The Role of CDKs and CDKIs in Murine Development


Abstract
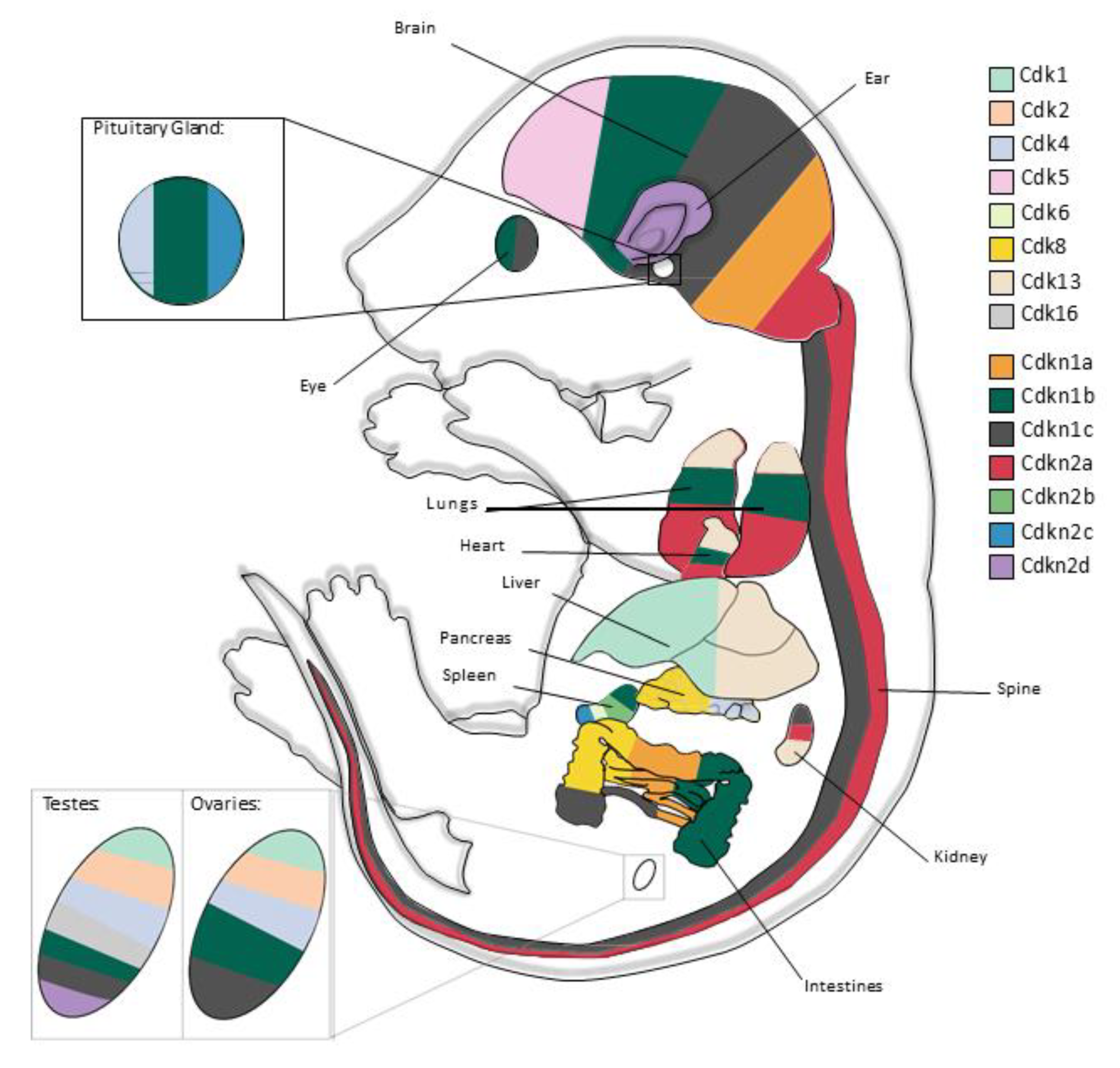
1. Introduction
2. Absolute Requirement of Cdk1
3. Embryo
4. Brain Development and Function
5. Pancreas
6. Fertility
7. In Vitro Studies
8. Discussion and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BAC | Bacterial Artificial Chromosomes |
| CDK | Cyclin-dependent kinase |
| CDKI | Cyclin-dependent kinase inhibitor |
| CDKi | CDK inhibitor compound |
| cKO | Conditional KO |
| dKO | Double KO |
| KO | Knock-out |
| tKO | Triple KO |
References
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK inhibitors: Cell cycle regulators and beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Suryadinata, R.; Sadowski, M.; Sarcevic, B. Control of cell cycle progression by phosphorylation of cyclin-dependent kinase (CDK) substrates. Biosci. Rep. 2010, 30, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Satyanarayana, A.; Kaldis, P. Mammalian cell-cycle regulation: Several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 2009, 28, 2925–2939. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Yang, Z.; Wang, S.; Li, Y.; Wei, H.; Tian, X.; Kan, Q. Recent development of CDK inhibitors: An overview of CDK/inhibitor co-crystal structures. Eur. J. Med. Chem. 2019, 164, 615–639. [Google Scholar] [CrossRef] [PubMed]
- Luh, F.Y.; Archer, S.J.; Domaille, P.J.; Smith, B.O.; Owen, D.; Brotherton, D.H.; Raine, A.R.; Xu, X.; Brizuela, L.; Brenner, S.L.; et al. Structure of the cyclin-dependent kinase inhibitor p19Ink4d. Nature 1997, 389, 999–1003. [Google Scholar] [CrossRef] [PubMed]
- Marassi, F. Utilizing NMR to study the structure of growth-inhibitory proteins. Methods Mol. Biol. 2003, 223, 3–15. [Google Scholar] [CrossRef]
- Martin, M.P.; Endicott, J.A.; Noble, M.E.M. Structure-based discovery of cyclin-dependent protein kinase inhibitors. Essays Biochem. 2017, 61, 439–452. [Google Scholar] [CrossRef]
- Wood, D.J.; Endicott, J.A. Structural insights into the functional diversity of the CDK-cyclin family. Open Biol. 2018, 8. [Google Scholar] [CrossRef]
- Malumbres, M.; Harlow, E.; Hunt, T.; Hunter, T.; Lahti, J.M.; Manning, G.; Morgan, D.O.; Tsai, L.H.; Wolgemuth, D.J. Cyclin-dependent kinases: A family portrait. Nat. Cell Biol. 2009, 11, 1275–1276. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef]
- Nakayama, K.; Nakayama, K. Cip/Kip cyclin-dependent kinase inhibitors: Brakes of the cell cycle engine during development. Bioessays 1998, 20, 1020–1029. [Google Scholar] [CrossRef]
- Chellappan, S.P.; Giordano, A.; Fisher, P.B. Role of cyclin-dependent kinases and their inhibitors in cellular differentiation and development. Curr. Top. Microbiol. Immunol. 1998, 227, 57–103. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Barberis, M.; De Gioia, L.; Ruzzene, M.; Sarno, S.; Coccetti, P.; Fantucci, P.; Vanoni, M.; Alberghina, L. The yeast cyclin-dependent kinase inhibitor Sic1 and mammalian p27Kip1 are functional homologues with a structurally conserved inhibitory domain. Biochem. J. 2005, 387, 639–647. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wallmann, A.; Kesten, C. Common functions of disordered proteins across evolutionary distant organisms. Int. J. Mol. Sci. 2020, 21, 2105. [Google Scholar] [CrossRef]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef]
- Risal, S.; Adhikari, D.; Liu, K. Animal models for studying the in vivo functions of cell cycle CDKs. Methods Mol. Biol. 2016, 1336, 155–166. [Google Scholar] [CrossRef]
- Santamaria, D.; Ortega, S. Cyclins and CDKS in development and cancer: Lessons from genetically modified mice. Front. Biosci. 2006, 11, 1164–1188. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004, 18, 2699–2711. [Google Scholar] [CrossRef]
- Kiyokawa, H.; Koff, A. Roles of cyclin-dependent kinase inhibitors: Lessons from knockout mice. Curr. Top. Microbiol. Immunol. 1998, 227, 105–120. [Google Scholar] [CrossRef]
- Santamaria, D.; Barriere, C.; Cerqueira, A.; Hunt, S.; Tardy, C.; Newton, K.; Caceres, J.F.; Dubus, P.; Malumbres, M.; Barbacid, M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007, 448, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Diril, M.K.; Ratnacaram, C.K.; Padmakumar, V.C.; Du, T.; Wasser, M.; Coppola, V.; Tessarollo, L.; Kaldis, P. Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 3826–3831. [Google Scholar] [CrossRef] [PubMed]
- Clement, T.M.; Inselman, A.L.; Goulding, E.H.; Willis, W.D.; Eddy, E.M. Disrupting cyclin dependent kinase 1 in spermatocytes causes late meiotic arrest and infertility in mice. Biol. Reprod. 2015, 93, 137. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, D.; Zheng, W.; Shen, Y.; Gorre, N.; Ning, Y.; Halet, G.; Kaldis, P.; Liu, K. Cdk1, but not Cdk2, is the sole Cdk that is essential and sufficient to drive resumption of meiosis in mouse oocytes. Hum. Mol. Genet. 2012, 21, 2476–2484. [Google Scholar] [CrossRef]
- Marlier, Q.; Jibassia, F.; Verteneuil, S.; Linden, J.; Kaldis, P.; Meijer, L.; Nguyen, L.; Vandenbosch, R.; Malgrange, B. Genetic and pharmacological inhibition of Cdk1 provides neuroprotection towards ischemic neuronal death. Cell Death Discov. 2018, 4, 43. [Google Scholar] [CrossRef]
- Ortega, S.; Prieto, I.; Odajima, J.; Martin, A.; Dubus, P.; Sotillo, R.; Barbero, J.L.; Malumbres, M.; Barbacid, M. Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat. Genet. 2003, 35, 25–31. [Google Scholar] [CrossRef]
- Berthet, C.; Aleem, E.; Coppola, V.; Tessarollo, L.; Kaldis, P. Cdk2 knockout mice are viable. Curr. Biol. 2003, 13, 1775–1785. [Google Scholar] [CrossRef]
- Ye, X.; Zhu, C.; Harper, J.W. A premature-termination mutation in the Mus musculus cyclin-dependent kinase 3 gene. Proc. Natl. Acad. Sci. USA 2001, 98, 1682–1686. [Google Scholar] [CrossRef]
- Rane, S.G.; Dubus, P.; Mettus, R.V.; Galbreath, E.J.; Boden, G.; Reddy, E.P.; Barbacid, M. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat. Genet. 1999, 22, 44–52. [Google Scholar] [CrossRef]
- Tsutsui, T.; Hesabi, B.; Moons, D.S.; Pandolfi, P.P.; Hansel, K.S.; Koff, A.; Kiyokawa, H. Targeted disruption of CDK4 delays cell cycle entry with enhanced p27(Kip1) activity. Mol. Cell Biol. 1999, 19, 7011–7019. [Google Scholar] [CrossRef] [PubMed]
- Moons, D.S.; Jirawatnotai, S.; Parlow, A.F.; Gibori, G.; Kineman, R.D.; Kiyokawa, H. Pituitary hypoplasia and lactotroph dysfunction in mice deficient for cyclin-dependent kinase-4. Endocrinology 2002, 143, 3001–3008. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, T.; Ward, J.M.; Huh, C.G.; Longenecker, G.; Veeranna; Pant, H.C.; Brady, R.O.; Martin, L.J.; Kulkarni, A.B. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc. Natl. Acad. Sci. USA 1996, 93, 11173–11178. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Li, Z.; Fu, W.Y.; Wang, J.H.; Fu, A.K.; Ip, N.Y. Pctaire1 interacts with p35 and is a novel substrate for Cdk5/p35. J. Biol. Chem. 2002, 277, 31988–31993. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Ohshima, T.; Hirasawa, M.; Pareek, T.K.; Bugge, T.H.; Morozov, A.; Fujieda, K.; Brady, R.O.; Kulkarni, A.B. Conditional deletion of neuronal cyclin-dependent kinase 5 in developing forebrain results in microglial activation and neurodegeneration. Am. J. Pathol. 2010, 176, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Benavides, D.R.; Quinn, J.J.; Zhong, P.; Hawasli, A.H.; DiLeone, R.J.; Kansy, J.W.; Olausson, P.; Yan, Z.; Taylor, J.R.; Bibb, J.A. Cdk5 modulates cocaine reward, motivation, and striatal neuron excitability. J. Neurosci. 2007, 27, 12967–12976. [Google Scholar] [CrossRef]
- Luo, F.; Zhang, J.; Burke, K.; Romito-DiGiacomo, R.R.; Miller, R.H.; Yang, Y. Oligodendrocyte-specific loss of Cdk5 disrupts the architecture of nodes of Ranvier as well as learning and memory. Exp. Neurol. 2018, 306, 92–104. [Google Scholar] [CrossRef]
- Luo, F.; Burke, K.; Kantor, C.; Miller, R.H.; Yang, Y. Cyclin-dependent kinase 5 mediates adult OPC maturation and myelin repair through modulation of Akt and GsK-3beta signaling. J. Neurosci. 2014, 34, 10415–10429. [Google Scholar] [CrossRef]
- He, X.; Takahashi, S.; Suzuki, H.; Hashikawa, T.; Kulkarni, A.B.; Mikoshiba, K.; Ohshima, T. Hypomyelination phenotype caused by impaired differentiation of oligodendrocytes in Emx1-cre mediated Cdk5 conditional knockout mice. Neurochem. Res. 2011, 36, 1293–1303. [Google Scholar] [CrossRef][Green Version]
- Ohshima, T.; Hirasawa, M.; Tabata, H.; Mutoh, T.; Adachi, T.; Suzuki, H.; Saruta, K.; Iwasato, T.; Itohara, S.; Hashimoto, M.; et al. Cdk5 is required for multipolar-to-bipolar transition during radial neuronal migration and proper dendrite development of pyramidal neurons in the cerebral cortex. Development 2007, 134, 2273–2282. [Google Scholar] [CrossRef]
- Kumazawa, A.; Mita, N.; Hirasawa, M.; Adachi, T.; Suzuki, H.; Shafeghat, N.; Kulkarni, A.B.; Mikoshiba, K.; Inoue, T.; Ohshima, T. Cyclin-dependent kinase 5 is required for normal cerebellar development. Mol. Cell Neurosci. 2013, 52, 97–105. [Google Scholar] [CrossRef]
- Malumbres, M.; Sotillo, R.; Santamaria, D.; Galan, J.; Cerezo, A.; Ortega, S.; Dubus, P.; Barbacid, M. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 2004, 118, 493–504. [Google Scholar] [CrossRef] [PubMed]
- McCleland, M.L.; Soukup, T.M.; Liu, S.D.; Esensten, J.H.; de Sousa e Melo, F.; Yaylaoglu, M.; Warming, S.; Roose-Girma, M.; Firestein, R. Cdk8 deletion in the Apc(Min) murine tumour model represses EZH2 activity and accelerates tumourigenesis. J. Pathol. 2015, 237, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Scotti, E.; Stoffel, M. CDK8 regulates insulin secretion and mediates postnatal and stress-induced expression of neuropeptides in pancreatic beta cells. Cell Rep. 2019, 28, 2892–2904. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Inoue, A.; Lahti, J.M.; Kidd, V.J. Failure to proliferate and mitotic arrest of CDK11(p110/p58)-null mutant mice at the blastocyst stage of embryonic cell development. Mol. Cell Biol. 2004, 24, 3188–3197. [Google Scholar] [CrossRef] [PubMed]
- Novakova, M.; Hampl, M.; Vrabel, D.; Prochazka, J.; Petrezselyova, S.; Prochazkova, M.; Sedlacek, R.; Kavkova, M.; Zikmund, T.; Kaiser, J.; et al. Mouse model of congenital heart defects, dysmorphic facial features and intellectual developmental disorders as a result of non-functional CDK13. Front. Cell Dev. Biol. 2019, 7, 155. [Google Scholar] [CrossRef]
- Mikolcevic, P.; Sigl, R.; Rauch, V.; Hess, M.W.; Pfaller, K.; Barisic, M.; Pelliniemi, L.J.; Boesl, M.; Geley, S. Cyclin-dependent kinase 16/PCTAIRE kinase 1 is activated by cyclin Y and is essential for spermatogenesis. Mol. Cell Biol. 2012, 32, 868–879. [Google Scholar] [CrossRef]
- Deng, C.; Zhang, P.; Harper, J.W.; Elledge, S.J.; Leder, P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 1995, 82, 675–684. [Google Scholar] [CrossRef]
- Qiu, J.; Takagi, Y.; Harada, J.; Rodrigues, N.; Moskowitz, M.A.; Scadden, D.T.; Cheng, T. Regenerative response in ischemic brain restricted by p21cip1/waf1. J. Exp. Med. 2004, 199, 937–945. [Google Scholar] [CrossRef]
- Yang, W.C.; Mathew, J.; Velcich, A.; Edelmann, W.; Kucherlapati, R.; Lipkin, M.; Yang, K.; Augenlicht, L.H. Targeted inactivation of the p21(WAF1/cip1) gene enhances Apc-initiated tumor formation and the tumor-promoting activity of a Western-style high-risk diet by altering cell maturation in the intestinal mucosal. Cancer Res. 2001, 61, 565–569. [Google Scholar]
- Martins, C.P.; Berns, A. Loss of p27(Kip1) but not p21(Cip1) decreases survival and synergizes with MYC in murine lymphomagenesis. EMBO J. 2002, 21, 3739–3748. [Google Scholar] [CrossRef]
- Fotedar, R.; Brickner, H.; Saadatmandi, N.; Rousselle, T.; Diederich, L.; Munshi, A.; Jung, B.; Reed, J.C.; Fotedar, A. Effect of p21waf1/cip1 transgene on radiation induced apoptosis in T cells. Oncogene 1999, 18, 3652–3658. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fero, M.L.; Rivkin, M.; Tasch, M.; Porter, P.; Carow, C.E.; Firpo, E.; Polyak, K.; Tsai, L.H.; Broudy, V.; Perlmutter, R.M.; et al. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell 1996, 85, 733–744. [Google Scholar] [CrossRef]
- Kiyokawa, H.; Kineman, R.D.; Manova-Todorova, K.O.; Soares, V.C.; Hoffman, E.S.; Ono, M.; Khanam, D.; Hayday, A.C.; Frohman, L.A.; Koff, A. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1). Cell 1996, 85, 721–732. [Google Scholar] [CrossRef]
- Nakayama, K.; Ishida, N.; Shirane, M.; Inomata, A.; Inoue, T.; Shishido, N.; Horii, I.; Loh, D.Y.; Nakayama, K. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell 1996, 85, 707–720. [Google Scholar] [CrossRef]
- Fero, M.L.; Randel, E.; Gurley, K.E.; Roberts, J.M.; Kemp, C.J. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature 1998, 396, 177–180. [Google Scholar] [CrossRef]
- Yan, Y.; Miao, D.; Yang, Z.; Zhang, D. Loss of p27(kip1) suppresses the myocardial senescence caused by estrogen deficiency. J. Cell. Biochem. 2019, 120, 13994–14003. [Google Scholar] [CrossRef]
- Lin, H.; Hu, G.X.; Dong, L.; Dong, Q.; Mukai, M.; Chen, B.B.; Holsberger, D.R.; Sottas, C.M.; Cooke, P.S.; Lian, Q.Q.; et al. Increased proliferation but decreased steroidogenic capacity in Leydig cells from mice lacking cyclin-dependent kinase inhibitor 1B. Biol. Reprod. 2009, 80, 1232–1238. [Google Scholar] [CrossRef]
- Nguyen, L.; Besson, A.; Heng, J.I.; Schuurmans, C.; Teboul, L.; Parras, C.; Philpott, A.; Roberts, J.M.; Guillemot, F. p27kip1 independently promotes neuronal differentiation and migration in the cerebral cortex. Genes Dev. 2006, 20, 1511–1524. [Google Scholar] [CrossRef]
- Qiu, J.; Takagi, Y.; Harada, J.; Topalkara, K.; Wang, Y.; Sims, J.R.; Zheng, G.; Huang, P.; Ling, Y.; Scadden, D.T.; et al. p27Kip1 constrains proliferation of neural progenitor cells in adult brain under homeostatic and ischemic conditions. Stem Cells 2009, 27, 920–927. [Google Scholar] [CrossRef]
- Mairet-Coello, G.; Tury, A.; Van Buskirk, E.; Robinson, K.; Genestine, M.; DiCicco-Bloom, E. p57(KIP2) regulates radial glia and intermediate precursor cell cycle dynamics and lower layer neurogenesis in developing cerebral cortex. Development 2012, 139, 475–487. [Google Scholar] [CrossRef]
- Geisen, C.; Karsunky, H.; Yücel, R.; Möröy, T. Loss of p27(Kip1) cooperates with cyclin E in T-cell lymphomagenesis. Oncogene 2003, 22, 1724–1729. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ul Quraish, R.; Sudou, N.; Nomura-Komoike, K.; Sato, F.; Fujieda, H. p27(KIP1) loss promotes proliferation and phagocytosis but prevents epithelial-mesenchymal transition in RPE cells after photoreceptor damage. Mol. Vis. 2016, 22, 1103–1121. [Google Scholar] [PubMed]
- Zhang, P.; Liégeois, N.J.; Wong, C.; Finegold, M.; Hou, H.; Thompson, J.C.; Silverman, A.; Harper, J.W.; DePinho, R.A.; Elledge, S.J. Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature 1997, 387, 151–158. [Google Scholar] [CrossRef]
- Yan, Y.; Frisen, J.; Lee, M.H.; Massague, J.; Barbacid, M. Ablation of the CDK inhibitor p57(Kip2) results in increased apoptosis and delayed differentiation during mouse development. Genes Dev. 1997, 11, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Nakayama, K.; Nakayama, K. Mice lacking a CDK inhibitor, p57(Kip2), exhibit skeletal abnormalities and growth retardation. J. Biochem. 2000, 127, 73–83. [Google Scholar] [CrossRef]
- Hiromura, K.; Haseley, L.A.; Zhang, P.; Monkawa, T.; Durvasula, R.; Petermann, A.T.; Alpers, C.E.; Mundel, P.; Shankland, S.J. Podocyte expression of the CDK-inhibitor p57 during development and disease. Kidney Int. 2001, 60, 2235–2246. [Google Scholar] [CrossRef]
- Mademtzoglou, D.; Alonso-Martin, S.; Chang, T.H.; Bismuth, K.; Drayton-Libotte, B.; Aurade, F.; Relaix, F. A p57 conditional mutant allele that allows tracking of p57-expressing cells. Genesis 2017, 55. [Google Scholar] [CrossRef]
- Tunster, S.J.; Van de Pette, M.; John, R.M. Fetal overgrowth in the Cdkn1c mouse model of Beckwith-Wiedemann syndrome. Dis. Model. Mech. 2011, 4, 814–821. [Google Scholar] [CrossRef]
- Imaizumi, Y.; Furutachi, S.; Watanabe, T.; Miya, H.; Kawaguchi, D.; Gotoh, Y. Role of the imprinted allele of the Cdkn1c gene in mouse neocortical development. Sci. Rep. 2020, 10, 1884. [Google Scholar] [CrossRef]
- Krimpenfort, P.; Quon, K.C.; Mooi, W.J.; Loonstra, A.; Berns, A. Loss of p16Ink4a confers susceptibility to metastatic melanoma in mice. Nature 2001, 413, 83–86. [Google Scholar] [CrossRef]
- Morrison, J.P.; Satoh, H.; Foley, J.; Horton, J.L.; Dunnick, J.K.; Kissling, G.E.; Malarkey, D.E. N-ethyl-N-nitrosourea (ENU)-induced meningiomatosis and meningioma in p16(INK4a)/p19(ARF) tumor suppressor gene-deficient mice. Toxicol. Pathol. 2007, 35, 780–787. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Bardeesy, N.; Lee, K.H.; Carrasco, D.; Castrillon, D.H.; Aguirre, A.J.; Wu, E.A.; Horner, J.W.; DePinho, R.A. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 2001, 413, 86–91. [Google Scholar] [CrossRef] [PubMed]
- An, S.; Chen, Y.; Gao, C.; Qin, B.; Du, X.; Meng, F.; Qi, Y. Inactivation of INK4a and ARF induces myocardial proliferation and improves cardiac repair following ischemia-reperfusion. Mol. Med. Rep. 2015, 12, 5911–5916. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Wolstein, J.M.; Pudasaini, B.; Plotkin, M. INK4a deletion results in improved kidney regeneration and decreased capillary rarefaction after ischemia-reperfusion injury. Am. J. Physiol. Renal Physiol. 2012, 302, F183–F191. [Google Scholar] [CrossRef] [PubMed]
- Sundar, I.K.; Rashid, K.; Gerloff, J.; Li, D.; Rahman, I. Genetic ablation of p16(INK4a) does not protect against cellular senescence in mouse models of chronic obstructive pulmonary disease/emphysema. Am. J. Respir. Cell Mol. Biol. 2018, 59, 189–199. [Google Scholar] [CrossRef]
- Kukuyan, A.M.; Sementino, E.; Kadariya, Y.; Menges, C.W.; Cheung, M.; Tan, Y.; Cai, K.Q.; Slifker, M.J.; Peri, S.; Klein-Szanto, A.J.; et al. Inactivation of Bap1 cooperates with losses of Nf2 and Cdkn2a to drive the development of pleural malignant mesothelioma in conditional mouse models. Cancer Res. 2019, 79, 4113–4123. [Google Scholar] [CrossRef]
- Novais, E.J.; Diekman, B.O.; Shapiro, I.M.; Risbud, M.V. p16(Ink4a) deletion in cells of the intervertebral disc affects their matrix homeostasis and senescence associated secretory phenotype without altering onset of senescence. Matrix Biol. 2019, 82, 54–70. [Google Scholar] [CrossRef]
- Latres, E.; Malumbres, M.; Sotillo, R.; Martín, J.; Ortega, S.; Martín-Caballero, J.; Flores, J.M.; Cordón-Cardo, C.; Barbacid, M. Limited overlapping roles of P15(INK4b) and P18(INK4c) cell cycle inhibitors in proliferation and tumorigenesis. EMBO J. 2000, 19, 3496–3506. [Google Scholar] [CrossRef]
- Franklin, D.S.; Godfrey, V.L.; Lee, H.; Kovalev, G.I.; Schoonhoven, R.; Chen-Kiang, S.; Su, L.; Xiong, Y. CDK inhibitors p18(INK4c) and p27(Kip1) mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes Dev. 1998, 12, 2899–2911. [Google Scholar] [CrossRef]
- Bai, F.; Pei, X.H.; Godfrey, V.L.; Xiong, Y. Haploinsufficiency of p18(INK4c) sensitizes mice to carcinogen-induced tumorigenesis. Mol. Cell. Biol. 2003, 23, 1269–1277. [Google Scholar] [CrossRef]
- Zindy, F.; van Deursen, J.; Grosveld, G.; Sherr, C.J.; Roussel, M.F. INK4d-deficient mice are fertile despite testicular atrophy. Mol. Cell. Biol. 2000, 20, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Gilles, L.; Guièze, R.; Bluteau, D.; Cordette-Lagarde, V.; Lacout, C.; Favier, R.; Larbret, F.; Debili, N.; Vainchenker, W.; Raslova, H. P19INK4D links endomitotic arrest and megakaryocyte maturation and is regulated by AML-1. Blood 2008, 111, 4081–4091. [Google Scholar] [CrossRef]
- Chen, P.; Zindy, F.; Abdala, C.; Liu, F.; Li, X.; Roussel, M.F.; Segil, N. Progressive hearing loss in mice lacking the cyclin-dependent kinase inhibitor Ink4d. Nat. Cell Biol. 2003, 5, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Satyanarayana, A.; Berthet, C.; Lopez-Molina, J.; Coppola, V.; Tessarollo, L.; Kaldis, P. Genetic substitution of Cdk1 by Cdk2 leads to embryonic lethality and loss of meiotic function of Cdk2. Development 2008, 135, 3389–3400. [Google Scholar] [CrossRef] [PubMed]
- Chotiner, J.Y.; Wolgemuth, D.J.; Wang, P.J. Functions of cyclins and CDKs in mammalian gametogenesis†. Biol. Reprod. 2019, 101, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Severance, A.L.; Latham, K.E. PLK1 regulates spindle association of phosphorylated eukaryotic translation initiation factor 4E-binding protein and spindle function in mouse oocytes. Am. J. Physiol. Cell Physiol. 2017, 313, C501–C515. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Koncicka, M.; Tetkova, A.; Jansova, D.; Del Llano, E.; Gahurova, L.; Kracmarova, J.; Prokesova, S.; Masek, T.; Pospisek, M.; Bruce, A.W.; et al. Increased expression of maturation promoting factor components speeds up meiosis in oocytes from aged females. Int. J. Mol. Sci. 2018, 19, 2841. [Google Scholar] [CrossRef]
- Levasseur, M.D.; Thomas, C.; Davies, O.R.; Higgins, J.M.G.; Madgwick, S. Aneuploidy in oocytes is prevented by sustained CDK1 activity through degron masking in cyclin B1. Dev. Cell 2019, 48, 672–684. [Google Scholar] [CrossRef]
- Zi, Z.; Zhang, Z.; Li, Q.; An, W.; Zeng, L.; Gao, D.; Yang, Y.; Zhu, X.; Zeng, R.; Shum, W.W.; et al. CCNYL1, but not CCNY, cooperates with CDK16 to regulate spermatogenesis in mouse. PLoS Genet. 2015, 11, e1005485. [Google Scholar] [CrossRef]
- Berthet, C.; Klarmann, K.D.; Hilton, M.B.; Suh, H.C.; Keller, J.R.; Kiyokawa, H.; Kaldis, P. Combined loss of Cdk2 and Cdk4 results in embryonic lethality and Rb hypophosphorylation. Dev. Cell 2006, 10, 563–573. [Google Scholar] [CrossRef]
- Barriere, C.; Santamaria, D.; Cerqueira, A.; Galan, J.; Martin, A.; Ortega, S.; Malumbres, M.; Dubus, P.; Barbacid, M. Mice thrive without Cdk4 and Cdk2. Mol. Oncol. 2007, 1, 72–83. [Google Scholar] [CrossRef]
- Zhang, P.; Wong, C.; DePinho, R.A.; Harper, J.W.; Elledge, S.J. Cooperation between the Cdk inhibitors p27(KIP1) and p57(KIP2) in the control of tissue growth and development. Genes Dev. 1998, 12, 3162–3167. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, Y.; Matsumoto, A.; Kanie, T.; Hara, E.; Nakayama, K.; Nakayama, K.I. Development of mice without Cip/Kip CDK inhibitors. Biochem. Biophys. Res. Commun. 2012, 427, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Susaki, E.; Nakayama, K.; Yamasaki, L.; Nakayama, K.I. Common and specific roles of the related CDK inhibitors p27 and p57 revealed by a knock-in mouse model. Proc. Natl. Acad. Sci. USA 2009, 106, 5192–5197. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, H.; Hatakeyama, S.; Nakayama, K.; Nagata, M.; Tomita, K.; Nakayama, K. Spatial and temporal expression patterns of the cyclin-dependent kinase (CDK) inhibitors p27Kip1 and p57Kip2 during mouse development. Anat. Embryol. 2001, 203, 77–87. [Google Scholar] [CrossRef]
- Hatada, I.; Mukai, T. Genomic imprinting of p57(Kip2), cyclin-dependent kinase inhibitor, in mouse. Nat. Genet. 1995, 11, 204–206. [Google Scholar] [CrossRef]
- Yan, Y.M.; Frisen, J.; Lee, M.H.; Massague, J.; Barbacid, M. Mice lacking p57(Kip2) display developmental defects in the gastrointestinal tract and in endochondral bone formation. Eur. J. Cell Biol. 1997, 72, 29. [Google Scholar]
- Takahashi, K.; Kobayashi, T.; Kanayama, N. p57(Kip2) regulates the proper development of labyrinthine and spongiotrophoblasts. Mol. Hum. Reprod. 2000, 6, 1019–1025. [Google Scholar] [CrossRef]
- Andrews, S.C.; Wood, M.D.; Tunster, S.J.; Barton, S.C.; Surani, M.A.; John, R.M. Cdkn1c (p57(Kip2)) is the major regulator of embryonic growth within its imprinted domain on mouse distal chromosome 7. BMC Dev. Biol. 2007, 7, 53. [Google Scholar] [CrossRef]
- Zindy, F.; den Besten, W.; Chen, B.; Rehg, J.E.; Latres, E.; Barbacid, M.; Pollard, J.W.; Sherr, C.J.; Cohen, P.E.; Roussel, M.F. Control of spermatogenesis in mice by the cyclin D-dependent kinase inhibitors p18(Ink4c) and p19(Ink4d). Mol. Cell Biol. 2001, 21, 3244–3255. [Google Scholar] [CrossRef]
- Hall, M.; Bates, S.; Peters, G. Evidence for different modes of action of cyclin-dependent kinase inhibitors: p15 and p16 bind to kinases, p21 and p27 bind to cyclins. Oncogene 1995, 11, 1581–1588. [Google Scholar] [PubMed]
- Shah, K.; Lahiri, D.K. Cdk5 activity in the brain—Multiple paths of regulation. J. Cell Sci. 2014, 127, 2391–2400. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Humbert, S.; Bronson, R.T.; Takahashi, S.; Kulkarni, A.B.; Li, E.; Tsai, L.H. p35 and p39 are essential for cyclin-dependent kinase 5 function during neurodevelopment. J. Neurosci. 2001, 21, 6758–6771. [Google Scholar] [CrossRef] [PubMed]
- Mita, N.; He, X.; Sasamoto, K.; Mishiba, T.; Ohshima, T. Cyclin-dependent kinase 5 regulates dendritic spine formation and maintenance of cortical neuron in the mouse brain. Cereb. Cortex 2016, 26, 967–976. [Google Scholar] [CrossRef]
- Sasamoto, K.; Nagai, J.; Nakabayashi, T.; He, X.; Ohshima, T. Cdk5 is required for the positioning and survival of GABAergic neurons in developing mouse striatum. Dev. Neurobiol. 2017, 77, 483–492. [Google Scholar] [CrossRef]
- Xu, B.; Kumazawa, A.; Kobayashi, S.; Hisanaga, S.I.; Inoue, T.; Ohshima, T. Cdk5 activity is required for Purkinje cell dendritic growth in cell-autonomous and non-cell-autonomous manners. Dev. Neurobiol. 2017, 77, 1175–1187. [Google Scholar] [CrossRef]
- Piccini, A.; Perlini, L.E.; Cancedda, L.; Benfenati, F.; Giovedi, S. Phosphorylation by PKA and Cdk5 mediates the early effects of Synapsin III in neuronal morphological maturation. J. Neurosci. 2015, 35, 13148–13159. [Google Scholar] [CrossRef]
- Qi, G.J.; Chen, Q.; Chen, L.J.; Shu, Y.; Bu, L.L.; Shao, X.Y.; Zhang, P.; Jiao, F.J.; Shi, J.; Tian, B. Phosphorylation of Connexin 43 by Cdk5 modulates neuronal migration during embryonic brain development. Mol. Neurobiol. 2016, 53, 2969–2982. [Google Scholar] [CrossRef]
- Westbury, J.; Watkins, M.; Ferguson-Smith, A.C.; Smith, J. Dynamic temporal and spatial regulation of the cdk inhibitor p57(kip2) during embryo morphogenesis. Mec. Dev. 2001, 109, 83–89. [Google Scholar] [CrossRef]
- Laukoter, S.; Beattie, R.; Pauler, F.M.; Amberg, N.; Nakayama, K.I.; Hippenmeyer, S. Imprinted Cdkn1c genomic locus cell-autonomously promotes cell survival in cerebral cortex development. Nat. Commun. 2020, 11, 195. [Google Scholar] [CrossRef]
- Colasante, G.; Simonet, J.C.; Calogero, R.; Crispi, S.; Sessa, A.; Cho, G.; Golden, J.A.; Broccoli, V. ARX regulates cortical intermediate progenitor cell expansion and upper layer neuron formation through repression of Cdkn1c. Cereb. Cortex 2015, 25, 322–335. [Google Scholar] [CrossRef]
- Kumar, S.; Biancotti, J.C.; Matalon, R.; de Vellis, J. Lack of aspartoacylase activity disrupts survival and differentiation of neural progenitors and oligodendrocytes in a mouse model of Canavan disease. J. Neurosci. Res. 2009, 87, 3415–3427. [Google Scholar] [CrossRef] [PubMed]
- Jansen, L.A.; Uhlmann, E.J.; Crino, P.B.; Gutmann, D.H.; Wong, M. Epileptogenesis and reduced inward rectifier potassium current in tuberous sclerosis complex-1-deficient astrocytes. Epilepsia 2005, 46, 1871–1880. [Google Scholar] [CrossRef]
- Smitherman, M.; Lee, K.; Swanger, J.; Kapur, R.; Clurman, B.E. Characterization and targeted disruption of murine Nup50, a p27(Kip1)-interacting component of the nuclear pore complex. Mol. Cell Biol. 2000, 20, 5631–5642. [Google Scholar] [CrossRef] [PubMed]
- Azzarelli, R.; Hurley, C.; Sznurkowska, M.K.; Rulands, S.; Hardwick, L.; Gamper, I.; Ali, F.; McCracken, L.; Hindley, C.; McDuff, F.; et al. Multi-site Neurogenin3 phosphorylation controls pancreatic endocrine differentiation. Dev. Cell 2017, 41, 274–286. [Google Scholar] [CrossRef] [PubMed]
- García-Reyes, B.; Kretz, A.L.; Ruff, J.P.; von Karstedt, S.; Hillenbrand, A.; Knippschild, U.; Henne-Bruns, D.; Lemke, J. The emerging role of cyclin-dependent kinases (CDKs) in pancreatic ductal adenocarcinoma. Int. J. Mol. Sci. 2018, 19, 3219. [Google Scholar] [CrossRef]
- Spaeth, J.M.; Hunter, C.S.; Bonatakis, L.; Guo, M.; French, C.A.; Slack, I.; Hara, M.; Fisher, S.E.; Ferrer, J.; Morrisey, E.E.; et al. The FOXP1, FOXP2 and FOXP4 transcription factors are required for islet alpha cell proliferation and function in mice. Diabetologia 2015, 58, 1836–1844. [Google Scholar] [CrossRef]
- Potikha, T.; Kassem, S.; Haber, E.P.; Ariel, I.; Glaser, B. p57Kip2 (cdkn1c): Sequence, splice variants and unique temporal and spatial expression pattern in the rat pancreas. Lab. Invest. 2005, 85, 364–375. [Google Scholar] [CrossRef][Green Version]
- Asahara, S.; Etoh, H.; Inoue, H.; Teruyama, K.; Shibutani, Y.; Ihara, Y.; Kawada, Y.; Bartolome, A.; Hashimoto, N.; Matsuda, T.; et al. Paternal allelic mutation at the Kcnq1 locus reduces pancreatic beta-cell mass by epigenetic modification of Cdkn1c. Proc. Natl. Acad. Sci. USA 2015, 112, 8332–8337. [Google Scholar] [CrossRef]
- Van de Pette, M.; Tunster, S.J.; McNamara, G.I.; Shelkovnikova, T.; Millership, S.; Benson, L.; Peirson, S.; Christian, M.; Vidal-Puig, A.; John, R.M. Cdkn1c boosts the development of brown adipose tissue in a murine model of Silver Russell Syndrome. PLoS Genet. 2016, 12, e1005916. [Google Scholar] [CrossRef]
- Rhee, K.; Wolgemuth, D.J. Cdk family genes are expressed not only in dividing but also in terminally differentiated mouse germ cells, suggesting their possible function during both cell division and differentiation. Dev. Dyn. 1995, 204, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Su, T.T.; Stumpff, J. Promiscuity rules? The dispensability of cyclin E and Cdk2. Sci. Signal. 2004, 2004, pe11. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Singh, P.; Patel, R.K.; Palmer, N.; Grenier, J.K.; Paduch, D.; Kaldis, P.; Grimson, A.; Schimenti, J.C. CDK2 kinase activity is a regulator of male germ cell fate. Development 2019, 146. [Google Scholar] [CrossRef]
- Palmer, N.; Talib, S.Z.A.; Kaldis, P. Diverse roles for CDK-associated activity during spermatogenesis. FEBS Lett. 2019, 593, 2925–2949. [Google Scholar] [CrossRef] [PubMed]
- Moniot, B.; Ujjan, S.; Champagne, J.; Hirai, H.; Aritake, K.; Nagata, K.; Dubois, E.; Nidelet, S.; Nakamura, M.; Urade, Y.; et al. Prostaglandin D2 acts through the Dp2 receptor to influence male germ cell differentiation in the foetal mouse testis. Development 2014, 141, 3561–3571. [Google Scholar] [CrossRef]
- Phelps, D.E.; Hsiao, K.M.; Li, Y.; Hu, N.; Franklin, D.S.; Westphal, E.; Lee, E.Y.; Xiong, Y. Coupled transcriptional and translational control of cyclin-dependent kinase inhibitor p18INK4c expression during myogenesis. Mol. Cell. Biol. 1998, 18, 2334–2343. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bayrak, A.; Oktay, K. The expression of cyclin-dependent kinase inhibitors p15, p16, p21, and p27 during ovarian follicle growth initiation in the mouse. Reprod. Biol. Endocrinol. 2003, 1, 41. [Google Scholar] [CrossRef]
- Feldmann, A.; Dimitrova, E.; Kenney, A.; Lastuvkova, A.; Klose, R.J. CDK-Mediator and FBXL19 prime developmental genes for activation by promoting atypical regulatory interactions. Nucleic Acids Res. 2020, 48, 2942–2955. [Google Scholar] [CrossRef]
- Ku, C.C.; Wuputra, K.; Kato, K.; Lin, W.H.; Pan, J.B.; Tsai, S.C.; Kuo, C.J.; Lee, K.H.; Lee, Y.L.; Lin, Y.C.; et al. Jdp2-deficient granule cell progenitors in the cerebellum are resistant to ROS-mediated apoptosis through xCT/Slc7a11 activation. Sci. Rep. 2020, 10, 4933. [Google Scholar] [CrossRef]
- Fu, W.Y.; Cheng, K.; Fu, A.K.; Ip, N.Y. Cyclin-dependent kinase 5-dependent phosphorylation of Pctaire1 regulates dendrite development. Neuroscience 2011, 180, 353–359. [Google Scholar] [CrossRef]
- Chen, X.Y.; Gu, X.T.; Saiyin, H.; Wan, B.; Zhang, Y.J.; Li, J.; Wang, Y.L.; Gao, R.; Wang, Y.F.; Dong, W.P.; et al. Brain-selective kinase 2 (BRSK2) phosphorylation on PCTAIRE1 negatively regulates glucose-stimulated insulin secretion in pancreatic beta-cells. J. Biol. Chem. 2012, 287, 30368–30375. [Google Scholar] [CrossRef] [PubMed]
- Poratti, M.; Marzaro, G. Third-generation CDK inhibitors: A review on the synthesis and binding modes of Palbociclib, Ribociclib and Abemaciclib. Eur. J. Med. Chem. 2019, 172, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Van de Pette, M.; Abbas, A.; Feytout, A.; McNamara, G.; Bruno, L.; To, W.K.; Dimond, A.; Sardini, A.; Webster, Z.; McGinty, J.; et al. Visualizing changes in Cdkn1c expression links early-life adversity to imprint mis-regulation in adults. Cell Rep. 2017, 18, 1090–1099. [Google Scholar] [CrossRef]
- Burd, C.E.; Sorrentino, J.A.; Clark, K.S.; Darr, D.B.; Krishnamurthy, J.; Deal, A.M.; Bardeesy, N.; Castrillon, D.H.; Beach, D.H.; Sharpless, N.E. Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell 2013, 152, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.J.; Safran, M.; Wei, W.; Sorensen, E.; Lassota, P.; Zhelev, N.; Neuberg, D.S.; Shapiro, G.; Kaelin, W.G., Jr. Bioluminescent imaging of Cdk2 inhibition in vivo. Nat. Med. 2004, 10, 643–648. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| CDK | Developmental Region | KO Phenotypes | References |
|---|---|---|---|
| Cdk1 | Whole Body | Mutant line with truncated protein–homozygous early embryonic lethal | [21] |
| Whole Body | KO–lethal at the 1-cell stage due to inability to complete cell division | [22] | |
| Testes | cKO in spermatocytes–cells arrest at prometaphase resulting in male infertility | [23] | |
| Ovaries | cKO in oocytes–females infertile due to failure to resume meiosis. Rescued with microinjection of Cdk1 | [24] | |
| Brain | cKO in Nex (postmitotic neurons of cortex and hippocampus)–normal brain development | [25] | |
| Liver | cKO in liver–failure to complete cell division but normal phenotype due to compensatory cell growth | [22] | |
| Cdk2 | Whole Body/ Testes/Ovaries | KO–develop normally but both sexes sterile. Spermatocytes and oocytes arrest at prophase I, oocytes slightly later than spermatocytes | [26] |
| Whole Body/ Testes/Ovaries | KO–normal development but both sexes infertile | [27] | |
| Ovaries | cKO in oocytes–develop normally | [24] | |
| Cdk3 | Whole Body | Null point mutation–normal development. Possible functional redundancy | [28] |
| Cdk4 | Whole Body/ Testes/Ovaries/ Endocrine System/ Pancreas | KO–reduced mutant numbers in heterozygous cross, suggesting some embryonic lethality. Decreased body size. Defective spermatogenesis reducing fertility. Females infertile due to deficit in hypothalamic-pituitary axis. Mice are insulin-deficient diabetic due to affected β-cells. | [29] |
| Whole Body/ Pancreas/ Testes/Ovaries | KO–mice are undersized and develop diabetes as above. Males show testicular atrophy due to defective proliferation of gonocytes and hypoplastic seminiferous tubules. Female infertility due to perturbed corpus luteum formation | [30] | |
| Whole Body/ Endocrine System | KO–decreased body size with small pituitaries, with particularly reduced lactotrophs resulting in female infertility | [31] | |
| Cdk5 | Brain | KO–>60% die in utero; altered brain development | [32] |
| Brain | KO–reduced phosphorylation of histone H1 by Cdk16 in the brain | [33] | |
| Brain | cKO in CaMKII (forebrain)–seizures, tremors, growth retardation, layer disruption and neurogenerative changes that progressed with age | [34] | |
| Brain | cKO in CaMKII–behavioural changes, increased susceptibility to cocaine | [35] | |
| Brain | cKO in CNP cells–hypomyelination and impaired learning and memory | [36] | |
| Brain | cKO in CNP cells–loss of myelin repair | [37] | |
| Brain | cKO in EMX1–impaired differentiation of oligodendrocytes | [38] | |
| Brain | cKO in EMX1–defective layer structure in cerebral cortex | [39] | |
| Brain | cKO in mid and hindbrain– small cerebellum and altered dendritic development | [40] | |
| Cdk6 | Whole Body/ Immune System | KO–undersized females. Underdeveloped thymus and spleen. Mild anaemia due to defects in haematopoiesis | [41] |
| Cdk7 | |||
| Cdk8 | Whole Body/ Intestine | KO–normal growth, but increased size and growth rate of intestinal tumours | [42] |
| Pancreas | cKO in β-cells–improved glucose tolerance due to increased insulin secretion. Increased cell death following stress conditions | [43] | |
| Cdk9 | Whole Body | KO–complete embryonic lethality from day E9.5 | * |
| Cdk10 | |||
| Cdk11 | Whole Body | KO–early embryonic lethal between days E3.5 and E4. Cells exhibit growth defects and mitotic arrest, resulting in blastocyte apoptosis | [44] |
| Cdk12 | |||
| Cdk13 | Whole Body/ Lung/Liver/ Kidney/Heart | Null mutant–developmental delay, growth retardation, underdeveloped lungs, liver and kidneys. Embryo lethal due to heart defect | [45] |
| Cdk14 | |||
| Cdk15 | |||
| Cdk16/PCTAIRE1 | Testes/Ovaries | cKO–failure of spermatogenesis. Female fertility normal | [46] |
| Cdk17/PCTAIRE2 | Whole Body/ Brain | Exon deletion–partial embryonic lethality. Increased vertical activity | * |
| Cdk18/PCTAIRE3 | Brain | Exon deletion–abnormal behavioural responses to light | * |
| Cdk19 | |||
| Cdk20 |
| CDKI | Protein | Developmental Region | KO Phenotypes | References |
|---|---|---|---|---|
| Cdkn1a | p21CIP1 | Whole Body | KO–no spontaneous tumorigenesis | [47] |
| Brain | KO–increased neural migration and differentiation in response to brain injury | [48] | ||
| Intestine | KO–increased susceptibility to intestinal tumours | [49] | ||
| Immune System | KO–no altered susceptibility to retrovirally induced lymphatic tumours | [50] | ||
| Immune System | cKO in T cells–hyposensitive to programmed cell death in response to radiation-induced DNA damage | [51] | ||
| Cdkn1b | p27KIP1 | Whole Body/Endocrine System/Ovaries | KO–oversized with larger internal organs, especially the pituitary, and female sterility due to impairment of the formation of corpora lutea | [52] |
| Whole Body/ Endocrine System/Ovaries | KO–oversized due to increased cell number and proliferation. Females infertile due to impaired luteal cell differentiation reflecting a disturbance in the hypothalamic–pituitary–ovarian axis | [53] | ||
| Whole Body/ Immune System/ Endocrine System/Eyes/ Ovaries | KO–oversized, especially in thymus, pituitary and adrenal glands and gonadal organs. Disturbed organization of retinas. Female sterility due to impaired development of ovarian follicles | [54] | ||
| Endocrine System/Intestine/ Lung | KO–increased susceptibility to pituitary and intestinal tumours, lesions of the female reproductive tract and lung adenomas following exposure to carcinogens | [55] | ||
| Heart/Ovaries | KO–protective delay of ageing and apoptosis of heart due to ovariectomy by upregulating antioxidant enzymes | [56] | ||
| Testes | KO–decreased testosterone production, despite increased proliferation of testosterone-producing cells | [57] | ||
| Brain | KO–altered neuron migration and differentiation | [58] | ||
| Brain | KO–increased neural progenitor cell proliferation under basal and injury conditions | [59] | ||
| Brain | KO–increased cell proliferation of lower layer neurons | [60] | ||
| Immune System | KO–increased susceptibility to retrovirally induced lymphatic tumours. Increased spleen proliferation | [50] | ||
| Immune System | KO–developed spontaneous T cell lymphomas | [61] | ||
| Eyes | KO–increased cell proliferation and poor repair response after photoreceptor damage | [62] | ||
| Cdkn1c | p57KIP2 | Whole Body/ Intestine/ Kidney/Spine/ Endocrine System/Eyes | KO–locational defects in small intestine and muscle. Neonatal lethality due to breathing difficulty from cleft palate. Undersized kidneys, delayed bone development and enlarged adrenal gland. Increased lens cell proliferation and apoptosis | [63] |
| Whole Body/ Intestine | KO–neonatal lethality, cleft palate, gastrointestinal abnormalities and short limbs. Increased apoptosis | [64] | ||
| Whole Body/ Spine/Testes/ Ovaries | KO–cleft palate and bone malformations. Most died within 24 hours of birth due to difficulties breathing. Surviving mice showed severe growth retardation. Both sexes were sexually immature | [65] | ||
| Whole Body/ Brain | KO–oversized embryo and macrocephaly, with increased cell proliferation, particularly of neuronal precursors and lower layer neurons. Decreased cell cycle length and increased cell cycle exit at days E14.5 and E16.5 | [60] | ||
| Kidney | KO–normal kidney differentiation and development | [66] | ||
| Whole Body/ Spine | cKO maternally–skeletal deformities, reduced body size and perinatal lethality | [67] | ||
| Whole Body | cKO maternally–perinatal lethality, overgrowth during early gestation and weaning, but constrained growth in late gestation | [68] | ||
| Brain | cKO paternally–decreased brain size and reduced number of neural progenitor cells during development | [69] | ||
| Cdkn2a | p16INK4a | Whole Body/ Testes/Ovaries | KO–no morphological or behavioural malformations. No significant increase in spontaneous tumours. Fertile | [70] |
| Brain | KO–increased susceptibility to CNS tumours and hydrocephaly following carcinogenic exposure | [71] | ||
| Immune System | KO–normal development, but with thymic hyperplasia | [72] | ||
| Heart | KO–increased cardiomyocyte proliferation and cardiac repair | [73] | ||
| Kidney | KO–increased kidney cell proliferation and decreased apoptosis in response to ischemia-reperfusion injury | [74] | ||
| Lung | KO–reduced inflammation in response to cigarette smoke, but no change to proinflammatory cytokine response | [75] | ||
| Lung | cKO in lung epithelia–reduction in inflammation response to cigarette smoke and suppression of proinflammatory cytokines, but no protective effect | [75] | ||
| Lung | cKO in pleura–no development of malignant mesotheliomas | [76] | ||
| Spine | cKO in intervertebral disc–no change in senescence, reduced apoptosis and altered matrix homeostasis | [77] | ||
| Cdkn2b | p15INK4b | Whole Body/ Testes/Ovaries/ Immune System | KO–viable and fertile with no morphological or behavioural abnormalities at birth. After two months of age, mice had atypical haematopoiesis and increased lymphocytes in the spleen | [78] |
| Cdkn2c | p18INK4c | Whole Body/ Endocrine System/ Immune System | KO–gigantism and organomegaly, particularly of the pituitary, spleen and thymus, due to altered cell cycle control. Altered cell cycle entry of resting B cells | [79] |
| Whole Body/ Testes/Ovaries/ Immune System/ Endocrine System | KO–viable and fertile with no morphological or behavioural abnormalities at birth. From six months of age mice developed splenomegaly, enlarged lymph nodes and increased incidence of pituitary tumours | [78] | ||
| Endocrine System/Immune System | KO–spontaneous pituitary and lymphatic tumours. Also observed in heterozygous mice, implying haploinsufficiency | [80] | ||
| Cdkn2d | p19INK4d | Whole Body/ Testes | KO–normal growth with no obvious malformations. Decreased, but not total loss of, male fertility due to testicular atrophy and increased apoptosis of germ cells | [81] |
| Immune System | KO–increase in mean ploidy level in bone marrow | [82] | ||
| Ears | KO–aberrant cell cycle entry and subsequent apoptosis in sensory hairs leading to hearing loss | [83] | ||
| Cdkn3 | Brain | Exon deletion–decreased grip strength | * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campbell, G.J.; Hands, E.L.; Van de Pette, M. The Role of CDKs and CDKIs in Murine Development. Int. J. Mol. Sci. 2020, 21, 5343. https://doi.org/10.3390/ijms21155343
Campbell GJ, Hands EL, Van de Pette M. The Role of CDKs and CDKIs in Murine Development. International Journal of Molecular Sciences. 2020; 21(15):5343. https://doi.org/10.3390/ijms21155343
Chicago/Turabian StyleCampbell, Grace Jean, Emma Langdale Hands, and Mathew Van de Pette. 2020. "The Role of CDKs and CDKIs in Murine Development" International Journal of Molecular Sciences 21, no. 15: 5343. https://doi.org/10.3390/ijms21155343
APA StyleCampbell, G. J., Hands, E. L., & Van de Pette, M. (2020). The Role of CDKs and CDKIs in Murine Development. International Journal of Molecular Sciences, 21(15), 5343. https://doi.org/10.3390/ijms21155343