta, A.B., Winchester, B., Eds.; Wiley-Blackwell: West Sussex, UK, 2012; pp. 166–173. [Google Scholar]
- Wraith, J.E.; Beck, M. Clinical Aspects and Clinical Diagnosis. In Lysosomal Storage Disorders—A Practial Guide, 1st ed.; Metha, A., Winchester, B., Eds.; Wiley-Blackwell: West Sussex, UK, 2012; pp. 13–19. [Google Scholar]
- Heese, B.A. Current strategies in the management of lysosomal storage diseases. Semin. Pediatr. Neurol. 2008, 15, 119–126. [Google Scholar] [CrossRef]
- Bley, A.E.; Giannikopoulos, O.A.; Hayden, D.; Kubilus, K.; Tifft, C.J.; Eichler, F.S. Natural history of infantile G(M2) gangliosidosis. Pediatrics 2011, 128, 1233–1241. [Google Scholar] [CrossRef]
- Coutinho, M.F.; Lacerda, L.; Alves, S. Glycosaminoglycan storage disorders: A review. Biochem. Res. Int. 2012, 2012, 471325. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Capece, G.; Cecio, A.; D’Auria, N.; Di Iorio, G.; Ronsisvalle, L.; Di Natale, P. Sanfilippo B syndrome (MPS III B): Case report with analysis of CSF mucopolysaccharides and conjunctival biopsy. J. Neurol. 1981, 225, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Gutierrez, M.A.; Ishimaru, T.; Peña, O.M.; Montaño, A.M.; Maeda, H.; Velez-Castrillon, S.; Nishioka, T.; Fachel, A.A.; Cooper, A.; et al. Heparan sulfate levels in mucopolysaccharidoses and mucolipidoses. J. Inherit. Metab. Dis. 2005, 28, 743–757. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Okamura, K.; Maeda, H.; Taketani, T.; Castrillon, S.V.; Gutierrez, M.A.; Nishioka, T.; Fachel, A.A.; Orii, K.O.; Grubb, J.H.; et al. Keratan sulphate levels in mucopolysaccharidoses and mucolipidoses. J. Inherit. Metab. Dis. 2005, 28, 187–202. [Google Scholar] [CrossRef]
- Munoz-Rojas, M.V.; Vieira, T.; Costa, R.; Fagondes, S.; John, A.; Jardim, L.B.; Vedolin, L.M.; Raymundo, M.; Dickson, P.I.; Kakkis, E.; et al. Intrathecal enzyme replacement therapy in a patient with mucopolysaccharidosis type I and symptomatic spinal cord compression. Am. J. Med. Genet. A 2008, 146A, 2538–2544. [Google Scholar] [CrossRef]
- Muenzer, J. Overview of the mucopolysaccharidoses. Rheumatology (Oxford) 2011, 50, 4–12. [Google Scholar] [CrossRef]
- Neufeld, E.U.; Muenzer, J. The Mucopolysaccharidoses. In The Metabolic Basis of Inherited Disease; Scriver, C.R., Ed.; McGraw-Hill: New York, NY, USA, 2001; pp. 3421–3452. [Google Scholar]
- Muenzer, J.; Wraith, J.E.; Clarke, L.A. Mucopolysaccharidosis I: Management and treatment guidelines. Pediatrics 2009, 123, 19–29. [Google Scholar] [CrossRef]
- Martin, R.; Beck, M.; Eng, C.; Giugliani, R.; Harmatz, P.; Muñoz, V.; Muenzer, J. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome). Pediatrics 2008, 121, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Heon-Roberts, R.; Nguyen, A.L.A.; Pshezhetsky, A.V. Molecular Bases of Neurodegeneration and Cognitive Decline, the Major Burden of Sanfilippo Disease. J. Clin. Med. 2020, 9, 344. [Google Scholar] [CrossRef]
- Paciotti, S.; Albi, E.; Parnetti, L.; Beccari, T. Lysosomal Ceramide Metabolism Disorders: Implications in Parkinson’s Disease. J. Clin. Med. 2020, 9, 594. [Google Scholar] [CrossRef]
- Aerts, J.M.F.G.; Kuo, C.L.; Lelieveld, L.T.; Boer, D.E.C.; van der Lienden, M.J.C.; Overkleeft, H.S.; Artola, M. Glycosphingolipids and lysosomal storage disorders as illustrated by gaucher disease. Curr. Opin. Chem. Biol. 2019, 53, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Elstein, D.; Zimran, A. Gaucher Disease. In Lysosomal Storage Disorders—A Practical Guide, 1st ed.; Mehta, A.B., Winchester, B., Eds.; Wiley-Blackwell: West Sussex, UK, 2012; pp. 49–57. [Google Scholar]
- Robak, L.A.; Jansen, I.E.; van Rooij, J.; Uitterlinden, A.G.; Kraaij, R.; Jankovic, J.; Heutink, P.; Shulman, J.M.; IPDGC. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 2017, 140, 3191–3203. [Google Scholar] [CrossRef] [PubMed]
- Blumenreich, S.; Barav, O.B.; Jenkins, B.J.; Futerman, A.H. Lysosomal Storage Disorders Shed Light on Lysosomal Dysfunction in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 4966. [Google Scholar] [CrossRef] [PubMed]
- Sun, A. Lysosomal storage disease overview. Ann. Transl. Med. 2018, 6, 476. [Google Scholar] [CrossRef] [PubMed]
- Mole, S.E.; Anderson, G.; Band, H.A.; Berkovic, S.F.; Cooper, J.D.; Kleine Holthaus, S.M.; McKay, T.R.; Medina, D.L.; Rahim, A.A.; Schulz, A.; et al. Clinical challenges and future therapeutic approaches for neuronal ceroid lipofuscinosis. Lancet. Neurol. 2019, 18, 107–116. [Google Scholar] [CrossRef]
- Cooper, D.C.; Williams, R.E. Neuronal Ceroid Lipofuscinoses. In Lysosomal Storage Disorders—A Practical Guide, 1st ed.; Mehta, A.B., Winchester, B., Eds.; Wiley-Blackwell: West Sussex, UK, 2012; pp. 3–12. [Google Scholar]
- Garbade, S.F.; Zielonka, M.; Mechler, K.; Kölker, S.; Hoffmann, G.F.; Staufner, C.; Mengel, E.; Ries, M. FDA orphan drug designations for lysosomal storage disorders—A cross-sectional analysis. PLoS ONE 2020, 15, e0230898. [Google Scholar] [CrossRef]
- Wraith, J.E.; Beck, M.; Lane, R.; van der Ploeg, A.; Shapiro, E.; Xue, Y.; Kakkis, E.D.; Guffon, N. Enzyme replacement therapy in patients who have mucopolysaccharidosis I and are younger than 5 years: Results of a multinational study of recombinant human alpha-L-iduronidase (laronidase). Pediatrics 2007, 120, 37–46. [Google Scholar] [CrossRef]
- Clarke, L.A.; Wraith, J.E.; Beck, M.; Kolodny, E.H.; Pastores, G.M.; Muenzer, J.; Rapoport, D.M.; Berger, K.I.; Sidman, M.; Kakkis, E.D.; et al. Long-term efficacy and safety of laronidase in the treatment of mucopolysaccharidosis I. Pediatrics 2009, 123, 229–240. [Google Scholar] [CrossRef]
- Muenzer, J.; Wraith, J.E.; Beck, M.; Giugliani, R.; Harmatz, P.; Eng, C.M.; Vellodi, A.; Martin, R.; Ramaswami, U.; Gucsavas-Calikoglu, M.; et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet. Med. 2006, 8, 465–473. [Google Scholar] [CrossRef]
- Harmatz, P.; Giugliani, R.; Schwartz, I.; Guffon, N.; Teles, E.L.; Miranda, M.C.; Wraith, J.E.; Beck, M.; Arash, L.; Scarpa, M.; et al. Enzyme replacement therapy for mucopolysaccharidosis VI: A phase 3, randomized, double-blind, placebo-controlled, multinational study of recombinant human N-acetylgalactosamine 4-sulfatase (recombinant human arylsulfatase B or rhASB) and follow-on, open-label extension study. J. Pediatr. 2006, 148, 533–539. [Google Scholar] [CrossRef]
- Muenzer, J.; Beck, M.; Giugliani, R.; Suzuki, Y.; Tylki-Szymanska, A.; Valayannopoulos, V.; Vellodi, A.; Wraith, J.E. Idursulfase treatment of Hunter syndrome in children younger than 6 years: Results from the Hunter Outcome Survey. Genet. Med. 2011, 13, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Muenzer, J.; Beck, M.; Eng, C.M.; Giugliani, R.; Harmatz, P.; Martin, R.; Ramaswami, U.; Vellodi, A.; Wraith, J.E.; Cleary, M.; et al. Long-term, open-labeled extension study of idursulfase in the treatment of Hunter syndrome. Genet. Med. 2011, 13, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Burton, B.K.; Whiteman, D.A.; Investigators, H. Incidence and timing of infusion-related reactions in patients with mucopolysaccharidosis type II (Hunter syndrome) on idursulfase therapy in the real-world setting: A perspective from the Hunter Outcome Survey (HOS). Mol. Genet. Metab. 2011, 103, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Sohar, I.; Wang, L.; Sleat, D.E.; Lobel, P. Systemic administration of tripeptidyl peptidase I in a mouse model of late infantile neuronal ceroid lipofuscinosis: Effect of glycan modification. PLoS ONE 2012, 7, e40509. [Google Scholar] [CrossRef]
- Veronese, F.M. Peptide and protein PEGylation: A review of problems and solutions. Biomaterials 2001, 22, 405–417. [Google Scholar] [CrossRef]
- Harris, J.M.; Chess, R.B. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2003, 2, 214–221. [Google Scholar] [CrossRef]
- Boado, R.J.; Lu, J.Z.; Hui, E.K.; Pardridge, W.M. Insulin receptor antibody-sulfamidase fusion protein penetrates the primate blood-brain barrier and reduces glycosoaminoglycans in Sanfilippo type A cells. Mol. Pharm. 2014, 11, 2928–2934. [Google Scholar] [CrossRef]
- Wang, D.; El-Amouri, S.S.; Dai, M.; Kuan, C.Y.; Hui, D.Y.; Brady, R.O.; Pan, D. Engineering a lysosomal enzyme with a derivative of receptor-binding domain of apoE enables delivery across the blood-brain barrier. Proc. Natl. Acad. Sci. USA 2013, 110, 2999–3004. [Google Scholar] [CrossRef]
- de Los Reyes, E.; Lehwald, L.; Augustine, E.F.; Berry-Kravis, E.; Butler, K.; Cormier, N.; Demarest, S.; Lu, S.; Madden, J.; Olaya, J.; et al. Intracerebroventricular Cerliponase Alfa for Neuronal Ceroid Lipofuscinosis Type 2 Disease: Clinical Practice Considerations From US Clinics. Pediatr. Neurol. 2020. [Google Scholar] [CrossRef]
- Stroobants, S.; Gerlach, D.; Matthes, F.; Hartmann, D.; Fogh, J.; Gieselmann, V.; D’Hooge, R.; Matzner, U. Intracerebroventricular enzyme infusion corrects central nervous system pathology and dysfunction in a mouse model of metachromatic leukodystrophy. Hum. Mol. Genet. 2011, 20, 2760–2769. [Google Scholar] [CrossRef][Green Version]
- Tsuji, D.; Akeboshi, H.; Matsuoka, K.; Yasuoka, H.; Miyasaki, E.; Kasahara, Y.; Kawashima, I.; Chiba, Y.; Jigami, Y.; Taki, T.; et al. Highly phosphomannosylated enzyme replacement therapy for GM2 gangliosidosis. Ann. Neurol. 2011, 69, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Kan, S.H.; Aoyagi-Scharber, M.; Le, S.Q.; Vincelette, J.; Ohmi, K.; Bullens, S.; Wendt, D.J.; Christianson, T.M.; Tiger, P.M.; Brown, J.R.; et al. Delivery of an enzyme-IGFII fusion protein to the mouse brain is therapeutic for mucopolysaccharidosis type IIIB. Proc. Natl. Acad. Sci. USA 2014, 111, 14870–14875. [Google Scholar] [CrossRef] [PubMed]
- Kan, S.H.; Le, S.; Vincellete, J.; Bullens, S.; Brown, J. Intracerebroventricular enzyme replacement therapy with glycosylation-independent lysosomal targeted NAGLU leads to widespread enzymatic activity, reduction of lysosomal storage and of secondary defects in brain of mice with Sanfilippo syndrome type B. Mol. Genet. Metab. 2014, 111, S59. [Google Scholar] [CrossRef]
- Specchio, N.; Pietrafusa, N.; Trivisano, M. Changing Times for CLN2 Disease: The Era of Enzyme Replacement Therapy. Clin. Risk Manag. 2020, 16, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.A.; Muller, V.J.; Hopwood, J.J. Stop-codon read-through for patients affected by a lysosomal storage disorder. Trends Mol. Med. 2006, 12, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Luddi, A.; Crifasi, L.; Capaldo, A.; Piomboni, P.; Costantino-Ceccarini, E. Suppression of galactocerebrosidase premature termination codon and rescue of galactocerebrosidase activity in twitcher cells. J. Neurosci. Res. 2016, 94, 1273–1283. [Google Scholar] [CrossRef]
- Banning, A.; Schiff, M.; Tikkanen, R. Amlexanox provides a potential therapy for nonsense mutations in the lysosomal storage disorder Aspartylglucosaminuria. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 668–675. [Google Scholar] [CrossRef]
- Matos, L.; Gonçalves, V.; Pinto, E.; Laranjeira, F.; Prata, M.J.; Jordan, P.; Desviat, L.R.; Pérez, B.; Alves, S. Functional analysis of splicing mutations in the IDS gene and the use of antisense oligonucleotides to exploit an alternative therapy for MPS II. Biochim. Biophys. Acta. 2015, 1852, 2712–2721. [Google Scholar] [CrossRef]
- Matos, L.; Duarte, A.J.; Ribeiro, D.; Chaves, J.; Amaral, O.; Alves, S. Correction of a Splicing Mutation Affecting an Unverricht-Lundborg Disease Patient by Antisense Therapy. Genes 2018, 9, 455. [Google Scholar] [CrossRef]
- Matos, L.; Vilela, R.; Rocha, M.; Santos, J.I.; Coutinho, M.F.; Gaspar, P.; Prata, M.J.; Alves, S. Development of an Antisense Oligonucleotide-Mediated Exon Skipping Therapeutic Strategy for Mucolipidosis II: Validation at RNA Level. Hum. Gene 2020, 31, 775–783. [Google Scholar] [CrossRef]
- Crivaro, A.N.; Mucci, J.M.; Bondar, C.M.; Ormazabal, M.E.; Ceci, R.; Simonaro, C.; Rozenfeld, P.A. Efficacy of pentosan polysulfate in in vitro models of lysosomal storage disorders: Fabry and Gaucher Disease. PLoS ONE 2019, 14, e0217780. [Google Scholar] [CrossRef] [PubMed]
- Gabig-Cimińska, M.; Jakóbkiewicz-Banecka, J.; Malinowska, M.; Kloska, A.; Piotrowska, E.; Chmielarz, I.; Moskot, M.; Węgrzyn, A.; Węgrzyn, G. Combined Therapies for Lysosomal Storage Diseases. Curr. Mol. Med. 2015, 15, 746–771. [Google Scholar] [CrossRef]
- Coutinho, M.F.; Santos, J.I.; Matos, L.; Alves, S. Genetic Substrate Reduction Therapy: A Promising Approach for Lysosomal Storage Disorders. Diseases 2016, 4, 33. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Rao, D.D.; Senzer, N.; Nemunaitis, J. RNA interference and cancer therapy. Pharm. Res. 2011, 28, 2983–2995. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, K.A.; Langer, R.; Anderson, D.G. Knocking down barriers: Advances in siRNA delivery. Nat. Rev. Drug Discov. 2009, 8, 129–138. [Google Scholar] [CrossRef]
- Aagaard, L.; Rossi, J.J. RNAi therapeutics: Principles, prospects and challenges. Adv. Drug Deliv. Rev. 2007, 59, 75–86. [Google Scholar] [CrossRef]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human MicroRNA targets. PloS Biol. 2004, 2, e363. [Google Scholar] [CrossRef]
- van de Water, F.M.; Boerman, O.C.; Wouterse, A.C.; Peters, J.G.; Russel, F.G.; Masereeuw, R. Intravenously administered short interfering RNA accumulates in the kidney and selectively suppresses gene function in renal proximal tubules. Drug Metab. Dispos. 2006, 34, 1393–1397. [Google Scholar] [CrossRef]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef]
- Guo, P.; Coban, O.; Snead, N.M.; Trebley, J.; Hoeprich, S.; Guo, S.; Shu, Y. Engineering RNA for targeted siRNA delivery and medical application. Adv. Drug Deliv. Rev. 2010, 62, 650–666. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.F.; Liu, Y.; Shen, S.; Zhu, Y.H.; Wang, J. Targeting glucose uptake of glioma cells by siRNA delivery with polymer nanoparticle. J. Control. Release 2015, 213, 23–24. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Guenthner-Biller, M.; Bourquin, C.; Ablasser, A.; Schlee, M.; Uematsu, S.; Noronha, A.; Manoharan, M.; Akira, S.; de Fougerolles, A.; et al. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat. Med. 2005, 11, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Birmingham, A.; Anderson, E.M.; Reynolds, A.; Ilsley-Tyree, D.; Leake, D.; Fedorov, Y.; Baskerville, S.; Maksimova, E.; Robinson, K.; Karpilow, J.; et al. 3′ UTR seed matches, but not overall identity, are associated with RNAi off-targets. Nat. Methods 2006, 3, 199–204. [Google Scholar] [CrossRef]
- Jackson, A.L.; Burchard, J.; Schelter, J.; Chau, B.N.; Cleary, M.; Lim, L.; Linsley, P.S. Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA 2006, 12, 1179–1187. [Google Scholar] [CrossRef]
- Jackson, A.L.; Linsley, P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010, 9, 57–67. [Google Scholar] [CrossRef]
- Zhang, B.; Mallapragada, S. The mechanism of selective transfection mediated by pentablock copolymers; part II: Nuclear entry and endosomal escape. Acta. Biomater. 2011, 7, 1580–1587. [Google Scholar] [CrossRef]
- Gomes, M.J.; Martins, S.; Sarmento, B. siRNA as a tool to improve the treatment of brain diseases: Mechanism, targets and delivery. Ageing Res. Rev. 2015, 21, 43–54. [Google Scholar] [CrossRef]
- Diaz-Font, A.; Chabás, A.; Grinberg, D.; Vilageliu, L. RNAi-mediated inhibition of the glucosylceramide synthase (GCS) gene: A preliminary study towards a therapeutic strategy for Gaucher disease and other glycosphingolipid storage diseases. Blood Cells Mol. Dis. 2006, 37, 197–203. [Google Scholar] [CrossRef]
- Zimran, A.; Elstein, D. Management of Gaucher disease: Enzyme replacement therapy. Pediatr. Endocrinol. Rev. 2014, 12, 82–87. [Google Scholar]
- Cox, T.; Lachmann, R.; Hollak, C.; Aerts, J.; van Weely, S.; Hrebícek, M.; Platt, F.; Butters, T.; Dwek, R.; Moyses, C.; et al. Novel oral treatment of Gaucher’s disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet 2000, 355, 1481–1485. [Google Scholar] [CrossRef]
- Venier, R.E.; Igdoura, S.A. Miglustat as a therapeutic agent: Prospects and caveats. J. Med. Genet. 2012, 49, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Nabizadeh, A.; Amani, B.; Kadivar, M.; Toroski, M.; Asl, A.A.; Bayazidi, Y.; Mojahedian, M.; Davari, M. The Clinical Efficacy of Imiglucerase versus Eliglustat in Patients with Gaucher’s Disease Type 1: A Systematic Review. J. Res. Pharm. Pr. 2018, 7, 171–177. [Google Scholar] [CrossRef]
- Wegrzyn, G.; Jakóbkiewicz-Banecka, J.; Gabig-Cimińska, M.; Piotrowska, E.; Narajczyk, M.; Kloska, A.; Malinowska, M.; Dziedzic, D.; Gołebiewska, I.; Moskot, M.; et al. Genistein: A natural isoflavone with a potential for treatment of genetic diseases. Biochem. Soc. Trans. 2010, 38, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Derrick-Roberts, A.L.; Marais, W.; Byers, S. Rhodamine B and 2-acetamido-1,3,6-tri-O-acetyl-4-deoxy-4-fluoro-D-glucopyranose (F-GlcNAc) inhibit chondroitin/dermatan and keratan sulphate synthesis by different mechanisms in bovine chondrocytes. Mol. Genet. Metab. 2012, 106, 214–220. [Google Scholar] [CrossRef]
- Dziedzic, D.; Wegrzyn, G.; Jakóbkiewicz-Banecka, J. Impairment of glycosaminoglycan synthesis in mucopolysaccharidosis type IIIA cells by using siRNA: A potential therapeutic approach for Sanfilippo disease. Eur. J. Hum. Genet. 2010, 18, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Dziedzic, D.; Narajczyk, M.; Gabig-Cimińska, M.; Jakóbkiewicz-Banecka, J. Simultaneous siRNA-mediated silencing of pairs of genes coding for enzymes involved in glycosaminoglycan synthesis. Acta Biochim. Pol. 2012, 59, 293–298. [Google Scholar] [CrossRef]
- Kaidonis, X.; Liaw, W.C.; Roberts, A.D.; Ly, M.; Anson, D.; Byers, S. Gene silencing of EXTL2 and EXTL3 as a substrate deprivation therapy for heparan sulphate storing mucopolysaccharidoses. Eur. J. Hum. Genet. 2010, 18, 194–199. [Google Scholar] [CrossRef]
- Chmielarz, I.; Gabig-Cimińska, M.; Malinowska, M.; Banecka-Majkutewicz, Z.; Węgrzyn, A.; Jakobkiewicz-Banecka, J. Comparison of siRNA-mediated silencing of glycosaminoglycan synthesis genes and enzyme replacement therapy for mucopolysaccharidosis in cell culture studies. Acta. Biochim. Pol. 2012, 59, 697–702. [Google Scholar] [CrossRef]
- Canals, I.; Benetó, N.; Cozar, M.; Vilageliu, L.; Grinberg, D. EXTL2 and EXTL3 inhibition with siRNAs as a promising substrate reduction therapy for Sanfilippo C syndrome. Sci. Rep. 2015, 5, 13654. [Google Scholar] [CrossRef]
- Benetó, N.; Cozar, M.; García-Morant, M.; Creus-Bachiller, E.; Vilageliu, L.; Grinberg, D.; Canals, I. Generation of two compound heterozygous HGSNAT-mutated lines from healthy induced pluripotent stem cells using CRISPR/Cas9 to model Sanfilippo C syndrome. Stem Cell Res. 2019, 41, 101616. [Google Scholar] [CrossRef] [PubMed]
- Benetó, N.; Cozar, M.; Castilla-Vallmanya, L.; Zetterdahl, O.G.; Sacultanu, M.; Segur-Bailach, E.; García-Morant, M.; Ribes, A.; Ahlenius, H.; Grinberg, D.; et al. Neuronal and Astrocytic Differentiation from Sanfilippo C Syndrome iPSCs for Disease Modeling and Drug Development. J. Clin. Med. 2020, 9, 644. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M. Emptying the stores: Lysosomal diseases and therapeutic strategies. Nat. Rev. Drug Discov. 2018, 17, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Vaishnaw, A.K.; Gollob, J.; Gamba-Vitalo, C.; Hutabarat, R.; Sah, D.; Meyers, R.; de Fougerolles, T.; Maraganore, J. A status report on RNAi therapeutics. Silence 2010, 1, 14. [Google Scholar] [CrossRef]
- Gomes-da-Silva, L.C.; Fonseca, N.A.; Moura, V.; Pedroso de Lima, M.C.; Simões, S.; Moreira, J.N. Lipid-based nanoparticles for siRNA delivery in cancer therapy: Paradigms and challenges. ACC Chem. Res. 2012, 45, 1163–1171. [Google Scholar] [CrossRef]
- Tatiparti, K.; Sau, S.; Kashaw, S.K.; Iyer, A.K. siRNA Delivery Strategies: A Comprehensive Review of Recent Developments. Nanomaterials 2017, 7, 77. [Google Scholar] [CrossRef]
- Akinc, A.; Zumbuehl, A.; Goldberg, M.; Leshchiner, E.S.; Busini, V.; Hossain, N.; Bacallado, S.A.; Nguyen, D.N.; Fuller, J.; Alvarez, R.; et al. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat. Biotechnol. 2008, 26, 561–569. [Google Scholar] [CrossRef]
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.; Stebbing, D.; Crosley, E.J.; Yaworski, E.; et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010, 28, 172–176. [Google Scholar] [CrossRef]
- Shi, B.; Keough, E.; Matter, A.; Leander, K.; Young, S.; Carlini, E.; Sachs, A.B.; Tao, W.; Abrams, M.; Howell, B.; et al. Biodistribution of small interfering RNA at the organ and cellular levels after lipid nanoparticle-mediated delivery. J. Histochem. Cytochem. 2011, 59, 727–740. [Google Scholar] [CrossRef]
- Wittrup, A.; Lieberman, J. Knocking down disease: A progress report on siRNA therapeutics. Nat. Rev. Genet. 2015, 16, 543–552. [Google Scholar] [CrossRef]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R.; et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Gooding, M.; Browne, L.P.; Quinteiro, F.M.; Selwood, D.L. siRNA delivery: From lipids to cell-penetrating peptides and their mimics. Chem. Biol. Drug Des. 2012, 80, 787–809. [Google Scholar] [CrossRef] [PubMed]
- Czech, M.P.; Aouadi, M.; Tesz, G.J. RNAi-based therapeutic strategies for metabolic disease. Nat. Rev. Endocrinol. 2011, 7, 473–484. [Google Scholar] [CrossRef]
- Coelho, T.; Adams, D.; Silva, A.; Lozeron, P.; Hawkins, P.N.; Mant, T.; Perez, J.; Chiesa, J.; Warrington, S.; Tranter, E.; et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N. Engl. J. Med. 2013, 369, 819–829. [Google Scholar] [CrossRef]
- Suhr, O.B.; Coelho, T.; Buades, J.; Pouget, J.; Conceicao, I.; Berk, J.; Schmidt, H.; Waddington-Cruz, M.; Campistol, J.M.; Bettencourt, B.R.; et al. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: A phase II multi-dose study. Orphanet J. Rare Dis. 2015, 10, 109. [Google Scholar] [CrossRef] [PubMed]
- Robbins, M.; Judge, A.; MacLachlan, I. siRNA and innate immunity. Oligonucleotides 2009, 19, 89–102. [Google Scholar] [CrossRef]
- Crooke, S.T.; Witztum, J.L.; Bennett, C.F.; Baker, B.F. RNA-Targeted Therapeutics. Cell Metab. 2018, 27, 714–739. [Google Scholar] [CrossRef]
- Xie, F.Y.; Woodle, M.C.; Lu, P.Y. Harnessing in vivo siRNA delivery for drug discovery and therapeutic development. Drug Discov. Today 2006, 11, 67–73. [Google Scholar] [CrossRef]
- Schiffelers, R.M.; Mixson, A.J.; Ansari, A.M.; Fens, M.H.; Tang, Q.; Zhou, Q.; Xu, J.; Molema, G.; Lu, P.Y.; Scaria, P.V.; et al. Transporting silence: Design of carriers for siRNA to angiogenic endothelium. J. Control. Release 2005, 109, 5–14. [Google Scholar] [CrossRef]
- Ishida, T.; Atobe, K.; Wang, X.; Kiwada, H. Accelerated blood clearance of PEGylated liposomes upon repeated injections: Effect of doxorubicin-encapsulation and high-dose first injection. J. Control. Release 2006, 115, 251–258. [Google Scholar] [CrossRef]
- Ishida, T.; Ichihara, M.; Wang, X.; Yamamoto, K.; Kimura, J.; Majima, E.; Kiwada, H. Injection of PEGylated liposomes in rats elicits PEG-specific IgM, which is responsible for rapid elimination of a second dose of PEGylated liposomes. J. Control. Release 2006, 112, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Song, L.Y.; Ahkong, Q.F.; Rong, Q.; Wang, Z.; Ansell, S.; Hope, M.J.; Mui, B. Characterization of the inhibitory effect of PEG-lipid conjugates on the intracellular delivery of plasmid and antisense DNA mediated by cationic lipid liposomes. Biochim. Biophys. Acta 2002, 1558, 1–13. [Google Scholar] [CrossRef]
- Mishra, S.; Webster, P.; Davis, M.E. PEGylation significantly affects cellular uptake and intracellular trafficking of non-viral gene delivery particles. Eur. J. Cell Biol. 2004, 83, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, D.V.; Blanchard, K.; Shaw, L.; Jensen, K.; Lockridge, J.A.; Dickinson, B.; McSwiggen, J.A.; Vargeese, C.; Bowman, K.; Shaffer, C.S.; et al. Activity of stabilized short interfering RNA in a mouse model of hepatitis B virus replication. Hepatology 2005, 41, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, D.V.; Lockridge, J.A.; Shaw, L.; Blanchard, K.; Jensen, K.; Breen, W.; Hartsough, K.; Machemer, L.; Radka, S.; Jadhav, V.; et al. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat. Biotechnol. 2005, 23, 1002–1007. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, T.S.; Lee, A.C.; Akinc, A.; Bramlage, B.; Bumcrot, D.; Fedoruk, M.N.; Harborth, J.; Heyes, J.A.; Jeffs, L.B.; John, M.; et al. RNAi-mediated gene silencing in non-human primates. Nature 2006, 441, 111–114. [Google Scholar] [CrossRef]
- Cannon, C.P.; Braunwald, E.; McCabe, C.H.; Rader, D.J.; Rouleau, J.L.; Belder, R.; Joyal, S.V.; Hill, K.A.; Pfeffer, M.A.; Skene, A.M.; et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N. Engl. J. Med. 2004, 350, 1495–1504. [Google Scholar] [CrossRef]
- Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Geick, A.; Hadwiger, P.; Harborth, J.; et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 2004, 432, 173–178. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Hensley, L.E.; Kagan, E.; Yu, E.Z.; Geisbert, J.B.; Daddario-DiCaprio, K.; Fritz, E.A.; Jahrling, P.B.; McClintock, K.; Phelps, J.R.; et al. Postexposure protection of guinea pigs against a lethal ebola virus challenge is conferred by RNA interference. J. Infect. Dis. 2006, 193, 1650–1657. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Lee, A.C.; Robbins, M.; Geisbert, J.B.; Honko, A.N.; Sood, V.; Johnson, J.C.; de Jong, S.; Tavakoli, I.; Judge, A.; et al. Postexposure protection of non-human primates against a lethal Ebola virus challenge with RNA interference: A proof-of-concept study. Lancet 2010, 375, 1896–1905. [Google Scholar] [CrossRef]
- Judge, A.D.; Robbins, M.; Tavakoli, I.; Levi, J.; Hu, L.; Fronda, A.; Ambegia, E.; McClintock, K.; MacLachlan, I. Confirming the RNAi-mediated mechanism of action of siRNA-based cancer therapeutics in mice. J. Clin. Investig. 2009, 119, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; South, V.J.; Zhang, Y.; Davide, J.P.; Farrell, L.; Kohl, N.E.; Sepp-Lorenzino, L.; Lobell, R.B. Induction of apoptosis by an inhibitor of the mitotic kinesin KSP requires both activation of the spindle assembly checkpoint and mitotic slippage. Cancer Cell 2005, 8, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Erikson, R.L. Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 5789–5794. [Google Scholar] [CrossRef] [PubMed]
- Steegmaier, M.; Hoffmann, M.; Baum, A.; Lénárt, P.; Petronczki, M.; Krssák, M.; Gürtler, U.; Garin-Chesa, P.; Lieb, S.; Quant, J.; et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol. 2007, 17, 316–322. [Google Scholar] [CrossRef]
- Adams, D.; Coelho, T.; Conceicao, I.; Cruz, M.W.n.; Schmidt, H.; Buades, J.; Campistol, J.; Pouget, J.; Berk, J.; Polydefkis, M.; et al. Phase 2 Open-Label Extension (OLE) Study of Patisiran, an Investigational RNA interference (RNAi) Therapeutic for the Treatment of Hereditary ATTR Amyloidosis with Polyneuropathy (S27.004). Neurology 2017, 88, S27.004. [Google Scholar]
- Adams, D.; Suhr, O.B.; Dyck, P.J.; Litchy, W.J.; Leahy, R.G.; Chen, J.; Gollob, J.; Coelho, T. Trial design and rationale for APOLLO, a Phase 3, placebo-controlled study of patisiran in patients with hereditary ATTR amyloidosis with polyneuropathy. BMC Neurol. 2017, 17, 181. [Google Scholar] [CrossRef]
- Hoy, S.M. Patisiran: First Global Approval. Drugs 2018, 78, 1625–1631. [Google Scholar] [CrossRef]
- Iyer, A.K.; Khaled, G.; Fang, J.; Maeda, H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov. Today 2006, 11, 812–818. [Google Scholar] [CrossRef]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef]
- Danhier, F. To exploit the tumor microenvironment: Since the EPR effect fails in the clinic, what is the future of nanomedicine? J. Control. Release 2016, 244, 108–121. [Google Scholar] [CrossRef]
- Mendonça, L.S.; Firmino, F.; Moreira, J.N.; Pedroso de Lima, M.C.; Simões, S. Transferrin receptor-targeted liposomes encapsulating anti-BCR-ABL siRNA or asODN for chronic myeloid leukemia treatment. Bioconjug. Chem. 2010, 21, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Savona, M.; Talpaz, M. Getting to the stem of chronic myeloid leukaemia. Nat. Rev. Cancer 2008, 8, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Ponka, P.; Lok, C.N. The transferrin receptor: Role in health and disease. Int. J. Biochem. Cell Biol. 1999, 31, 1111–1137. [Google Scholar] [CrossRef]
- Li, H.; Qian, Z.M. Transferrin/transferrin receptor-mediated drug delivery. Med. Res. Rev. 2002, 22, 225–250. [Google Scholar] [CrossRef] [PubMed]
- Daniels, T.R.; Delgado, T.; Rodriguez, J.A.; Helguera, G.; Penichet, M.L. The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin. Immunol. 2006, 121, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Daniels, T.R.; Delgado, T.; Helguera, G.; Penichet, M.L. The transferrin receptor part II: Targeted delivery of therapeutic agents into cancer cells. Clin. Immunol. 2006, 121, 159–176. [Google Scholar] [CrossRef]
- Savage, D.G.; Antman, K.H. Imatinib mesylate--a new oral targeted therapy. N. Engl. J. Med. 2002, 346, 683–693. [Google Scholar] [CrossRef]
- Walz, C.; Sattler, M. Novel targeted therapies to overcome imatinib mesylate resistance in chronic myeloid leukemia (CML). Crit. Rev. Oncol. Hematol. 2006, 57, 145–164. [Google Scholar] [CrossRef]
- Mendonça, L.S.; Moreira, J.N.; de Lima, M.C.; Simões, S. Co-encapsulation of anti-BCR-ABL siRNA and imatinib mesylate in transferrin receptor-targeted sterically stabilized liposomes for chronic myeloid leukemia treatment. Biotechnol. Bioeng. 2010, 107, 884–893. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Christian, S.; Pilch, J.; Akerman, M.E.; Porkka, K.; Laakkonen, P.; Ruoslahti, E. Nucleolin expressed at the cell surface is a marker of endothelial cells in angiogenic blood vessels. J. Cell Biol. 2003, 163, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Porkka, K.; Laakkonen, P.; Hoffman, J.A.; Bernasconi, M.; Ruoslahti, E. A fragment of the HMGN2 protein homes to the nuclei of tumor cells and tumor endothelial cells in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 7444–7449. [Google Scholar] [CrossRef] [PubMed]
- Gomes-da-Silva, L.C.; Fernández, Y.; Abasolo, I.; Schwartz, S.; Ramalho, J.S.; Pedroso de Lima, M.C.; Simões, S.; Moreira, J.N. Efficient intracellular delivery of siRNA with a safe multitargeted lipid-based nanoplatform. Nanomedicine (London) 2013, 8, 1397–1413. [Google Scholar] [CrossRef] [PubMed]
- Gomes-da-Silva, L.C.; Santos, A.O.; Bimbo, L.M.; Moura, V.; Ramalho, J.S.; Pedroso de Lima, M.C.; Simões, S.; Moreira, J.N. Toward a siRNA-containing nanoparticle targeted to breast cancer cells and the tumor microenvironment. Int. J. Pharm. 2012, 434, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Gomes-da-Silva, L.C.; Ramalho, J.S.; Pedroso de Lima, M.C.; Simões, S.; Moreira, J.N. Impact of anti-PLK1 siRNA-containing F3-targeted liposomes on the viability of both cancer and endothelial cells. Eur. J. Pharm. Biopharm. 2013, 85, 356–364. [Google Scholar] [CrossRef]
- Moura, V.; Lacerda, M.; Figueiredo, P.; Corvo, M.L.; Cruz, M.E.; Soares, R.; de Lima, M.C.; Simões, S.; Moreira, J.N. Targeted and intracellular triggered delivery of therapeutics to cancer cells and the tumor microenvironment: Impact on the treatment of breast cancer. Breast Cancer Res. Treat. 2012, 133, 61–73. [Google Scholar] [CrossRef]
- Fonseca, N.A.; Gomes-da-Silva, L.C.; Moura, V.; Simões, S.; Moreira, J.N. Simultaneous active intracellular delivery of doxorubicin and C6-ceramide shifts the additive/antagonistic drug interaction of non-encapsulated combination. J. Control. Release 2014, 196, 122–131. [Google Scholar] [CrossRef]
- Fonseca, N.A.; Rodrigues, A.S.; Rodrigues-Santos, P.; Alves, V.; Gregório, A.C.; Valério-Fernandes, Â.; Gomes-da-Silva, L.C.; Rosa, M.S.; Moura, V.; Ramalho-Santos, J.; et al. Nucleolin overexpression in breast cancer cell sub-populations with different stem-like phenotype enables targeted intracellular delivery of synergistic drug combination. Biomaterials 2015, 69, 76–88. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef]
- Costa, P.M.; Cardoso, A.L.; Mendonça, L.S.; Serani, A.; Custódia, C.; Conceição, M.; Simões, S.; Moreira, J.N.; Pereira de Almeida, L.; Pedroso de Lima, M.C. Tumor-targeted Chlorotoxin-coupled Nanoparticles for Nucleic Acid Delivery to Glioblastoma Cells: A Promising System for Glioblastoma Treatment. Mol. Nucleic Acids 2013, 2, e100. [Google Scholar] [CrossRef]
- Costa, P.M.; Cardoso, A.L.; Custódia, C.; Cunha, P.; Pereira de Almeida, L.; Pedroso de Lima, M.C. MiRNA-21 silencing mediated by tumor-targeted nanoparticles combined with sunitinib: A new multimodal gene therapy approach for glioblastoma. J. Control. Release 2015, 207, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, C.; Desviat, L.R.; Smedsrod, B.; Pietri-Rouxel, F.; Denti, M.A.; Disterer, P.; Lorain, S.; Nogales-Gadea, G.; Sardone, V.; Anwar, R.; et al. Delivery is key: Lessons learnt from developing splice-switching antisense therapies. Embo. Mol. Med. 2017, 9, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Gabathuler, R. Approaches to transport therapeutic drugs across the blood-brain barrier to treat brain diseases. Neurobiol. Dis. 2010, 37, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Simões, S.; Slepushkin, V.; Gaspar, R.; de Lima, M.C.; Düzgüneş, N. Gene delivery by negatively charged ternary complexes of DNA, cationic liposomes and transferrin or fusigenic peptides. Gene Ther. 1998, 5, 955–964. [Google Scholar] [CrossRef]
- Simões, S.; Slepushkin, V.; Pires, P.; Gaspar, R.; de Lima, M.P.; Düzgüneş, N. Mechanisms of gene transfer mediated by lipoplexes associated with targeting ligands or pH-sensitive peptides. Gene Ther. 1999, 6, 1798–1807. [Google Scholar] [CrossRef]
- Simões, S.; Slepushkin, V.; Pretzer, E.; Dazin, P.; Gaspar, R.; Pedroso de Lima, M.C.; Düzgüneş, N. Transfection of human macrophages by lipoplexes via the combined use of transferrin and pH-sensitive peptides. J. Leukoc. Biol. 1999, 65, 270–279. [Google Scholar] [CrossRef]
- Tros de Ilarduya, C.; Düzgüneş, N. Efficient gene transfer by transferrin lipoplexes in the presence of serum. Biochim. Biophys. Acta 2000, 1463, 333–342. [Google Scholar] [CrossRef]
- da Cruz, M.T.; Simões, S.; de Lima, M.C. Improving lipoplex-mediated gene transfer into C6 glioma cells and primary neurons. Exp. Neurol. 2004, 187, 65–75. [Google Scholar] [CrossRef]
- da Cruz, M.T.; Cardoso, A.L.; de Almeida, L.P.; Simões, S.; de Lima, M.C. Tf-lipoplex-mediated NGF gene transfer to the CNS: Neuronal protection and recovery in an excitotoxic model of brain injury. Gene 2005, 12, 1242–1252. [Google Scholar] [CrossRef]
- Cardoso, A.L.; Simões, S.; de Almeida, L.P.; Pelisek, J.; Culmsee, C.; Wagner, E.; Pedroso de Lima, M.C. siRNA delivery by a transferrin-associated lipid-based vector: A non-viral strategy to mediate gene silencing. J. Gene Med. 2007, 9, 170–183. [Google Scholar] [CrossRef]
- Cardoso, A.L.; Simões, S.; de Almeida, L.P.; Plesnila, N.; Pedroso de Lima, M.C.; Wagner, E.; Culmsee, C. Tf-lipoplexes for neuronal siRNA delivery: A promising system to mediate gene silencing in the CNS. J. Control. Release 2008, 132, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.L.; Costa, P.; de Almeida, L.P.; Simões, S.; Plesnila, N.; Culmsee, C.; Wagner, E.; de Lima, M.C. Tf-lipoplex-mediated c-Jun silencing improves neuronal survival following excitotoxic damage in vivo. J. Control. Release 2010, 142, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Son, S.; Hwang, D.W.; Singha, K.; Jeong, J.H.; Park, T.G.; Lee, D.S.; Kim, W.J. RVG peptide tethered bioreducible polyethylenimine for gene delivery to brain. J. Control. Release 2011, 155, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Huey, R.; Hawthorne, S.; McCarron, P. The potential use of rabies virus glycoprotein-derived peptides to facilitate drug delivery into the central nervous system: A mini review. J. Drug Target. 2017, 25, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.; Zhao, Z.; Gao, F.; Zhang, M. Cellular uptake mechanism and therapeutic utility of a novel peptide in targeted-delivery of proteins into neuronal cells. Pharm. Res. 2013, 30, 2108–2117. [Google Scholar] [CrossRef] [PubMed]
- Oswald, M.; Geissler, S.; Goepferich, A. Targeting the Central Nervous System (CNS): A Review of Rabies Virus-Targeting Strategies. Mol. Pharm. 2017, 14, 2177–2196. [Google Scholar] [CrossRef]
- Cantin, E.M.; Rossi, J.J. Molecular medicine: Entry granted. Nature 2007, 448, 33–34. [Google Scholar] [CrossRef]
- Kumar, P.; Wu, H.; McBride, J.L.; Jung, K.E.; Kim, M.H.; Davidson, B.L.; Lee, S.K.; Shankar, P.; Manjunath, N. Transvascular delivery of small interfering RNA to the central nervous system. Nature 2007, 448, 39–43. [Google Scholar] [CrossRef]
- Lewis, P.; Fu, Y.; Lentz, T.L. Rabies virus entry at the neuromuscular junction in nerve-muscle cocultures. Muscle Nerve 2000, 23, 720–730. [Google Scholar] [CrossRef]
- Hislop, J.N.; Islam, T.A.; Eleftheriadou, I.; Carpentier, D.C.; Trabalza, A.; Parkinson, M.; Schiavo, G.; Mazarakis, N.D. Rabies virus envelope glycoprotein targets lentiviral vectors to the axonal retrograde pathway in motor neurons. J. Biol. Chem. 2014, 289, 16148–16163. [Google Scholar] [CrossRef]
- Kwon, E.J.; Lasiene, J.; Jacobson, B.E.; Park, I.K.; Horner, P.J.; Pun, S.H. Targeted nonviral delivery vehicles to neural progenitor cells in the mouse subventricular zone. Biomaterials 2010, 31, 2417–2424. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Ke, W.; Liu, Y.; Wu, D.; Feng, L.; Jiang, C.; Pei, Y. Gene therapy using lactoferrin-modified nanoparticles in a rotenone-induced chronic Parkinson model. J. Neurol. Sci. 2010, 290, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.J.; Wei, K.C.; Ma, C.C.; Yang, S.Y.; Chen, J.P. Dual targeted delivery of doxorubicin to cancer cells using folate-conjugated magnetic multi-walled carbon nanotubes. Colloids Surf. B. Biointerfaces 2012, 89, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.Q.; Lv, Q.; Li, L.M.; Tang, X.J.; Li, F.Z.; Hu, Y.L.; Han, M. Glioma targeting and blood-brain barrier penetration by dual-targeting doxorubincin liposomes. Biomaterials 2013, 34, 5628–5639. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Loh, X.J. Recent advances of using polyhydroxyalkanoate-based nanovehicles as therapeutic delivery carriers. Nanomed. Nanobiotechnol. 2016, 9, e1429. [Google Scholar] [CrossRef] [PubMed]
- Byeon, H.J.; Thao, l.Q.; Lee, S.; Min, S.Y.; Lee, E.S.; Shin, B.S.; Choi, H.G.; Youn, Y.S. Doxorubicin-loaded nanoparticles consisted of cationic- and mannose-modified-albumins for dual-targeting in brain tumors. J. Control. Release 2016, 225, 301–313. [Google Scholar] [CrossRef]
- Shevtsov, M.A.; Nikolaev, B.P.; Yakovleva, L.Y.; Dobrodumov, A.V.; Zhakhov, A.V.; Mikhrina, A.L.; Pitkin, E.; Parr, M.A.; Rolich, V.I.; Simbircev, A.S.; et al. Recombinant interleukin-1 receptor antagonist conjugated to superparamagnetic iron oxide nanoparticles for theranostic targeting of experimental glioblastoma. Neoplasia 2015, 17, 32–42. [Google Scholar] [CrossRef]
- Meng, J.; Agrahari, V.; Youm, I. Advances in Targeted Drug Delivery Approaches for the Central Nervous System Tumors: The Inspiration of Nanobiotechnology. J. Neuroimmune Pharm. 2017, 12, 84–98. [Google Scholar] [CrossRef]
- Conceição, M.; Mendonça, L.; Nóbrega, C.; Gomes, C.; Costa, P.; Hirai, H.; Moreira, J.N.; Lima, M.C.; Manjunath, N.; Pereira de Almeida, L. Intravenous administration of brain-targeted stable nucleic acid lipid particles alleviates Machado-Joseph disease neurological phenotype. Biomaterials 2016, 82, 124–137. [Google Scholar] [CrossRef]
- Santos, A.O.; da Silva, L.C.; Bimbo, L.M.; de Lima, M.C.; Simões, S.; Moreira, J.N. Design of peptide-targeted liposomes containing nucleic acids. Biochim. Biophys. Acta 2010, 1798, 433–441. [Google Scholar] [CrossRef]
- Borel, F.; Kay, M.A.; Mueller, C. Recombinant AAV as a platform for translating the therapeutic potential of RNA interference. Mol. Ther. 2014, 22, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Kleine Holthaus, S.M.; Smith, A.J.; Mole, S.E.; Ali, R.R. Gene Therapy Approaches to Treat the Neurodegeneration and Visual Failure in Neuronal Ceroid Lipofuscinoses. Adv. Exp. Med. Biol. 2018, 1074, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Haney, M.J.; Zhao, Y.; Jin, Y.S.; Batrakova, E.V. Extracellular Vesicles as Drug Carriers for Enzyme Replacement Therapy to Treat CLN2 Batten Disease: Optimization of Drug Administration Routes. Cells 2020, 9, 1273. [Google Scholar] [CrossRef]
- Banks, W.A.; Farr, S.A.; Butt, W.; Kumar, V.B.; Franko, M.W.; Morley, J.E. Delivery across the blood-brain barrier of antisense directed against amyloid beta: Reversal of learning and memory deficits in mice overexpressing amyloid precursor protein. J. Pharm. Exp. Ther. 2001, 297, 1113–1121. [Google Scholar]
- Lee, H.J.; Boado, R.J.; Braasch, D.A.; Corey, D.R.; Pardridge, W.M. Imaging gene expression in the brain in vivo in a transgenic mouse model of Huntington’s disease with an antisense radiopharmaceutical and drug-targeting technology. J. Nucl. Med. 2002, 43, 948–956. [Google Scholar]
- Kozlu, S.; Caban, S.; Yerlikaya, F.; Fernandez-Megia, E.; Novoa-Carballal, R.; Riguera, R.; Yemisci, M.; Gursoy-Ozdemir, Y.; Dalkara, T.; Couvreur, P.; et al. An aquaporin 4 antisense oligonucleotide loaded, brain targeted nanoparticulate system design. Pharmazie 2014, 69, 340–345. [Google Scholar]
- Matos, L.; Canals, I.; Dridi, L.; Choi, Y.; Prata, M.J.; Jordan, P.; Desviat, L.R.; Pérez, B.; Pshezhetsky, A.V.; Grinberg, D.; et al. Therapeutic strategies based on modified U1 snRNAs and chaperones for Sanfilippo C splicing mutations. Orphanet J. Rare Dis. 2014, 9, 180. [Google Scholar] [CrossRef]
- Di Paolo, D.; Brignole, C.; Pastorino, F.; Carosio, R.; Zorzoli, A.; Rossi, M.; Loi, M.; Pagnan, G.; Emionite, L.; Cilli, M.; et al. Neuroblastoma-targeted nanoparticles entrapping siRNA specifically knockdown ALK. Mol. Ther. 2011, 19, 1131–1140. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coutinho, M.F.; Santos, J.I.; S. Mendonça, L.; Matos, L.; Prata, M.J.; S. Jurado, A.; Pedroso de Lima, M.C.; Alves, S. Lysosomal Storage Disease-Associated Neuropathy: Targeting Stable Nucleic Acid Lipid Particle (SNALP)-Formulated siRNAs to the Brain as a Therapeutic Approach. Int. J. Mol. Sci. 2020, 21, 5732. https://doi.org/10.3390/ijms21165732
Coutinho MF, Santos JI, S. Mendonça L, Matos L, Prata MJ, S. Jurado A, Pedroso de Lima MC, Alves S. Lysosomal Storage Disease-Associated Neuropathy: Targeting Stable Nucleic Acid Lipid Particle (SNALP)-Formulated siRNAs to the Brain as a Therapeutic Approach. International Journal of Molecular Sciences. 2020; 21(16):5732. https://doi.org/10.3390/ijms21165732
Chicago/Turabian StyleCoutinho, Maria Francisca, Juliana Inês Santos, Liliana S. Mendonça, Liliana Matos, Maria João Prata, Amália S. Jurado, Maria C. Pedroso de Lima, and Sandra Alves. 2020. "Lysosomal Storage Disease-Associated Neuropathy: Targeting Stable Nucleic Acid Lipid Particle (SNALP)-Formulated siRNAs to the Brain as a Therapeutic Approach" International Journal of Molecular Sciences 21, no. 16: 5732. https://doi.org/10.3390/ijms21165732
APA StyleCoutinho, M. F., Santos, J. I., S. Mendonça, L., Matos, L., Prata, M. J., S. Jurado, A., Pedroso de Lima, M. C., & Alves, S. (2020). Lysosomal Storage Disease-Associated Neuropathy: Targeting Stable Nucleic Acid Lipid Particle (SNALP)-Formulated siRNAs to the Brain as a Therapeutic Approach. International Journal of Molecular Sciences, 21(16), 5732. https://doi.org/10.3390/ijms21165732