N-Glycoproteins Have a Major Role in MGL Binding to Colorectal Cancer Cell Lines: Associations with Overall Proteome Diversity

 , ,
, ,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
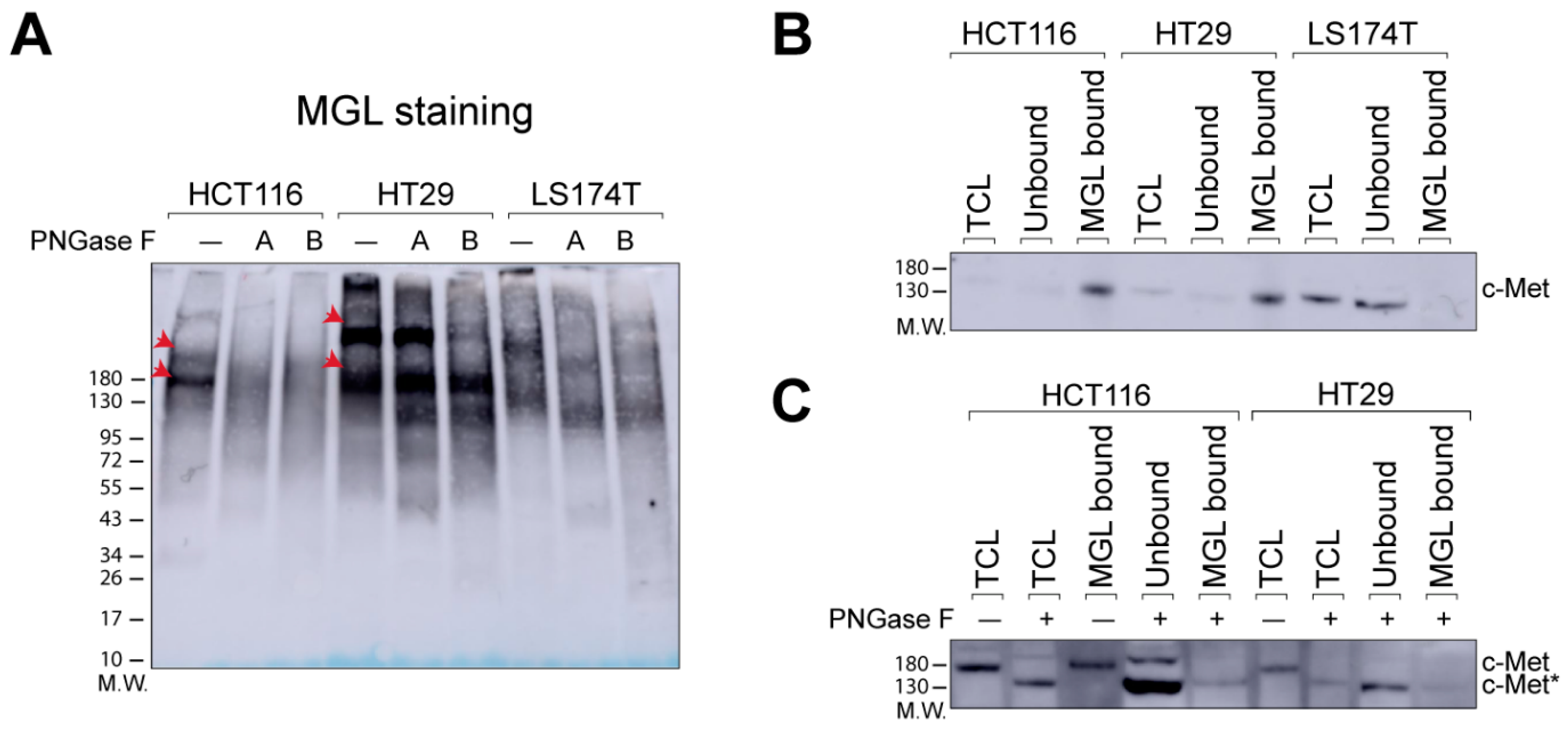
2.1. N-Glycans Are Important for MGL-Binding in CRC Cell Lines
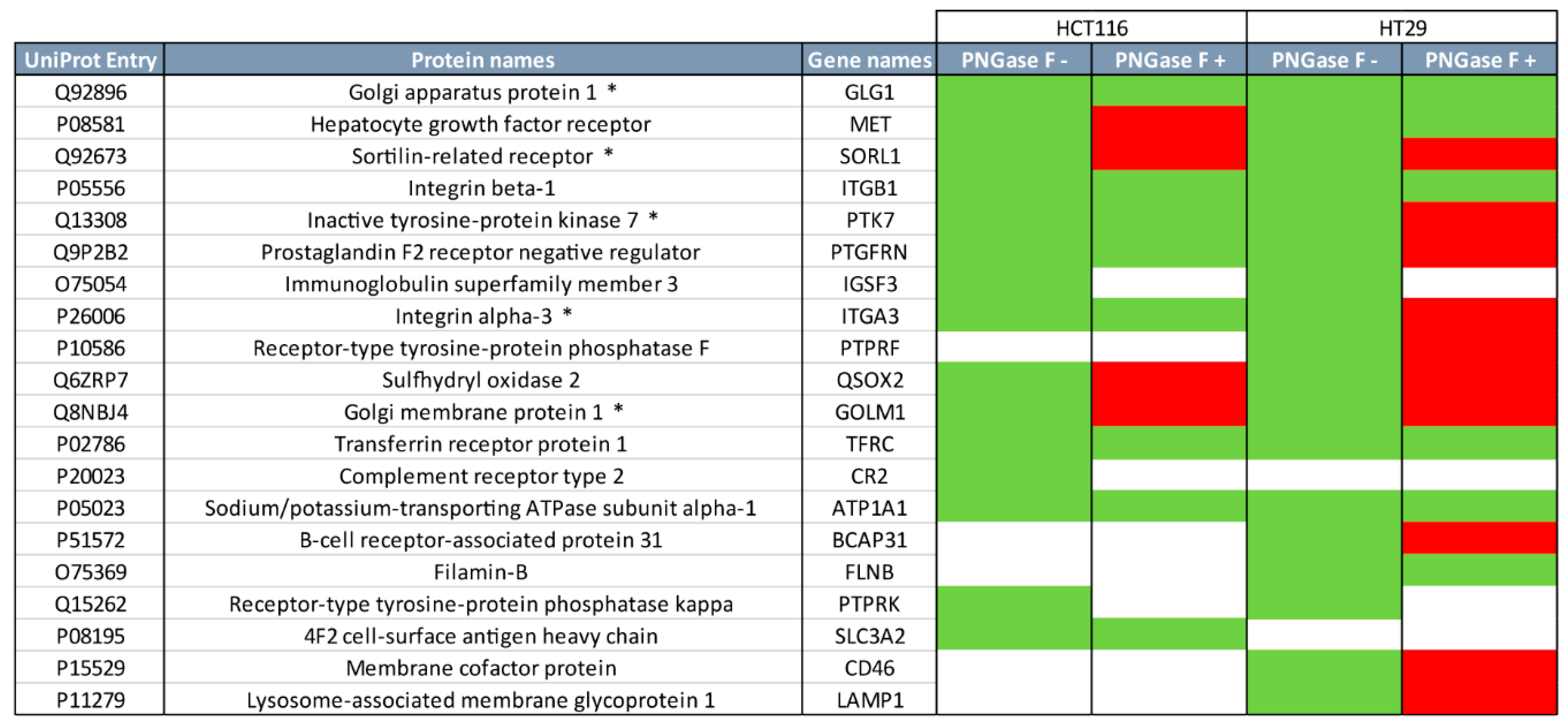
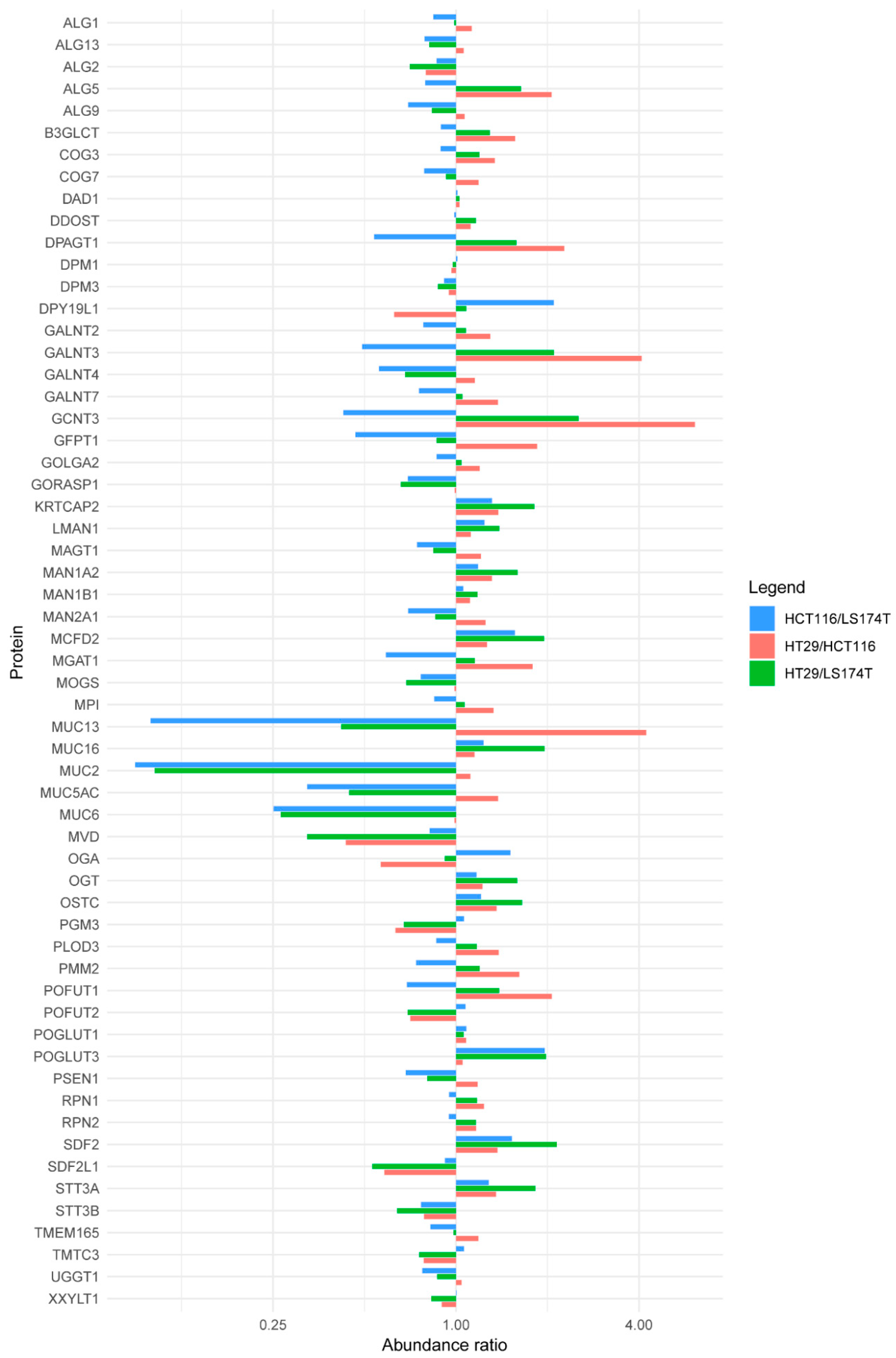
2.2. Quantitative Proteomics Provides Insights into Glycosylation Mechanisms Involved in High MGL Binding
3. Materials and Methods
3.1. Cell Lines Culture and Lysis
3.2. Lectins and Antibodies
3.3. Pull-Down Assay and PNGase F Treatment
3.4. SDS-PAGE and Western/Lectin Blot
3.5. SDS-PAGE and NanoLC-MS/MS Analysis
3.6. Quantitative Proteomics Using TMT Labeling
3.7. Data Availability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MGL | Macrophage galactose-type C-type lectin |
| CRC | Colorectal cancer |
| TACA | Tumor-Associated Carbohydrate Antigens |
| CRD | Carbohydrate recognition domain |
| TMT | Tandem Mass Tag |
| Fc | Fragment crystallizable |
| mAb | Monoclonal antibody |
| HRP | Horseradish peroxidase |
| LC | Liquid chromatography |
| MS | Mass Spectrometry |
References
- Munkley, J.; Elliott, D.J. Hallmarks of glycosylation in cancer. Oncotarget 2016, 7, 35478–35489. [Google Scholar] [CrossRef] [PubMed]
- Stowell, S.R.; Ju, T.; Cummings, R.D. Protein glycosylation in cancer. Annu. Rev. Pathol. 2015, 10, 473–510. [Google Scholar] [CrossRef] [PubMed]
- Varki, A. Biological roles of glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef] [PubMed]
- Blanas, A.; Sahasrabudhe, N.M.; Rodriguez, E.; van Kooyk, Y.; van Vliet, S.J. Fucosylated Antigens in Cancer: An Alliance toward Tumor Progression, Metastasis, and Resistance to Chemotherapy. Front. Oncol. 2018, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Holst, S.; Wuhrer, M.; Rombouts, Y. Glycosylation characteristics of colorectal cancer. Adv. Cancer Res. 2015, 126, 203–256. [Google Scholar] [CrossRef]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef]
- Beckwith, D.M.; Cudic, M. Tumor-associated O-glycans of MUC1: Carriers of the glyco-code and targets for cancer vaccine design. Semin. Immunol. 2020. [Google Scholar] [CrossRef]
- Cervoni, G.E.; Cheng, J.J.; Stackhouse, K.A.; Heimburg-Molinaro, J.; Cummings, R.D. O-glycan recognition and function in mice and human cancers. Biochem. J. 2020, 477, 1541–1564. [Google Scholar] [CrossRef]
- Silva, M.L.S. Lectin biosensors in cancer glycan biomarker detection. Adv. Clin. Chem. 2019, 93, 1–61. [Google Scholar] [CrossRef]
- Ju, T.; Wang, Y.; Aryal, R.P.; Lehoux, S.D.; Ding, X.; Kudelka, M.R.; Cutler, C.; Zeng, J.; Wang, J.; Sun, X.; et al. Tn and sialyl-Tn antigens, aberrant O-glycomics as human disease markers. Proteom. Clin. Appl. 2013, 7, 618–631. [Google Scholar] [CrossRef]
- Hirano, K.; Matsuda, A.; Shirai, T.; Furukawa, K. Expression of LacdiNAc groups on N-glycans among human tumors is complex. Biomed. Res. Int. 2014, 2014, 981627. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Kenny, D.T.; Skoog, E.C.; Padra, M.; Adamczyk, B.; Vitizeva, V.; Thorell, A.; Venkatakrishnan, V.; Linden, S.K.; Karlsson, N.G. Structural Diversity of Human Gastric Mucin Glycans. Mol. Cell Proteom. 2017, 16, 743–758. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, S.J.; Gringhuis, S.I.; Geijtenbeek, T.B.; van Kooyk, Y. Regulation of effector T cells by antigen-presenting cells via interaction of the C-type lectin MGL with CD45. Nat. Immunol. 2006, 7, 1200–1208. [Google Scholar] [CrossRef] [PubMed]
- Marcelo, F.; Supekar, N.; Corzana, F.; van der Horst, J.C.; Vuist, I.M.; Live, D.; Boons, G.P.H.; Smith, D.F.; van Vliet, S.J. Identification of a secondary binding site in human macrophage galactose-type lectin by microarray studies: Implications for the molecular recognition of its ligands. J. Biol. Chem. 2019, 294, 1300–1311. [Google Scholar] [CrossRef] [PubMed]
- Lenos, K.; Goos, J.A.; Vuist, I.M.; den Uil, S.H.; Delis-van Diemen, P.M.; Belt, E.J.; Stockmann, H.B.; Bril, H.; de Wit, M.; Carvalho, B.; et al. MGL ligand expression is correlated to BRAF mutation and associated with poor survival of stage III colon cancer patients. Oncotarget 2015, 6, 26278–26290. [Google Scholar] [CrossRef] [PubMed]
- Pirro, M.; Rombouts, Y.; Stella, A.; Neyrolles, O.; Burlet-Schiltz, O.; van Vliet, S.J.; de Ru, A.H.; Mohammed, Y.; Wuhrer, M.; van Veelen, P.A.; et al. Characterization of Macrophage Galactose-type Lectin (MGL) ligands in colorectal cancer cell lines. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129513. [Google Scholar] [CrossRef]
- Holst, S.; Deuss, A.J.; van Pelt, G.W.; van Vliet, S.J.; Garcia-Vallejo, J.J.; Koeleman, C.A.; Deelder, A.M.; Mesker, W.E.; Tollenaar, R.A.; Rombouts, Y.; et al. N-glycosylation Profiling of Colorectal Cancer Cell Lines Reveals Association of Fucosylation with Differentiation and Caudal Type Homebox 1 (CDX1)/Villin mRNA Expression. Mol. Cell Proteom. 2016, 15, 124–140. [Google Scholar] [CrossRef]
- Madunic, K.; Zhang, T.; Mayboroda, O.A.; Holst, S.; Stavenhagen, K.; Jin, C.; Karlsson, N.G.; Lageveen-Kammeijer, G.S.M.; Wuhrer, M. Colorectal cancer cell lines show striking diversity of their O-glycome reflecting the cellular differentiation phenotype. Cell Mol. Life Sci. 2020. [Google Scholar] [CrossRef]
- Parizadeh, S.M.; Jafarzadeh-Esfehani, R.; Fazilat-Panah, D.; Hassanian, S.M.; Shahidsales, S.; Khazaei, M.; Parizadeh, S.M.R.; Ghayour-Mobarhan, M.; Ferns, G.A.; Avan, A. The potential therapeutic and prognostic impacts of the c-MET/HGF signaling pathway in colorectal cancer. Iubmb Life 2019, 71, 802–811. [Google Scholar] [CrossRef]
- Berg, K.C.G.; Eide, P.W.; Eilertsen, I.A.; Johannessen, B.; Bruun, J.; Danielsen, S.A.; Bjornslett, M.; Meza-Zepeda, L.A.; Eknaes, M.; Lind, G.E.; et al. Multi-omics of 34 colorectal cancer cell lines—A resource for biomedical studies. Mol. Cancer 2017, 16, 116. [Google Scholar] [CrossRef]
- Sahasrabudhe, N.M.; Lenos, K.; van der Horst, J.C.; Rodriguez, E.; van Vliet, S.J. Oncogenic BRAFV600E drives expression of MGL ligands in the colorectal cancer cell line HT29 through N-acetylgalactosamine-transferase 3. Biol. Chem. 2018, 399, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, K.; Noda, K.; Furukawa, Y.; Ohshima, K.; Uchiyama, A.; Nakagawa, T.; Taniguchi, N.; Daigo, Y.; Nakamura, Y.; Hayashi, N.; et al. Deficiency of GMDS leads to escape from NK cell-mediated tumor surveillance through modulation of TRAIL signaling. Gastroenterology 2009, 137, 188–198.e2. [Google Scholar] [CrossRef] [PubMed]
- Beatson, R.; Maurstad, G.; Picco, G.; Arulappu, A.; Coleman, J.; Wandell, H.H.; Clausen, H.; Mandel, U.; Taylor-Papadimitriou, J.; Sletmoen, M.; et al. The Breast Cancer-Associated Glycoforms of MUC1, MUC1-Tn and sialyl-Tn, Are Expressed in COSMC Wild-Type Cells and Bind the C-Type Lectin MGL. PLoS ONE 2015, 10, e0125994. [Google Scholar] [CrossRef] [PubMed]
- Kurze, A.K.; Buhs, S.; Eggert, D.; Oliveira-Ferrer, L.; Muller, V.; Niendorf, A.; Wagener, C.; Nollau, P. Immature O-glycans recognized by the macrophage glycoreceptor CLEC10A (MGL) are induced by 4-hydroxy-tamoxifen, oxidative stress and DNA-damage in breast cancer cells. Cell Commun. Signal. 2019, 17, 107. [Google Scholar] [CrossRef] [PubMed]
- Saeland, E.; Belo, A.I.; Mongera, S.; van Die, I.; Meijer, G.A.; van Kooyk, Y. Differential glycosylation of MUC1 and CEACAM5 between normal mucosa and tumour tissue of colon cancer patients. Int. J. Cancer 2012, 131, 117–128. [Google Scholar] [CrossRef]
- Boyd, M.; Hansen, M.; Jensen, T.G.; Perearnau, A.; Olsen, A.K.; Bram, L.L.; Bak, M.; Tommerup, N.; Olsen, J.; Troelsen, J.T. Genome-wide analysis of CDX2 binding in intestinal epithelial cells (Caco-2). J. Biol. Chem. 2010, 285, 25115–25125. [Google Scholar] [CrossRef]
- Lauc, G.; Essafi, A.; Huffman, J.E.; Hayward, C.; Knezevic, A.; Kattla, J.J.; Polasek, O.; Gornik, O.; Vitart, V.; Abrahams, J.L.; et al. Genomics meets glycomics-the first GWAS study of human N-Glycome identifies HNF1alpha as a master regulator of plasma protein fucosylation. PLoS Genet. 2010, 6, e1001256. [Google Scholar] [CrossRef]
- Pinto, R.; Barros, R.; Pereira-Castro, I.; Mesquita, P.; da Costa, L.T.; Bennett, E.P.; Almeida, R.; David, L. CDX2 homeoprotein is involved in the regulation of ST6GalNAc-I gene in intestinal metaplasia. Lab. Invest. 2015, 95, 718–727. [Google Scholar] [CrossRef]
- Holst, S.; Wilding, J.L.; Koprowska, K.; Rombouts, Y.; Wuhrer, M. N-Glycomic and Transcriptomic Changes Associated with CDX1 mRNA Expression in Colorectal Cancer Cell Lines. Cells 2019, 8, 273. [Google Scholar] [CrossRef]
- Conze, T.; Carvalho, A.S.; Landegren, U.; Almeida, R.; Reis, C.A.; David, L.; Soderberg, O. MUC2 mucin is a major carrier of the cancer-associated sialyl-Tn antigen in intestinal metaplasia and gastric carcinomas. Glycobiology 2010, 20, 199–206. [Google Scholar] [CrossRef]
- Westen, A.A.; Kraaijenbrink, T.; Robles de Medina, E.A.; Harteveld, J.; Willemse, P.; Zuniga, S.B.; van der Gaag, K.J.; Weiler, N.E.; Warnaar, J.; Kayser, M.; et al. Comparing six commercial autosomal STR kits in a large Dutch population sample. Forensic. Sci. Int. Genet. 2014, 10, 55–63. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, S.J.; van Liempt, E.; Saeland, E.; Aarnoudse, C.A.; Appelmelk, B.; Irimura, T.; Geijtenbeek, T.B.; Blixt, O.; Alvarez, R.; van Die, I.; et al. Carbohydrate profiling reveals a distinctive role for the C-type lectin MGL in the recognition of helminth parasites and tumor antigens by dendritic cells. Int. Immunol. 2005, 17, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Pirro, M.; Schoof, E.; van Vliet, S.J.; Rombouts, Y.; Stella, A.; de Ru, A.; Mohammed, Y.; Wuhrer, M.; van Veelen, P.A.; Hensbergen, P.J. Glycoproteomic Analysis of MGL-Binding Proteins on Acute T-Cell Leukemia Cells. J. Proteome Res. 2019, 18, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Paulo, J.A.; Gygi, S.P. Nicotine-induced protein expression profiling reveals mutually altered proteins across four human cell lines. Proteomics 2017, 17. [Google Scholar] [CrossRef]
- McAlister, G.C.; Nusinow, D.P.; Jedrychowski, M.P.; Wuhr, M.; Huttlin, E.L.; Erickson, B.K.; Rad, R.; Haas, W.; Gygi, S.P. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 2014, 86, 7150–7158. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Csordas, A.; Del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, 11033. [Google Scholar] [CrossRef]
- Saeland, E.; van Vliet, S.J.; Backstrom, M.; van den Berg, V.C.; Geijtenbeek, T.B.; Meijer, G.A.; van Kooyk, Y. The C-type lectin MGL expressed by dendritic cells detects glycan changes on MUC1 in colon carcinoma. Cancer Immunol. Immunother. 2007, 56, 1225–1236. [Google Scholar] [CrossRef]
- van Vliet, S.J.; Bay, S.; Vuist, I.M.; Kalay, H.; Garcia-Vallejo, J.J.; Leclerc, C.; van Kooyk, Y. MGL signaling augments TLR2-mediated responses for enhanced IL-10 and TNF-alpha secretion. J. Leukoc Biol. 2013, 94, 315–323. [Google Scholar] [CrossRef]
- Diniz, A.; Coelho, H.; Dias, J.S.; van Vliet, S.J.; Jimenez-Barbero, J.; Corzana, F.; Cabrita, E.J.; Marcelo, F. The Plasticity of the Carbohydrate Recognition Domain Dictates the Exquisite Mechanism of Binding of Human Macrophage Galactose-Type Lectin. Chemistry 2019, 25, 13945–13955. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pirro, M.; Mohammed, Y.; van Vliet, S.J.; Rombouts, Y.; Sciacca, A.; de Ru, A.H.; Janssen, G.M.C.; Tjokrodirijo, R.T.N.; Wuhrer, M.; van Veelen, P.A.; et al. N-Glycoproteins Have a Major Role in MGL Binding to Colorectal Cancer Cell Lines: Associations with Overall Proteome Diversity. Int. J. Mol. Sci. 2020, 21, 5522. https://doi.org/10.3390/ijms21155522
Pirro M, Mohammed Y, van Vliet SJ, Rombouts Y, Sciacca A, de Ru AH, Janssen GMC, Tjokrodirijo RTN, Wuhrer M, van Veelen PA, et al. N-Glycoproteins Have a Major Role in MGL Binding to Colorectal Cancer Cell Lines: Associations with Overall Proteome Diversity. International Journal of Molecular Sciences. 2020; 21(15):5522. https://doi.org/10.3390/ijms21155522
Chicago/Turabian StylePirro, Martina, Yassene Mohammed, Sandra J. van Vliet, Yoann Rombouts, Agnese Sciacca, Arnoud H. de Ru, George M. C. Janssen, Rayman T. N. Tjokrodirijo, Manfred Wuhrer, Peter A. van Veelen, and et al. 2020. "N-Glycoproteins Have a Major Role in MGL Binding to Colorectal Cancer Cell Lines: Associations with Overall Proteome Diversity" International Journal of Molecular Sciences 21, no. 15: 5522. https://doi.org/10.3390/ijms21155522
APA StylePirro, M., Mohammed, Y., van Vliet, S. J., Rombouts, Y., Sciacca, A., de Ru, A. H., Janssen, G. M. C., Tjokrodirijo, R. T. N., Wuhrer, M., van Veelen, P. A., & Hensbergen, P. J. (2020). N-Glycoproteins Have a Major Role in MGL Binding to Colorectal Cancer Cell Lines: Associations with Overall Proteome Diversity. International Journal of Molecular Sciences, 21(15), 5522. https://doi.org/10.3390/ijms21155522