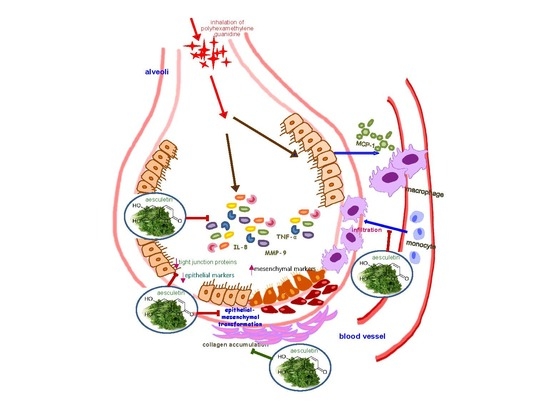
Aesculetin Attenuates Alveolar Injury and Fibrosis Induced by Close Contact of Alveolar Epithelial Cells with Blood-Derived Macrophages via IL-8 Signaling


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Release of Inflammatory Cytokines/CRP/PDGF of Monocyte-Derived Macrophages
2.2. Blockade of Alveolar Epithelial–Mesenchymal Transformation (EMT) by Aesculetin
2.3. Inhibition of mCM-Induced Alveolar Injury by Aesculetin
2.4. Involvement of IL-8 in Induction of EMT and Disruption of the Epithelial Barrier
2.5. Inhibition of Inflammatory Cell Infiltration into PHMG-Exposed Airways by Aesculetin
2.6. Blockade of PHMG-Induced and IL-8-Mediated Airway EMT and Fibrosis by Aesculetin
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Preparation of Conditioned Media
4.3. Alveolar Epithelial A549 Cell Culture and Viability
4.4. Enzyme-Linked Immunosorbent Assay (ELISA)
4.5. Western Blot Analysis
4.6. Immunocytochemistry
4.7. Animal Experiments
4.8. Immunohistochemical Staining
4.9. H&E Staining
4.10. Masson Trichrome Staining
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| COPD | chronic obstructive pulmonary disease |
| CRP | C-reactive protein |
| CXCR2 | CXC-chemokine receptor 2 |
| ECM | extracellular matrix |
| EMT | epithelial-mesenchymal transition |
| IFN-γ | interferon-γ |
| IL | interleukin |
| LPS | lipopolysaccharide |
| mCM | macrophage conditioned media |
| MMP-9 | matrix metalloproteinase-9 |
| MT-1 MMP | membrane type-1 matrix metalloproteinase |
| PDGF | platelet-derived growth factor |
| PHMG | polyhexamethylene guanidine |
| α-SMA | α-smooth muscle actin |
| TIMP | tissue inhibitor of metalloproteinases |
| TNF-α | tumor necrosis factor-α |
References
- Moldoveanu, B.; Otmishi, P.; Jani, P.; Walker, J.; Sarmiento, X.; Guardiola, J.; Saad, M.; Yu, J. Inflammatory mechanisms in the lung. J. Inflamm. Res. 2009, 2, 1–11. [Google Scholar] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2017, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed]
- Robb, C.T.; Regan, K.H.; Dorward, D.A.; Rossi, A.G. Key mechanisms governing resolution of lung inflammation. Semin. Immunopathol. 2016, 38, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.A. Acute lung injury: How the lung inflammatory response works. Eur. Respir. J. 2003, 22, 22s–23s. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, M.; Moochhala, S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J. Pathol. 2004, 202, 145–156. [Google Scholar] [CrossRef]
- Potey, P.M.D.; Rossi, A.G.; Lucas, C.D.; Dorward, D.A. Neutrophils in the initiation and resolution of acute pulmonary inflammation: Understanding biological function and therapeutic potential. J. Pathol. 2019, 247, 672–685. [Google Scholar] [CrossRef]
- Liu, J.; Pang, Z.; Wang, G.; Guan, X.; Fang, K.; Wang, Z.; Wang, F. Advanced role of neutrophils in common respiratory diseases. J. Immunol. Res. 2017, 2017, 6710278. [Google Scholar] [CrossRef]
- Barnes, P.J. Similarities and differences in inflammatory mechanisms of asthma and COPD. Breathe 2011, 7, 229–238. [Google Scholar] [CrossRef]
- Kapellos, T.S.; Bassler, K.; Aschenbrenner, A.C.; Fujii, W.; Schultze, J.L. Dysregulated functions of lung macrophage populations in COPD. J. Immunol. Res. 2018, 2018, 2349045. [Google Scholar] [CrossRef]
- Byrne, A.J.; Mathie, S.A.; Gregory, L.G.; Lloyd, C.M. Pulmonary macrophages: Key players in the innate defense of the airways. Thorax 2015, 70, 1189–1196. [Google Scholar] [CrossRef]
- Byrne, A.J.; Maher, T.M.; Lloyd, C.M. Pulmonary Macrophages: A new therapeutic pathway in fibrosing lung disease? Trends Mol. Med. 2016, 22, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Bringardner, B.D.; Baran, C.P.; Eubank, T.D.; Marsh, C.B. The role of inflammation in the pathogenesis of idiopathic pulmonary fibrosis. Antioxid. Redox Signal. 2008, 10, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Klingberg, F.; Hinz, B.; White, E.S. The myofibroblast matrix: Implications for tissue repair and fibrosis. J. Pathol. 2013, 229, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Herold, S.; Mayer, K.; Lohmeyer, J. Acute lung injury: How macrophages orchestrate resolution of inflammation and tissue Repair. Front. Immunol. 2011, 2, 65. [Google Scholar] [CrossRef] [PubMed]
- Chambers, R.C.; Paul, F.; Mercer, P. Mechanisms of alveolar epithelial injury, repair, and fibrosis. Ann. Am. Thorac. Soc. 2015, 12, s16–s20. [Google Scholar] [CrossRef]
- Hewitt, R.J.; Molyneaux, P.L. The respiratory microbiome in idiopathic pulmonary fibrosis. Ann. Transl. Med. 2017, 5, 250. [Google Scholar] [CrossRef]
- Camelo, A.; Dunmore, R.; Sleeman, M.A.; Clarke, D.L. The epithelium in idiopathic pulmonary fibrosis: Breaking the barrier. Front. Pharmacol. 2013, 4, 173. [Google Scholar] [CrossRef]
- Impellizzeri, D.; Talero, E.; Siracusa, R.; Alcaide, A.; Cordaro, M.; Maria Zubelia, J.; Bruschetta, G.; Crupi, R.; Esposito, E.; Cuzzocrea, S.; et al. Protective effect of polyphenols in an inflammatory process associated with experimental pulmonary fibrosis in mice. Br. J. Nutr. 2015, 114, 853–865. [Google Scholar] [CrossRef]
- Day, B.J. Antioxidants as potential therapeutics for lung fibrosis. Antioxid. Redox Signal. 2008, 10, 355–370. [Google Scholar] [CrossRef]
- Bahri, S.; Ben Ali, R.; Gasmi, K.; Mlika, M.; Fazaa, S.; Ksouri, R.; Serairi, R.; Jameleddine, S.; Shlyonsky, V. Prophylactic and curative effect of rosemary leaves extract in a bleomycin model of pulmonary fibrosis. Pharm. Biol. 2017, 55, 462–471. [Google Scholar] [CrossRef]
- Duan, J.; Shi, J.; Ma, X.; Xuan, Y.; Li, P.; Wang, H.; Fan, Y.; Gong, H.; Wang, L.; Pang, Y.; et al. Esculetin inhibits proliferation, migration, and invasion of clear cell renal cell carcinoma cells. Biomed. Pharmacother. 2020, 125, 110031. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.S.; Lee, W.; Jung, Y.; Lee, J.H.; Lee, S.; Eom, D.W.; Jeon, Y.; Yoo, H.H.; Jin, M.J.; Song, K.I.; et al. Protective effect of esculin on streptozotocin-induced diabetic renal damage in mice. J. Agric. Food Chem. 2014, 62, 2069–2076. [Google Scholar] [CrossRef] [PubMed]
- Witaicenis, A.; Seito, L.N.; Di Stasi, L.C. Intestinal anti-inflammatory activity of esculetin and 4-methylesculetin in the trinitrobenzenesulphonic acid model of rat colitis. Chem. Biol. Interact. 2010, 186, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Liu, F.C.; Tsai, C.N.; Chou, A.H.; Liao, C.C.; Yu, H.P. Esculetin ameliorates lipopolysaccharide-induced acute lung injury in mice via modulation of the AKT/ERK/NF-κB and RORγt/IL-17 pathways. Inflammation 2020, 43, 962–974. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lee, K.; Lee, E.J.; Lee, S.Y.; In, K.H.; Kim, H.K.; Kang, M.S. Humidifier disinfectant-associated interstitial lung disease in an animal model induced by polyhexamethylene guanidine aerosol. Am. J. Respir. Crit. Care Med. 2014, 190, 706–708. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Lee, Y.C.; Kim, D.I.; Park, H.J.; Kim, S.H. Inhaled PHMG can induce pulmonary fibrosis through mitochondrial ROS generation. Eur. Respir. J. 2015, 46, PA3046. [Google Scholar]
- Kim, M.S.; Kim, S.H.; Jeon, D.; Kim, H.Y.; Lee, K. Changes in expression of cytokines in polyhexamethylene guanidine-induced lung fibrosis in mice: Comparison of bleomycin-induced lung fibrosis. Toxicology 2018, 393, 185–192. [Google Scholar] [CrossRef]
- Driscoll, K.E. Macrophage inflammatory proteins: Biology and role in pulmonary inflammation. Exp. Lung Res. 1994, 20, 473–490. [Google Scholar] [CrossRef]
- Mauad, T.; Dolhnikoff, M. Pathologic similarities and differences between asthma and chronic obstructive pulmonary disease. Curr. Opin. Pulm. Med. 2008, 14, 31–38. [Google Scholar] [CrossRef]
- Di Stefano, A.; Capelli, A.; Lusuardi, M.; Balbo, P.; Vecchio, C.; Maestrelli, P.; Mapp, C.E.; Fabbri, L.M.; Donner, C.F.; Saetta, M. Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am. J. Respir. Crit. Care Med. 1998, 158, 1277–1285. [Google Scholar] [CrossRef]
- O’Donnell, R.; Breen, D.; Wilson, S.; Djukanovic, R. Inflammatory cells in the airways in COPD. Thorax 2006, 61, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Wallace, W.A.; Fitch, P.M.; Simpson, A.J.; Howie, S.E. Inflammation-Associated remodelling and fibrosis in the lung—A process and an end point. Int. J. Exp. Pathol. 2007, 88, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Alveolar macrophages as orchestrators of COPD. COPD 2004, 1, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Y.; Wu, G.; Xiong, W.; Gu, W.; Wang, C.Y. Macrophages: Friend or foe in idiopathic pulmonary fibrosis? Respir. Res. 2018, 19, 170. [Google Scholar] [CrossRef] [PubMed]
- Findlay, E.G.; Hussell, T. Macrophage-Mediated inflammation and disease: A focus on the lung. Mediators Inflamm. 2012, 2012, 140937. [Google Scholar]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Salazar, L.M.; Herrera, A.M. Fibrotic response of tissue remodeling in COPD. Lung 2011, 189, 101–109. [Google Scholar] [CrossRef]
- Karvonen, H.M.; Lehtonen, S.T.; Harju, T.; Sormunen, R.T.; Lappi-Blanco, E.; Mäkinen, J.M.; Laitakari, K.; Johnson, S.; Kaarteenaho, R.L. Myofibroblast expression in airways and alveoli is affected by smoking and COPD. Respir. Res. 2013, 14, 84. [Google Scholar] [CrossRef]
- Milara, J.; Peiro, T.; Serrano, A.; Cortijo, J. Epithelial to mesen-chymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax 2013, 68, 410–420. [Google Scholar] [CrossRef]
- Sohal, S.S.; Soltani, A.; Reid, D.; Ward, C.; Wills, K.E.; Muller, H.K.; Walters, E.H. A randomized controlled trial of inhaled corticosteroids (ICS) on markers of epithelial-mesenchymal transition (EMT) in large airway samples in COPD: An exploratory proof of concept study. Int. J. Chron. Obstruct. Pulmon. Dis. 2014, 9, 533–542. [Google Scholar] [CrossRef]
- Avila-Carrasco, L.; Majano, P.; Sánchez-Toméro, J.A.; Selgas, R.; López-Cabrera, M.; Aguilera, A.; Mateo, G.G. Natural plants compounds as modulators of epithelial-to-mesenchymal transition. Front. Pharmacol. 2019, 10, 715. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Sarkar, N.; Bishayee, A.; Sinha, D. Dietary phytochemicals in the regulation of epithelial to mesenchymal transition and associated enzymes: A promising anticancer therapeutic approach. Semin. Cancer Biol. 2019, 56, 196–218. [Google Scholar] [CrossRef] [PubMed]
- Herrero, R.; Sanchez, G.; Lorente, J.A. New insights into the mechanisms of pulmonary edema in acute lung injury. Ann. Transl. Med. 2018, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.W.; Lee, G.H.; Pham, H.T.; Choi, J.H.; Jeong, H.G. Polyhexamethylene guanidine phosphate damages tight junctions and the F-actin architecture by activating calpain-1 via the P2RX7/Ca2+ signaling pathway. Cells 2019, 9, 59. [Google Scholar] [CrossRef]
- Jeong, M.H.; Kim, H.R.; Park, Y.J.; Chung, K.H. Akt and Notch pathways mediate polyhexamethylene guanidine phosphate-induced epithelial-mesenchymal transition via ZEB2. Toxicol. Appl. Pharmacol. 2019, 380, 114691. [Google Scholar] [CrossRef]
- Kim, H.Y.; Kim, M.S.; Kim, S.H.; Joen, D.; Lee, K. Protective effects of nintedanib against polyhexamethylene guanidine phosphate-induced lung fibrosis in mice. Molecules 2018, 23, 1974. [Google Scholar] [CrossRef]
- Wollin, L.; Distler, J.H.W.; Redente, E.F.; Riches, D.W.H.; Stowasser, S.; Schlenker-Herceg, R.; Maher, T.M.; Kolb, M. Potential of nintedanib in treatment of progressive fibrosing interstitial lung diseases. Eur. Respir. J. 2019, 54, 1900161. [Google Scholar] [CrossRef]
- Wang, X.; Liu, M.; Zhu, M.J.; Shi, L.; Liu, L.; Zhao, Y.L.; Cheng, L.; Gu, Y.J.; Zhou, M.Y.; Chen, L.; et al. Resveratrol protects the integrity of alveolar epithelial barrier via SIRT1/PTEN/p-Akt pathway in methamphetamine-induced chronic lung injury. Cell. Prolif. 2020, 53, e12773. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, S.Y.; Kim, Y.-H.; Kang, M.-K.; Lee, E.-J.; Kim, D.Y.; Oh, H.; Kim, S.-I.; Na, W.; Kang, Y.-H. Aesculetin Attenuates Alveolar Injury and Fibrosis Induced by Close Contact of Alveolar Epithelial Cells with Blood-Derived Macrophages via IL-8 Signaling. Int. J. Mol. Sci. 2020, 21, 5518. https://doi.org/10.3390/ijms21155518
Oh SY, Kim Y-H, Kang M-K, Lee E-J, Kim DY, Oh H, Kim S-I, Na W, Kang Y-H. Aesculetin Attenuates Alveolar Injury and Fibrosis Induced by Close Contact of Alveolar Epithelial Cells with Blood-Derived Macrophages via IL-8 Signaling. International Journal of Molecular Sciences. 2020; 21(15):5518. https://doi.org/10.3390/ijms21155518
Chicago/Turabian StyleOh, Su Yeon, Yun-Ho Kim, Min-Kyung Kang, Eun-Jung Lee, Dong Yeon Kim, Hyeongjoo Oh, Soo-Il Kim, Woojin Na, and Young-Hee Kang. 2020. "Aesculetin Attenuates Alveolar Injury and Fibrosis Induced by Close Contact of Alveolar Epithelial Cells with Blood-Derived Macrophages via IL-8 Signaling" International Journal of Molecular Sciences 21, no. 15: 5518. https://doi.org/10.3390/ijms21155518
APA StyleOh, S. Y., Kim, Y.-H., Kang, M.-K., Lee, E.-J., Kim, D. Y., Oh, H., Kim, S.-I., Na, W., & Kang, Y.-H. (2020). Aesculetin Attenuates Alveolar Injury and Fibrosis Induced by Close Contact of Alveolar Epithelial Cells with Blood-Derived Macrophages via IL-8 Signaling. International Journal of Molecular Sciences, 21(15), 5518. https://doi.org/10.3390/ijms21155518