Targeting the DNA Damage Response to Overcome Cancer Drug Resistance in Glioblastoma

Abstract
1. Introduction
2. Glioblastoma Multiforme
3. The DNA Damage Response in Mediating Resistance in Glioblastoma
3.1. Single Strand Break Repair Systems
3.2. Double Strand Break Repair Systems
4. Temozolomide Treatment and Cell Death
TMZ and CNUs Affect Cell Cycle and Death
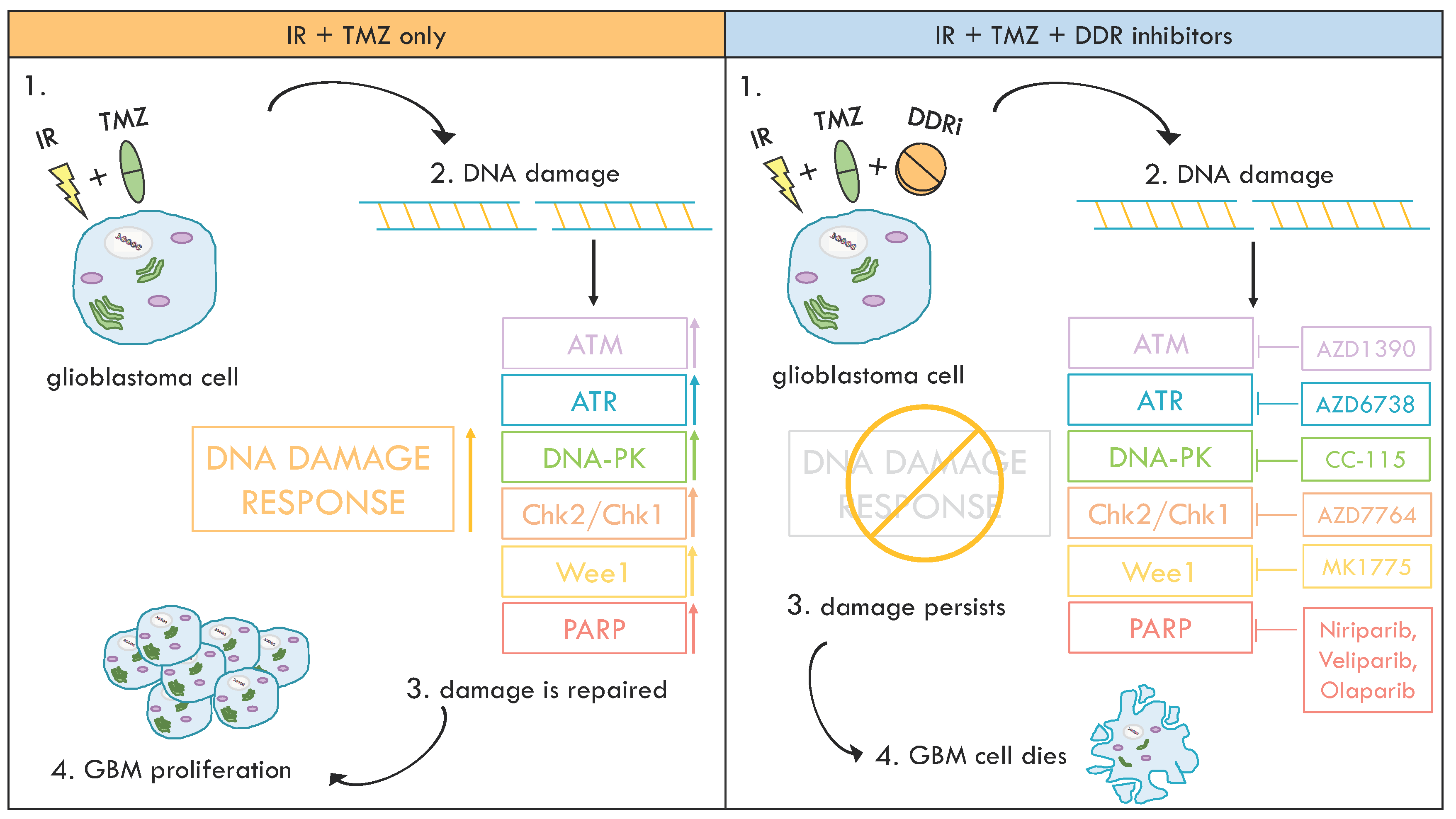
5. Inhibition of DDR Kinases to Overcome Therapy Resistance in GBM
5.1. ATM
5.2. ATR
5.3. DNA-PK
5.4. PARP
6. Conclusions

{kind=link}
| Kinase | Inhibitor | Clinical Trial Phase | End Date | Drug Combinatory Strategy | Characteristics | BBB Permeability | Tumors/Cell Lines | Reference |
|---|---|---|---|---|---|---|---|---|
| ATM | KU55933 | Pre-clinical | - | +IR +etoposide phosphate | Poor aqueous solubility and poor bioavailability | no | Human cervical cancer (HeLa), Human osteosarcoma (U2OS) | [33,94] |
| KU60019 | Pre-clinical | - | - | Poor aqueous solubility and poor bioavailability | no | Human glioma (U87, U1241) | [34] | |
| CP466722 | Pre-clinical | - | +temozolomide | Improved aqueous solubility and bioavailability | no | Human breast cancer cell line (MCF7), fibroblasts (HFF), A-T cells, GBM12 glioblastoma xenograft cell lines | [35] | |
| KU59403 | Pre-clinical | - | +irinotecan +etoposide phosphate | Improved aqueous solubility and bioavailability | no | Human colon cancer (HCT116, SW620), Human osteosarcoma (U2OS), Human breast cancer (MDA-MB-231) | [41] | |
| AZ32 | Pre-clinical | - | +IR | Good bioavailability | yes | Human glioma (U87, LN18, T98G, …, A172) | [36] | |
| AZD0156 | Phase-I NCT02588105 clinicaltrials.gov | 30 April 2020 | +olaparib +irinotecan | - | yes (poor) | Various metastatic solid tumours (including gastric adenocarcinoma, colorectal cancer) | [95] | |
| AZD1390 | Phase-I NCT03423628 clinicaltrials.gov | 05 April 2022 | +IR | Good bioavailability | yes | Primary and recurrent glioblastoma multiforme | [38] | |
| ATR | VE-821 | Pre-clinical | - | +cisplatin | - | yes (poor) | Hamster ovarian cells (AA8), hamster lung fibroblasts (V79), human glioblastoma multiforme (M059J) | [47] |
| NU-6027 | Pre-clinical | - | +cisplatin +PARPi +hydroxyurea | - | unclear | Breast cancer, pancreatic cancer, ovarian cancer | [96] | |
| AZ20 | Pre-clinical | - | monotherapy | Poor aqueous solubility | yes (poor) | Colorectal adenocarcinoma cell line (HT29), glioblastoma CSCs | [55,97] | |
| VX-970 | Phase-I NCT02157792 clinicaltrials.gov | 11 March 2020, 30 April 2025 | monotherapy +cisplatin | - | unclear | Advanced solid tumors | [50,51] | |
| BAY1895344 | Phase-I NCT03188965 clinicaltrials.gov | 25 March 2022 | monotherapy | - | unclear | Solid cancers and lymphomas | [52] | |
| AZD6738 | Phase-II NCT03682289 clinicaltrials.gov | 19 March 2023 | +olaparib | Good oral bioavailability | yes (good) | Renal and pancreatic carcinoma, glioma initiating cells | [49,53] | |
| DNAPK | M3814 | Phase-I NCT02316197 NCT02516813 clinicaltrials.gov | 19 December 2020 | monotherapy +IR +cisplatin +doxorubicin | Orally bioavailable | unclear | CLL and solid tumors | [69,70] |
| CC-115 | Phase-I NCT02977780 clinicaltrials.gov | May 2022 | +neratinib +temozolomide | - | yes (good) | Glioblastoma multiforme | [73] | |
| Chk2 | PV1019 | Pre-clinical | - | +IR +topotecan | Good bioavailability | yes (good) | Human breast cancer (MCF7), Human ovarian cancer (OVCAR-3,-4,-5,-8), Human glioblastoma (U251) | [43] |
| CCT241533 | Pre-clinical | - | +bleomycin +olaparib +IR | Good bioavailability | yes (good) | Human colorectal cancer (HT-29), human breast cancer (MCF7), human glioblastoma (U87MG), human ovarian cancer line (OVCAR-3,5) | [44] | |
| Chk2/Chk1 | AZD7762 | Phase-I NCT00473616 clinicaltrials.gov | February 2011, terminated due to cardiotoxicity | +irinotecan | - | unclear | Solid advanced tumors, glioblastoma primary isolates (pre-clinical) | [45] |
| Chk1 | LY2606368 | Phase-I NCT04023669 clinicaltrials.gov | June 2026 | +gemcitabine +cyclophosphamide | Good oral bioavailability | unclear | Advanced solid tumors, medulloblastoma | [60] |
| SRA737 | Phase-I/II NCT02797964 NCT02797977 clinicaltrials.gov | 28 October 2019 | monotherapy +cisplatin +gemcitabine | Good oral bioavailability | unclear | Advanced solid tumors, Non-Hodgkin’s lymphoma | [59] | |
| Wee1 | MK-1775 | Phase-I NCT02207010 NCT01849146 clinicaltrials.gov | May 2018, 28 September 2020 | monotherapy +IR +temozolomide | Good oral bioavailability | yes (good) | Recurrent glioblastoma, Glioblastoma xenografts, Human glioblastoma cell line (U251, U87MG, T98G) | [66,67,68] |
| PARP | Rucaparib | Phase II NCT01891344 Phase III NCT01968213 clinicaltrials.gov | 31 October 2021 June 2020 | monotherapy | Good oral availability | yes (poor) | Ovarian cancer, Epithelial ovarian cancer, Fallopian tube cancer | [83,84,98,99] |
| Niraparib | Phase I NCT01294735 Phase II NCT03307785 | May 2012 February 2020 | monotherapy +temozolomide +bevaciumab +carboplatin | Good bioavailability | yes (good) | Melanoma, Glioblastoma Multiforme, Metastatic solid tumors | [85] | |
| Veliparib | Phase I NCT01514201 Phase II NCT03581292 Phase III NCT02152982 | 28 March 2018, 29 October 2024, 14 January 2021 | +IR +temozolomide | Good oral bioavailability | yes (good) | Newly diagnosed Glioblastoma and glioma | [86] | |
| Olaparib | Phase II NCT03233204 Phase II NCT02974621 Phase IINCT 03212274 Phase I NCT03212742 | 30 September 2024, 31 May 2020, 31 July 2020, 30 June 2022 | monotherapy +bevacizumab +IR +temozolomide | Good oral bioavailability | yes (poor) | Non-hodgkin lymphoma, Advanced solid tumours, Glioma | [89,90] |
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 9-1-1 | Rad9-HUS1-Rad1 |
| ATM | Ataxia-telangiectasia-mutated |
| ATR | Ataxia-telangiectasia- and Rad3-related |
| BAK | Bcl2 antagonist/killer |
| BAX | Bcl2 associated X protein |
| BBB | Blood–brain barrier |
| BRCA1 | Breast cancer 1 |
| CDC25A | Cell division cycle 25A |
| CDK1 | Cyclin dependent kinase 1 |
| CHK1 | Checkpoint kinase 1 |
| CHK2 | Checkpoint kinase 2 |
| CNU | Chloroethylating nitrosoureas |
| DDR | DNA damage response |
| DDRi | DNA damage response inhibitors |
| DNA-PK | DNA-dependent protein kinase |
| DSB | Double-strand break |
| GBM | Glioblastoma multiforme |
| GSC | Glioblastoma stem cell |
| HR | Homologous recombination |
| IR | Ionizing radiation |
| MCM8 | Minichromosome maintenance 8 |
| MCM9 | Minichromosome maintenance 9 |
| MGMT | O-6-Methylguanine DNA-methyltransferase |
| MLH1 | MutL homologue 1 |
| MMR | Mismatch repair |
| MRN | Mre11-Rad50-Nbs1 |
| MSH2 | MutS homologue 2 |
| MSH6 | MutS homologue 6 |
| NSC | Neural stem cell |
| NHEJ | Non-homologous end-joining |
| PARP | Poly-ADP-ribose polymerase |
| PIKK | Phosphatidylinositol 3-kinase related kinase |
| PMS2 | PMS1 homolog 2 |
| PTEN | Phosphatase and tensin homologue |
| PUMA | P53 upregulated modulator of apoptosis |
| ROS | Reactive oxygen species |
| SSB | Single-strand break |
| TMZ | Temozolomide |
| XRCC1 | X-ray repair cross-complementing 1 |
References
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Pearlman, A.H.; Hsieh, P. DNA mismatch repair and the DNA damage response. DNA Repair (Amst) 2016, 38, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B.; Christmann, M. DNA repair in personalized brain cancer therapy with temozolomide and nitrosoureas. DNA Repair (Amst) 2019, 78, 128–141. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Behjati, S.; Gundem, G.; Wedge, D.C.; Roberts, N.D.; Tarpey, P.S.; Cooke, S.L.; Van Loo, P.; Alexandrov, L.B.; Ramakrishna, M.; Davies, H.; et al. Mutational signatures of ionizing radiation in second malignancies. Nat. Commun. 2016, 7, 12605. [Google Scholar] [CrossRef]
- Zhang, J.; Stevens, M.F.; Bradshaw, T.D. Temozolomide: Mechanisms of action, repair and resistance. Curr. Mol. Pharm. 2012, 5, 102–114. [Google Scholar] [CrossRef]
- Ochs, K.; Kaina, B. Apoptosis induced by DNA damage O6-methylguanine is Bcl-2 and caspase-9/3 regulated and Fas/caspase-8 independent. Cancer Res. 2000, 60, 5815–5824. [Google Scholar]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef]
- Quiros, S.; Roos, W.P.; Kaina, B. Processing of O6-methylguanine into DNA double-strand breaks requires two rounds of replication whereas apoptosis is also induced in subsequent cell cycles. Cell Cycle 2010, 9, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Nikolova, T.; Roos, W.P.; Krämer, O.H.; Strik, H.M.; Kaina, B. Chloroethylating nitrosoureas in cancer therapy: DNA damage, repair and cell death signaling. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Yoshimoto, K.; Mizoguchi, M.; Hata, N.; Murata, H.; Hatae, R.; Amano, T.; Nakamizo, A.; Sasaki, T. Complex DNA repair pathways as possible therapeutic targets to overcome temozolomide resistance in glioblastoma. Front. Oncol. 2012, 2, 186. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, G.; Bozek, D.A.; Rajakulendran, N.; Monteiro, V.; Ahmadi, M.; Steinhart, Z.; Kushida, M.M.; Yu, H.; Coutinho, F.J.; Cavalli, F.M.G.; et al. Genome-Wide CRISPR-Cas9 Screens Expose Genetic Vulnerabilities and Mechanisms of Temozolomide Sensitivity in Glioblastoma Stem Cells. Cell Rep. 2019, 27, 971–986.e979. [Google Scholar] [CrossRef] [PubMed]
- Felsberg, J.; Thon, N.; Eigenbrod, S.; Hentschel, B.; Sabel, M.C.; Westphal, M.; Schackert, G.; Kreth, F.W.; Pietsch, T.; Löffler, M.; et al. Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int. J. Cancer 2011, 129, 659–670. [Google Scholar] [CrossRef]
- Shinsato, Y.; Furukawa, T.; Yunoue, S.; Yonezawa, H.; Minami, K.; Nishizawa, Y.; Ikeda, R.; Kawahara, K.; Yamamoto, M.; Hirano, H.; et al. Reduction of MLH1 and PMS2 confers temozolomide resistance and is associated with recurrence of glioblastoma. Oncotarget 2013, 4, 2261–2270. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef]
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Langerak, P.; Russell, P. Regulatory networks integrating cell cycle control with DNA damage checkpoints and double-strand break repair. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 3562–3571. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Nikolova, T.; Quiros, S.; Naumann, S.C.; Kiedron, O.; Zdzienicka, M.Z.; Kaina, B. Brca2/Xrcc2 dependent HR, but not NHEJ, is required for protection against O(6)-methylguanine triggered apoptosis, DSBs and chromosomal aberrations by a process leading to SCEs. DNA Repair (Amst) 2009, 8, 72–86. [Google Scholar] [CrossRef] [PubMed]
- Eich, M.; Roos, W.P.; Nikolova, T.; Kaina, B. Contribution of ATM and ATR to the resistance of glioblastoma and malignant melanoma cells to the methylating anticancer drug temozolomide. Mol. Cancer 2013, 12, 2529–2540. [Google Scholar] [CrossRef]
- Barciszewska, A.M.; Gurda, D.; Głodowicz, P.; Nowak, S.; Naskręt-Barciszewska, M.Z. A New Epigenetic Mechanism of Temozolomide Action in Glioma Cells. PLoS ONE 2015, 10, e0136669. [Google Scholar] [CrossRef]
- Hirose, Y.; Berger, M.S.; Pieper, R.O. p53 effects both the duration of G2/M arrest and the fate of temozolomide-treated human glioblastoma cells. Cancer Res. 2001, 61, 1957–1963. [Google Scholar]
- Matt, S.; Hofmann, T.G. The DNA damage-induced cell death response: A roadmap to kill cancer cells. Cell Mol. Life Sci. 2016, 73, 2829–2850. [Google Scholar] [CrossRef]
- Roos, W.P.; Batista, L.F.; Naumann, S.C.; Wick, W.; Weller, M.; Menck, C.F.; Kaina, B. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene 2007, 26, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Tomicic, M.T.; Meise, R.; Aasland, D.; Berte, N.; Kitzinger, R.; Krämer, O.H.; Kaina, B.; Christmann, M. Apoptosis induced by temozolomide and nimustine in glioblastoma cells is supported by JNK/c-Jun-mediated induction of the BH3-only protein BIM. Oncotarget 2015, 6, 33755–33768. [Google Scholar] [CrossRef]
- Stagni, V.; Oropallo, V.; Fianco, G.; Antonelli, M.; Cina, I.; Barila, D. Tug of War between Survival and Death: Exploring ATM Function in Cancer. Int. J. Mol. Sci. 2014, 15, 5388–5409. [Google Scholar] [CrossRef]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef]
- Zannini, L.; Delia, D.; Buscemi, G. CHK2 kinase in the DNA damage response and beyond. J. Mol. Cell Biol. 2014, 6, 442–457. [Google Scholar] [CrossRef] [PubMed]
- Hickson, I.; Zhao, Y.; Richardson, C.J.; Green, S.J.; Martin, N.M.; Orr, A.I.; Reaper, P.M.; Jackson, S.P.; Curtin, N.J.; Smith, G.C. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004, 64, 9152–9159. [Google Scholar] [CrossRef] [PubMed]
- Golding, S.E.; Rosenberg, E.; Valerie, N.; Hussaini, I.; Frigerio, M.; Cockcroft, X.F.; Chong, W.Y.; Hummersone, M.; Rigoreau, L.; Menear, K.A.; et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol. Cancer 2009, 8, 2894–2902. [Google Scholar] [CrossRef] [PubMed]
- Rainey, M.D.; Charlton, M.E.; Stanton, R.V.; Kastan, M.B. Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation. Cancer Res. 2008, 68, 7466–7474. [Google Scholar] [CrossRef] [PubMed]
- Karlin, J.; Allen, J.; Ahmad, S.F.; Hughes, G.; Sheridan, V.; Odedra, R.; Farrington, P.; Cadogan, E.B.; Riches, L.C.; Garcia-Trinidad, A.; et al. Orally Bioavailable and Blood-Brain Barrier-Penetrating ATM Inhibitor (AZ32) Radiosensitizes Intracranial Gliomas in Mice. Mol. Cancer 2018, 17, 1637–1647. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Shelat, A.A.; Guy, R.K.; Kastan, M.B. Development of a cell-based, high-throughput screening assay for ATM kinase inhibitors. J. Biomol. Screen. 2014, 19, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Durant, S.T.; Zheng, L.; Wang, Y.; Chen, K.; Zhang, L.; Zhang, T.; Yang, Z.; Riches, L.; Trinidad, A.G.; Fok, J.H.L.; et al. The brain-penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Sci. Adv. 2018, 4, eaat1719. [Google Scholar] [CrossRef]
- Bakr, A.; Oing, C.; Köcher, S.; Borgmann, K.; Dornreiter, I.; Petersen, C.; Dikomey, E.; Mansour, W.Y. Involvement of ATM in homologous recombination after end resection and RAD51 nucleofilament formation. Nucleic Acids Res. 2015, 43, 3154–3166. [Google Scholar] [CrossRef]
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Oldreive, C.; Petermann, E.; Stewart, G.; Brown, J.; Lau, A.; Pratt, G.; et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood 2016, 127, 582–595. [Google Scholar] [CrossRef]
- Batey, M.A.; Zhao, Y.; Kyle, S.; Richardson, C.; Slade, A.; Martin, N.M.; Lau, A.; Newell, D.R.; Curtin, N.J. Preclinical evaluation of a novel ATM inhibitor, KU59403, in vitro and in vivo in p53 functional and dysfunctional models of human cancer. Mol. Cancer 2013, 12, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Biddlestone-Thorpe, L.; Sajjad, M.; Rosenberg, E.; Beckta, J.M.; Valerie, N.C.; Tokarz, M.; Adams, B.R.; Wagner, A.F.; Khalil, A.; Gilfor, D.; et al. ATM kinase inhibition preferentially sensitizes p53-mutant glioma to ionizing radiation. Clin. Cancer Res. 2013, 19, 3189–3200. [Google Scholar] [CrossRef] [PubMed]
- Jobson, A.G.; Lountos, G.T.; Lorenzi, P.L.; Llamas, J.; Connelly, J.; Cerna, D.; Tropea, J.E.; Onda, A.; Zoppoli, G.; Kondapaka, S.; et al. Cellular inhibition of checkpoint kinase 2 (Chk2) and potentiation of camptothecins and radiation by the novel Chk2 inhibitor PV1019 [7-nitro-1H-indole-2-carboxylic acid {4-[1-(guanidinohydrazone)-ethyl]-phenyl}-amide]. J. Pharm. Exp. 2009, 331, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Anderson, V.E.; Walton, M.I.; Eve, P.D.; Boxall, K.J.; Antoni, L.; Caldwell, J.J.; Aherne, W.; Pearl, L.H.; Oliver, A.W.; Collins, I.; et al. CCT241533 is a potent and selective inhibitor of CHK2 that potentiates the cytotoxicity of PARP inhibitors. Cancer Res. 2011, 71, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Dai, Y.; Grant, S.; Dent, P. Enhancing CHK1 inhibitor lethality in glioblastoma. Cancer Biol. 2012, 13, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Yazinski, S.A.; Zou, L. Functions, Regulation, and Therapeutic Implications of the ATR Checkpoint Pathway. Annu. Rev. Genet. 2016, 50, 155–173. [Google Scholar] [CrossRef]
- Charrier, J.D.; Durrant, S.J.; Golec, J.M.; Kay, D.P.; Knegtel, R.M.; MacCormick, S.; Mortimore, M.; O’Donnell, M.E.; Pinder, J.L.; Reaper, P.M.; et al. Discovery of potent and selective inhibitors of ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase as potential anticancer agents. J. Med. Chem. 2011, 54, 2320–2330. [Google Scholar] [CrossRef]
- Middleton, F.K.; Patterson, M.J.; Elstob, C.J.; Fordham, S.; Herriott, A.; Wade, M.A.; McCormick, A.; Edmondson, R.; May, F.E.; Allan, J.M.; et al. Common cancer-associated imbalances in the DNA damage response confer sensitivity to single agent ATR inhibition. Oncotarget 2015, 6, 32396–32409. [Google Scholar] [CrossRef]
- Foote, K.M.; Nissink, J.W.M.; McGuire, T.; Turner, P.; Guichard, S.; Yates, J.W.T.; Lau, A.; Blades, K.; Heathcote, D.; Odedra, R.; et al. Discovery and Characterization of AZD6738, a Potent Inhibitor of Ataxia Telangiectasia Mutated and Rad3 Related (ATR) Kinase with Application as an Anticancer Agent. J. Med. Chem. 2018, 61, 9889–9907. [Google Scholar] [CrossRef]
- Fokas, E.; Prevo, R.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; Cornelissen, B.; Vallis, K.A.; Hammond, E.M.; Olcina, M.M.; Gillies McKenna, W.; et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012, 3, e441. [Google Scholar] [CrossRef]
- Karnitz, L.M.; Zou, L. Molecular Pathways: Targeting ATR in Cancer Therapy. Clin. Cancer Res. 2015, 21, 4780–4785. [Google Scholar] [CrossRef]
- Wengner, A.M.; Siemeister, G.; Lücking, U.; Lefranc, J.; Wortmann, L.; Lienau, P.; Bader, B.; Bömer, U.; Moosmayer, D.; Eberspächer, U.; et al. The Novel ATR Inhibitor BAY 1895344 Is Efficacious as Monotherapy and Combined with DNA Damage-Inducing or Repair-Compromising Therapies in Preclinical Cancer Models. Mol. Cancer 2020, 19, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Fròsina, G.; Profumo, A.; Marubbi, D.; Marcello, D.; Ravetti, J.L.; Daga, A. ATR kinase inhibitors NVP-BEZ235 and AZD6738 effectively penetrate the brain after systemic administration. Radiat. Oncol. 2018, 13, 76. [Google Scholar] [CrossRef] [PubMed]
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.D.; Maccormick, S.; Charlton, P.A.; Golec, J.M.; Pollard, J.R. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430. [Google Scholar] [CrossRef]
- Foote, K.M.; Blades, K.; Cronin, A.; Fillery, S.; Guichard, S.S.; Hassall, L.; Hickson, I.; Jacq, X.; Jewsbury, P.J.; McGuire, T.M.; et al. Discovery of 4-{4-[(3R)-3-Methylmorpholin-4-yl]-6-[1-(methylsulfonyl)cyclopropyl]pyrimidin-2-yl}-1H-indole (AZ20): A potent and selective inhibitor of ATR protein kinase with monotherapy in vivo antitumor activity. J. Med. Chem. 2013, 56, 2125–2138. [Google Scholar] [CrossRef] [PubMed]
- Menezes, D.L.; Holt, J.; Tang, Y.; Feng, J.; Barsanti, P.; Pan, Y.; Ghoddusi, M.; Zhang, W.; Thomas, G.; Holash, J.; et al. A synthetic lethal screen reveals enhanced sensitivity to ATR inhibitor treatment in mantle cell lymphoma with ATM loss-of-function. Mol. Cancer Res. 2015, 13, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.L.; Wijnhoven, P.W.G.; Ramos-Montoya, A.; Wilson, Z.; Illuzzi, G.; Falenta, K.; Jones, G.N.; James, N.; Chabbert, C.D.; Stott, J.; et al. Combined PARP and ATR inhibition potentiates genome instability and cell death in ATM-deficient cancer cells. Oncogene 2020. [Google Scholar] [CrossRef]
- Sanjiv, K.; Hagenkort, A.; Calderón-Montaño, J.M.; Koolmeister, T.; Reaper, P.M.; Mortusewicz, O.; Jacques, S.A.; Kuiper, R.V.; Schultz, N.; Scobie, M.; et al. Cancer-Specific Synthetic Lethality between ATR and CHK1 Kinase Activities. Cell Rep. 2016, 17, 3407–3416. [Google Scholar] [CrossRef]
- Plummer, E.R.; Kristeleit, R.S.; Cojocaru, E.; Haris, N.; Carter, L.; Jone, R.H.; Blagden, S.P.; Evans, T.R.J.; Arkenau, H.T.; Sarker, D.; et al. A first-in-human phase I/II trial of SRA737 (a Chk1 inhibitor) in subjects with advanced cancer. J. Clin. Oncol. 2019, 37, 3094. [Google Scholar] [CrossRef]
- Hong, D.; Infante, J.; Janku, F.; Jones, S.; Nguyen, L.M.; Burris, H.; Naing, A.; Bauer, T.M.; Piha-Paul, S.; Johnson, F.M.; et al. Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients With Advanced Cancer. J. Clin. Oncol. 2016, 34, 1764–1771. [Google Scholar] [CrossRef]
- Bukhari, A.B.; Lewis, C.W.; Pearce, J.J.; Luong, D.; Chan, G.K.; Gamper, A.M. Inhibiting Wee1 and ATR kinases produces tumor-selective synthetic lethality and suppresses metastasis. J. Clin. Investig. 2019, 129, 1329–1344. [Google Scholar] [CrossRef] [PubMed]
- Carrassa, L.; Chilà, R.; Lupi, M.; Ricci, F.; Celenza, C.; Mazzoletti, M.; Broggini, M.; Damia, G. Combined inhibition of Chk1 and Wee1: In vitro synergistic effect translates to tumor growth inhibition in vivo. Cell Cycle 2012, 11, 2507–2517. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kumagai, A.; Dunphy, W.G. Positive regulation of Wee1 by Chk1 and 14-3-3 proteins. Mol. Biol. Cell 2001, 12, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Mayor-Ruiz, C.; Lafarga, V.; Murga, M.; Vega-Sendino, M.; Ortega, S.; Fernandez-Capetillo, O. A Genome-wide CRISPR Screen Identifies CDC25A as a Determinant of Sensitivity to ATR Inhibitors. Mol. Cell 2016, 62, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Aarts, M.; Sharpe, R.; Garcia-Murillas, I.; Gevensleben, H.; Hurd, M.S.; Shumway, S.D.; Toniatti, C.; Ashworth, A.; Turner, N.C. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2012, 2, 524–539. [Google Scholar] [CrossRef] [PubMed]
- Pokorny, J.L.; Calligaris, D.; Gupta, S.K.; Iyekegbe, D.O.; Mueller, D.; Bakken, K.K.; Carlson, B.L.; Schroeder, M.A.; Evans, D.L.; Lou, Z.; et al. The Efficacy of the Wee1 Inhibitor MK-1775 Combined with Temozolomide Is Limited by Heterogeneous Distribution across the Blood-Brain Barrier in Glioblastoma. Clin. Cancer Res. 2015, 21, 1916–1924. [Google Scholar] [CrossRef]
- Sanai, N.; Li, J.; Boerner, J.; Stark, K.; Wu, J.; Kim, S.; Derogatis, A.; Mehta, S.; Dhruv, H.D.; Heilbrun, L.K.; et al. Phase 0 Trial of AZD1775 in First-Recurrence Glioblastoma Patients. Clin. Cancer Res. 2018, 24, 3820–3828. [Google Scholar] [CrossRef]
- Alexander, B.; Supko, J.; Agar, N.; Ahluwalia, M.; Desai, A.; Dietrich, J.; Kaley, T.; Peereboom, D.; Takebe, N.; Desideri, S.; et al. ACTR-14. Phase i study of azd1775 with radiation therapy (RT) and temozolomide (TMZ) in patients with newly diagnosed glioblastoma (GBM) and evaluation of intratumoral drug distribution (idd) in patients with recurrent GBM. Neuro Oncol. 2018, 20, vi13–vi14. [Google Scholar] [CrossRef][Green Version]
- Biau, J.; Chautard, E.; Verrelle, P.; Dutreix, M. Altering DNA Repair to Improve Radiation Therapy: Specific and Multiple Pathway Targeting. Front. Oncol. 2019, 9, 1009. [Google Scholar] [CrossRef]
- Mohiuddin, I.S.; Kang, M.H. DNA-PK as an Emerging Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 635. [Google Scholar] [CrossRef]
- Riabinska, A.; Daheim, M.; Herter-Sprie, G.S.; Winkler, J.; Fritz, C.; Hallek, M.; Thomas, R.K.; Kreuzer, K.A.; Frenzel, L.P.; Monfared, P.; et al. Therapeutic targeting of a robust non-oncogene addiction to PRKDC in ATM-defective tumors. Sci. Transl. Med. 2013, 5, 189ra178. [Google Scholar] [CrossRef] [PubMed]
- Kantidze, O.L.; Velichko, A.K.; Luzhin, A.V.; Petrova, N.V.; Razin, S.V. Synthetically Lethal Interactions of ATM, ATR, and DNA-PKcs. Trends Cancer 2018, 4, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Munster, P.; Mita, M.; Mahipal, A.; Nemunaitis, J.; Massard, C.; Mikkelsen, T.; Cruz, C.; Paz-Ares, L.; Hidalgo, M.; Rathkopf, D.; et al. First-In-Human Phase I Study Of A Dual mTOR Kinase And DNA-PK Inhibitor (CC-115) In Advanced Malignancy. Cancer Manag. Res. 2019, 11, 10463–10476. [Google Scholar] [CrossRef]
- Amé, J.C.; Spenlehauer, C.; de Murcia, G. The PARP superfamily. Bioessays 2004, 26, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. XRCC1 protein; Form and function. DNA Repair (Amst) 2019, 81, 102664. [Google Scholar] [CrossRef] [PubMed]
- Nomura, F.; Yaguchi, M.; Togawa, A.; Miyazaki, M.; Isobe, K.; Miyake, M.; Noda, M.; Nakai, T. Enhancement of poly-adenosine diphosphate-ribosylation in human hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2000, 15, 529–535. [Google Scholar] [CrossRef]
- Ossovskaya, V.; Koo, I.C.; Kaldjian, E.P.; Alvares, C.; Sherman, B.M. Upregulation of Poly (ADP-Ribose) Polymerase-1 (PARP1) in Triple-Negative Breast Cancer and Other Primary Human Tumor Types. Genes Cancer 2010, 1, 812–821. [Google Scholar] [CrossRef]
- Galia, A.; Calogero, A.E.; Condorelli, R.; Fraggetta, F.; La Corte, A.; Ridolfo, F.; Bosco, P.; Castiglione, R.; Salemi, M. PARP-1 protein expression in glioblastoma multiforme. Eur. J. Histochem. 2012, 56, e9. [Google Scholar] [CrossRef]
- de Murcia, J.M.; Niedergang, C.; Trucco, C.; Ricoul, M.; Dutrillaux, B.; Mark, M.; Oliver, F.J.; Masson, M.; Dierich, A.; LeMeur, M.; et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc. Natl. Acad. Sci. USA 1997, 94, 7303–7307. [Google Scholar] [CrossRef]
- Dziadkowiec, K.N.; Gąsiorowska, E.; Nowak-Markwitz, E.; Jankowska, A. PARP inhibitors: Review of mechanisms of action and BRCA1/2 mutation targeting. Prz. Menopauzalny 2016, 15, 215–219. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Kristeleit, R.S.; Oaknin, A.; Ray-Coquard, I.; Leary, A.; Balmaña, J.; Drew, Y.; Oza, A.M.; Shapira-Frommer, R.; Domchek, S.M.; Cameron, T.; et al. Antitumor activity of the poly(ADP-ribose) polymerase inhibitor rucaparib as monotherapy in patients with platinum-sensitive, relapsed. Int. J. Gynecol. Cancer 2019, 29, 1396–1404. [Google Scholar] [CrossRef]
- Parrish, K.E.; Cen, L.; Murray, J.; Calligaris, D.; Kizilbash, S.; Mittapalli, R.K.; Carlson, B.L.; Schroeder, M.A.; Sludden, J.; Boddy, A.V.; et al. Efficacy of PARP Inhibitor Rucaparib in Orthotopic Glioblastoma Xenografts Is Limited by Ineffective Drug Penetration into the Central Nervous System. Mol. Cancer 2015, 14, 2735–2743. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Mikule, K.; Wang, Z.; Poon, G.; Vaidyanathan, A.; Smith, G.; Zhang, Z.Y.; Hanke, J.; Ramaswamy, S.; Wang, J. A comparative pharmacokinetic study of PARP inhibitors demonstrates favorable properties for niraparib efficacy in preclinical tumor models. Oncotarget 2018, 9, 37080–37096. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Kizilbash, S.H.; Carlson, B.L.; Mladek, A.C.; Boakye-Agyeman, F.; Bakken, K.K.; Pokorny, J.L.; Schroeder, M.A.; Decker, P.A.; Cen, L.; et al. Delineation of MGMT Hypermethylation as a Biomarker for Veliparib-Mediated Temozolomide-Sensitizing Therapy of Glioblastoma. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Gupta, S.K.; Smith, E.J.; Mladek, A.C.; Tian, S.; Decker, P.A.; Kizilbash, S.H.; Kitange, G.J.; Sarkaria, J.N. PARP Inhibitors for Sensitization of Alkylation Chemotherapy in Glioblastoma: Impact of Blood-Brain Barrier and Molecular Heterogeneity. Front. Oncol. 2018, 8, 670. [Google Scholar] [CrossRef]
- Higuchi, F.; Nagashima, H.; Ning, J.; Koerner, M.V.A.; Wakimoto, H.; Cahill, D.P. Restoration of Temozolomide Sensitivity by PARP Inhibitors in Mismatch Repair Deficient Glioblastoma is Independent of Base Excision Repair. Clin. Cancer Res. 2020, 26, 1690–1699. [Google Scholar] [CrossRef]
- Lesueur, P.; Lequesne, J.; Grellard, J.M.; Dugué, A.; Coquan, E.; Brachet, P.E.; Geffrelot, J.; Kao, W.; Emery, E.; Berro, D.H.; et al. Phase I/IIa study of concomitant radiotherapy with olaparib and temozolomide in unresectable or partially resectable glioblastoma: OLA-TMZ-RTE-01 trial protocol. BMC Cancer 2019, 19, 198. [Google Scholar] [CrossRef]
- Hanna, C.; Kurian, K.M.; Williams, K.; Watts, C.; Jackson, A.; Carruthers, R.; Strathdee, K.; Cruickshank, G.; Dunn, L.; Erridge, S.; et al. Pharmacokinetics, safety and tolerability of olaparib and temozolomide for recurrent glioblastoma: Results of the phase I OPARATIC trial. Neuro-Oncology 2020. [Google Scholar] [CrossRef]
- Middleton, M.R.; Friedlander, P.; Hamid, O.; Daud, A.; Plummer, R.; Falotico, N.; Chyla, B.; Jiang, F.; McKeegan, E.; Mostafa, N.M.; et al. Randomized phase II study evaluating veliparib (ABT-888) with temozolomide in patients with metastatic melanoma. Ann. Oncol. 2015, 26, 2173–2179. [Google Scholar] [CrossRef] [PubMed]
- Fulton, B.; Short, S.C.; James, A.; Nowicki, S.; McBain, C.; Jefferies, S.; Kelly, C.; Stobo, J.; Morris, A.; Williamson, A.; et al. PARADIGM-2: Two parallel phase I studies of olaparib and radiotherapy or olaparib and radiotherapy plus temozolomide in patients with newly diagnosed glioblastoma, with treatment stratified by MGMT status. Clin. Transl. Radiat. Oncol. 2018, 8, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, M.; Gan, H.; Wang, H.; Lee, J.H.; Fang, D.; Kitange, G.J.; He, L.; Hu, Z.; Parney, I.F.; et al. A novel enhancer regulates MGMT expression and promotes temozolomide resistance in glioblastoma. Nat. Commun. 2018, 9, 2949. [Google Scholar] [CrossRef] [PubMed]
- Nadkarni, A.; Shrivastav, M.; Mladek, A.C.; Schwingler, P.M.; Grogan, P.T.; Chen, J.; Sarkaria, J.N. ATM inhibitor KU-55933 increases the TMZ responsiveness of only inherently TMZ sensitive GBM cells. J. Neurooncol. 2012, 110, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Riches, L.C.; Trinidad, A.G.; Hughes, G.; Jones, G.N.; Hughes, A.M.; Thomason, A.G.; Gavine, P.; Cui, A.; Ling, S.; Stott, J.; et al. Pharmacology of the ATM Inhibitor AZD0156: Potentiation of Irradiation and Olaparib Responses Preclinically. Mol. Cancer 2020, 19, 13–25. [Google Scholar] [CrossRef]
- Peasland, A.; Wang, L.Z.; Rowling, E.; Kyle, S.; Chen, T.; Hopkins, A.; Cliby, W.A.; Sarkaria, J.; Beale, G.; Edmondson, R.J.; et al. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br. J. Cancer 2011, 105, 372–381. [Google Scholar] [CrossRef]
- Ning, J.; Wakimoto, H.; Martuza, R.; Rabkin, S. ATR inhibitors synergize with PARP inhibitors in killing glioblastoma stem cells and treating glioblastoma. Cancer Res. 2017, 77, AM2017-1122. [Google Scholar]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferri, A.; Stagni, V.; Barilà, D. Targeting the DNA Damage Response to Overcome Cancer Drug Resistance in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 4910. https://doi.org/10.3390/ijms21144910
Ferri A, Stagni V, Barilà D. Targeting the DNA Damage Response to Overcome Cancer Drug Resistance in Glioblastoma. International Journal of Molecular Sciences. 2020; 21(14):4910. https://doi.org/10.3390/ijms21144910
Chicago/Turabian StyleFerri, Alessandra, Venturina Stagni, and Daniela Barilà. 2020. "Targeting the DNA Damage Response to Overcome Cancer Drug Resistance in Glioblastoma" International Journal of Molecular Sciences 21, no. 14: 4910. https://doi.org/10.3390/ijms21144910
APA StyleFerri, A., Stagni, V., & Barilà, D. (2020). Targeting the DNA Damage Response to Overcome Cancer Drug Resistance in Glioblastoma. International Journal of Molecular Sciences, 21(14), 4910. https://doi.org/10.3390/ijms21144910