Abstract
Current evidence indicates that postischemic brain injury is associated with the accumulation of folding proteins, such as amyloid and tau protein, in the intra- and extracellular spaces of neuronal cells. In this review, we summarize protein changes associated with Alzheimer’s disease and their gene expression (amyloid protein precursor and tau protein) after brain ischemia, and their roles in the postischemic period. Recent advances in understanding the postischemic mechanisms in development of neurodegeneration have revealed dysregulation of amyloid protein precursor, α-, β- and γ-secretase and tau protein genes. Reduced expression of the α-secretase gene after brain ischemia with recirculation causes neuronal cells to be less resistant to injury. We present the latest data that Alzheimer’s disease-related proteins and their genes play a crucial role in postischemic neurodegeneration. Understanding the underlying processes of linking Alzheimer’s disease-related proteins and their genes in development of postischemic neurodegeneration will provide the most significant goals to date for therapeutic development.
1. Introduction
Brain ischemia is one of the most common forms of neurodegeneration, with a series of pathological molecular processes that occur during and after ischemia and gradually spread to various brain structures. New data suggest that there is a similarity between neuropathology developed in the postischemic brain and Alzheimer’s disease [1,2,3,4,5,6,7,8]. Both ischemic stroke in human and experimental ischemic brain episodes are life-threatening pathological events with development of Alzheimer’s disease-type dementia after ischemia [9,10,11,12,13,14,15]. Current studies indicate that ischemia-reperfusion brain injury can be involved in development of Alzheimer’s disease neuropathology [3,16]. First, ischemic stroke and Alzheimer’s disease have the same risk factors like age, hyperlipidemia, hypertension and diabetes. Second, the postischemic brain generates a unique pattern of disappearance of neuronal cells in the CA1 area of the hippocampus with serious general brain atrophy, which is similar to the atrophy noted in Alzheimer’s disease [17,18,19,20,21]. Third, neuroinflammatory reactions have an important role in the progress of the postischemic brain and Alzheimer’s disease [22,23]. Fourth, the data suggest that postischemic brain injury with recirculation can trigger the neuropathology of folding proteins characteristic of Alzheimer’s disease by generation and accumulation of amyloid [19,24,25,26]. Finally, investigations proved that tau protein pathology also played a significant role in progression after ischemic brain neurodegeneration [27,28,29,30,31,32,33,34,35,36,37,38]. In this review, we present changes in expression of genes involved in the amyloidogenic metabolism of the amyloid protein precursor, which are associated with the production of amyloid in the postischemic brain. In addition, we pay attention to whether the amyloid is involved in death of neurons in the CA1 and CA3 areas of the hippocampus and the medial temporal cortex postischemia. Also, we take into account the importance of ischemic changes in gene expression of tau protein during clinical onset, progression and maturation of brain neurodegeneration after ischemia. Below we summarize the latest data that Alzheimer’s disease-related proteins, like amyloid and tau protein, and their genes, play a fundamental role in postischemic neurodegeneration. Progress in understanding new key processes induced by brain ischemia with recirculation, like changes in the genotype and phenotype of the Alzheimer’s disease type, which are not yet fully explained, may help develop strategies for prevention and treatment against neurodegeneration induced by ischemia.
2. Dysregulation of Amyloid Processing Genes in Global Cerebral Ischemia due to Cardiac Arrest in Rats
In the CA1 area of the hippocampus, gene expression of the amyloid protein precursor was below the control value within 2 days after ischemia and 7 and 30 days postischemia the expression of the amyloid protein precursor gene was above the control values (Table 1) [39]. In the CA3 region, 2, 7 and 30 days following ischemia, the expression of the amyloid protein precursor gene was above the control values (Table 1) [38]. In the medial temporal cortex, gene expression of the amyloid protein precursor was below the control value within 2 days after ischemia and 7 and 30 days postischemia the expression of the amyloid protein precursor gene was above the control values (Table 1) [40].

Table 1.
Changes in the expression of the Alzheimer’s disease-associated amyloid protein precursor gene in different brain structures at different times after experimental brain ischemia.
The expression of the β-secretase gene went above the control values in the hippocampal CA1 area 2–7 days postischemia but 30 days following ischemia the β-secretase gene expression was below the control value (Table 2) [39]. The expression of the β-secretase gene was below the control values in the hippocampal CA3 area 2–7 days following ischemia. On the contrary, 30 days postischemia, β-secretase gene expression was above control (Table 2) [38]. The expression of the β-secretase gene was above the control value in the medial temporal cortex 2 days postischemia but 7–30 days postischemia gene expression was reduced (Table 2) [40].
Table 2.
Changes in the expression of the Alzheimer’s disease-associated β-secretase gene in different brain structures at different times after experimental brain ischemia.
In the CA1 region of the hippocampus, expression of the presenilin 1 gene increased 2–7 days after ischemia but 30 days postischemia the expression of this gene was below the control values (Table 3) [39]. In the CA3 area of the hippocampus, expression of the presenilin 1 gene increased 2–7 days postischemia, and was below the control values 30 days after ischemia (Table 3) [38]. In the medial temporal cortex, the expression of the presenilin 1 gene oscillated around the control values during 2, 7 and 30 days postischemia (Table 3) [41].
Table 3.
Changes in the expression of the Alzheimer’s disease-associated presenilin 1 gene in different brain structures at different times after experimental brain ischemia.
In the CA1 area of the hippocampus, expression of the presenilin 2 gene increased 2–7 days postischemia but, in contrast, 30 days following ischemia the expression of this gene was below the control values (Table 4) [39]. In the CA3 field of the hippocampus, expression of the presenilin 2 gene decreased 2–7 days after ischemia. On the contrary, 30 days postischemia, the expression of this gene increased above the control values (Table 4) [38]. In the medial temporal cortex, the expression of the presenilin 2 gene was above the control value on day 2 postischemia (Table 4) [41]. In contrast, the expression of this gene oscillated around the control values during 7–30 days postischemia (Table 4) [41].
Table 4.
Changes in the expression of the Alzheimer’s disease-associated presenilin 2 gene in different brain structures at different times after experimental brain ischemia.
In the CA3 region 2, 7 and 30 days postischemia, the expression of the α-secretase gene was below the control values (Table 5) [38].
Table 5.
Changes in the expression of the Alzheimer’s disease-associated α-secretase gene in the CA3 of the hippocampus at different times after experimental brain ischemia.
3. Amyloid Staining in Experimental Postischemic Brain
Postischemic brain injury, with a survival period up to 2 years, revealed brain parenchyma staining to amyloid. The staining was found in intra- and extracellular spaces [18,21,24,42,43,44,45,46,47,48,49,50,51,52,53,54,55]. Amyloid was noted in neurons and neuroglial cells [18,47,50,56,57,58,59,60]. Astrocytes with massive accumulation of amyloid might be involved in the development of glial scar [18,50,58,59,60]. Additionally, reactive astrocytes with huge accumulation of amyloid might be involved in repair of postischemic tissue with associated astrocyte death [18,24,50,61,62].
Staining for amyloid has been noted in subcortical white matter and the periventricular area postischemia [19,63,64]. The more intense injury of postischemic white matter is, the more widespread staining of amyloid in the brain parenchyma occurs [65]. It is assumed that the above kind of alterations are responsible for the development of leukoaraiosis after an ischemic brain episode [64]. Extracellular deposits of amyloid ranged from very small dots to typical diffuse amyloid plaques [18,19,20,24,48,50,66,67,68,69,70,71]. Multifocal diffuse amyloid plaques were observed in the ischemic hippocampus, cortex, corpus callosum and around the lateral ventricles. The accumulation of diffuse amyloid plaques in response to ischemia-reperfusion brain injury in rats was not transient, since it has been documented that these plaques transform into senile amyloid plaques during one year after an ischemic episode [72].
The accumulation of amyloid inside neuronal cells and astrocytes underscores the possible importance of amyloid in the occurrence of postischemic neurodegeneration [24,45,59,60,67,68]. In addition, these accumulations may influence synaptic disintegration and turn on further retrograde neuronal death after ischemia. These facts indicate that gradual postischemic amyloid accumulation may be responsible for additional neurodegenerative mechanisms that could worsen the outcome during recirculation by continuous neuronal death [9,19,21,47,48,54,55,73,74,75]. Postischemia amyloid is generated as a product of neuronal death [44] and as a final point shows its own neurotoxic effects. Amyloid is a neurotoxic particle and triggers intracellular mechanisms in neurons and neuroglial cells that cause additional neuronal and neuroglial cell injury or death after ischemia [55,76].
4. Dysregulation of Tau Protein Gene in Global Cerebral Ischemia due to Cardiac Arrest in Rats
In the CA1 field of the hippocampus, gene expression of the tau protein was above the control value within 2 days postischemia and 7–30 days postischemia the expression of the above gene was below the control values (Table 6) [8,37]. In the CA3 area of hippocampus, its expression was opposite (Table 6) [8,38].
Table 6.
Changes in the expression of the Alzheimer’s disease-associated tau protein gene in different brain structures at different times after experimental brain ischemia.
5. Tau Protein Staining in Experimental Postischemic Brain
Massive staining of tau protein at neurons was found in the ischemic hippocampus and brain cortex [52,77,78]. Tau protein staining was also noted in astrocytes, microglia and oligodendrocytes, postischemia [31,33,79,80,81]. The above observations indicated that neuronal and neuroglial cells display abnormalities in tau protein after an ischemic brain episode [79], which may illustrate a prime pathological stage of the ischemic processes in these cells [80]. Another study showed that tau protein itself can inhibit the transport of amyloid in the way of the neuron body at axons and dendrites, leading to amyloid accumulation in the body of neurons [82]. Available evidence shows that postischemia, hyperphosphorylated tau protein dominates in neurons and goes along with apoptosis [28,31,33,83,84]. The above-mentioned data indicate that neuronal apoptosis after ischemic brain injury is straightway connected with tau protein hyperphosphorylation. Other observations revealed that transient brain ischemia was engaged in a neurofibrillary tangle-like formation postischemia [28,83,84]. The above data provide a neuropathological basis for neurodegeneration following brain ischemia with recirculation [28].
6. Amyloid and Tau Protein in Postischemic Human Brain and Plasma
In autopsy studies of human postischemic brains, the relationship between ischemia and the accumulation of amyloid was observed [25,85,86,87]. Studies have documented diffuse and senile amyloid plaques in the hippocampus and cortex [25,85,86,87]. The neurons’ staining for amyloid depended on the brain area. Cortical and hippocampal neurons were the most intensely stained. In contrast, the staining of dentate gyrus neuronal cells appeared to be weak. Some neuronal cells were also labeled with antibodies against tau-1 [86]. Ependymal and epithelial cells were stained intensively for amyloid. Examined brain vessels of gray and white matter were surrounded by amyloid deposits. The deposits had mainly a cuffs shape. In postischemic brains, weak staining for amyloid was noted around blood–brain barrier vessels [86]. Amyloid around the blood–brain barrier microvessels indicated that it originated from serum. Data from a clinical study showing that plasma amyloid had been raised in patients following brain ischemia supported the above suggestion [26,88,89]. According to another study, β-amyloid peptide 1–40 and 1–42 were documented in the human postischemic hippocampus [25]. This strong staining of different amyloid forms may contribute to the development of postischemic neurodegeneration.
Observations of patients for a period of 4 days postischemia showed an increase in blood amyloid [26]. The increase correlated with clinical outcome after ischemic brain injury [26]. The results support the notion that ischemic brain with reperfusion may play a key role in the amyloidogenic processing of amyloid protein precursor. Tau protein was evident in plasma postischemia in humans with two peaks at 2 and 4 days, and most likely indicated the progression of neuron changes during recirculation [29]. Observed bimodal elevation of tau protein in blood is consistent with two types of neuronal death: firstly via necrosis and secondly by apoptosis [30]. It seems likely that the profiles reflect a time course of primary and secondary postischemic neuronal alterations [30]. The above observations suggest that tau protein in human blood has the potential to be used as a predictor for the neurological outcome postischemia [29,30]. Other data revealed that transient focal cerebral ischemia in humans was involved in the development of neurofibrillary tangles [27].
7. Neuropathophysiology in Postischemic Brain
After ischemia with reperfusion, a massive release of excitatory amino acids, and an intracellular overload of calcium in the hippocampus, were documented [90]. The release of glutamate from presynaptic endings, and its deficient reuptake, triggered an increase of glutamate in the extracellular space of the hippocampus [90]. As a consequence of the above-mentioned process, glutamate receptors were excessively stimulated, leading to an enormous influx of calcium to the neuronal cells by calcium channels [90]. Consequently, calcium was released from intracellular compartments into the neuronal cytoplasm. Subsequently, intracellular calcium could activate miscellaneous enzymes, necessary for survival or death of neurons. For instance phospholipases, endonucleases, nitric oxide synthase and proteases are activated via calcium and, as an ending effect of this course of action, injury to the nucleus, membranes and cytoplasm organelles, with loss of neuronal cells, was observed. The above-mentioned pathological pathways play a part in neuronal damage or death postischemia. As a rule, neuronal death after an ischemic brain episode was considered as necrotic, but eventually the neuronal cells die as a result of apoptosis. Necrosis happens due to deficit of energy and abnormal osmotic homeostasis and involves a huge number of neurons in the brain parenchyma. The postischemic neurons swell as they take up an overload of water and rupture of the cytoplasmic membrane takes place, which results in an outflow of neuronal contents into the neighboring tissue. DNA cleavage in necrotic neurons is a delayed phase occurring by mechanisms needing serine proteases. The quick drop of energy in neurons, and glucose uptake postischemia, are responsible for necrosis. Apoptosis has been noted in neurons of the CA1 region of the hippocampus 4 days after brain ischemia with reperfusion. Postischemia, caspase-3 plays a key role in the death of neurons [91,92,93,94]. The connection of autophagy and mitophagy with apoptosis should be suggested, too [91,92,93,94]. Delayed neuronal death after postischemic injury is controlled by apoptotic routes. Presently, one more neuronal cell death pathway called necroptosis was documented after brain ischemia with reperfusion. In this mechanism, postischemic neurons exhibited features of both necrotic and apoptotic processes. Another process of neuronal death is called autophagic-programmed cell death. In this event, autophagosomes and autolysosomes are found in dying neuronal cells. Recent evidence indicated that autophagy and mitophagy play a significant role in postischemic brain injury [91,92,93,94].
8. Neuropathology in Postischemic Brain
The death of neurons in the CA1 hippocampal area develops 2–7 days following ischemia and is called delayed neuronal death. Extending survival following brain ischemia injury, e.g., up to 2 years, triggers alterations in neuronal cells in the hippocampal areas with nonselective sensitivity to ischemia, e.g., in the CA3 area [19]. In contrast, changes in the striatum, mainly of medium-sized neurons, are noted primarily in the dorsolateral area. In the postischemic cortex, layers 3, 5 and 6 presented numerous neuronal changes [18,67,68]. Borderline zones of the brain cortex were also a region of intense neuronal changes after ischemia. Between 6–24 months following ischemia insult of brain, in addition to localized neuronal death various types of pathological neuronal injuries were observed. The first took the form of chronic neuronal change. Other changes were acute neuronal modifications postischemia and were present in those regions of brain parenchyma which were not involved in early changes, e.g., sectors CA2, CA3 and CA4 of the hippocampus [19]. Disappearance of neuronal cells in the CA1 hippocampal area, with a decrease in acetylcholine level, was found following focal brain ischemia. This suggests that neuronal death may also result from failure of neuronal stimulation and cholinergic transmission [90].
In the regions of massive neuronal damage, an intense response of microglia and astrocytes was observed [18,22,23,67,68,95]. Additionally, postischemic astrocytes in the hippocampal CA1 region showed an enhanced response to cytokines [95]. This evidence indicates that the rise in neuroinflammatory mediators in astrocytes is directly related to the selective sensitivity of neurons to brain injury as a result of an ischemic episode [95]. The above data imply that neuronal cells in sensitive regions of the ischemic brain are targets for interleukin-1β produced by astrocytes. This is explained by the intensified expression of the neural interleukin-1 receptor. Also, interleukin-1β has been proved to play an important role in the development of alterations in brain cells and the development of edema after brain injury due to an ischemic episode with reperfusion. Postischemic inflammatory mediators can trigger a self-sustaining cycle that leads postischemic pathology to neurodegeneration. In brain ischemia, interleukin-1 is a key player stimulating neurons to amyloidogenic processing of the amyloid protein precursor along with the release of neuroinflammatory mediators. These processes cause abnormality in the functioning of neurons and, in the end, their death with irreversible disruption of the neural network. The death of neurons results from, among others causes, neuroinflammatory mediators, which additionally cause neuronal pathology. This process activates microglia causing further strengthening, which leads to self-propagation of the inflammatory cycle. Moreover, strong evidence was provided that the amyloid which is produced following brain ischemia [19,24,50] promotes the release of inflammatory mediators by microglia. In the hippocampus, activation of neuroglial cells precedes neuronal injury and neuronal death, and lasts long after an ischemic episode [22,23].
There is plenty evidence of abnormal synaptic activity following experimental brain ischemia [96,97]. This is supported by ultrastructural observations in the hippocampal CA1 region following ischemia [97]. Other studies have revealed that brain ischemia triggers the activity of synaptic autophagy, which is associated with the death of neurons in the hippocampal CA1 area following transient ischemia [98,99]. What is more, a reduction in excitatory synaptic transmission was noted after brain ischemia in the hippocampal CA1 subfield [90]. The rise in intracellular calcium following an ischemic episode activates calpain activity in neurons, and calpain target proteins are present in glutaminergic and GABAergic synapses. In ischemia-reperfusion brain injury, calpain cleaves pre- and postsynaptic proteins. Calpain-related cleavage of proteins contributes to the loss of neuronal cells in ischemic brain parenchyma [100].
The effect of brain ischemia on the permeability of the blood-brain barrier has been widely studied for many years, and vice versa. Ischemia-reperfusion brain injury triggers a number of changes that increase the permeability of the blood-brain barrier to cellular and noncellular blood elements (e.g., platelets, amyloid) and lead to opening of tight junctions and diffuse leakage of blood elements through the necrotic wall of the blood vessel [63,70,101,102,103,104]. In postischemic damage to the blood-brain barrier, two unusual features deserve attention. One is important because of the chronic effects of extravasated amyloid [105,106] in the development of neurodegeneration, and the other relates to the leakage of platelets, which causes massive toxic, mechanical and rapid destruction of brain parenchyma [107]. It has long been known that platelets continuously produce a neurotoxic amyloid. The ability of the amyloid to cross the ischemic blood-brain barrier leads to its location of neurotoxic effects on specific neuronal populations, which may then lead to subsequent increased production of amyloid in brain parenchyma. Circulating amyloid is delivered to ischemic brain tissue and, therefore, contributes to brain amyloidosis, vasoconstriction and the development of cerebral amyloid angiopathy following an ischemic-reperfusion brain episode [25,26,28,85,86,87,104,105,108,109].
Animal and human brains respond to brain ischemia by inducing neuroinflammation [22,23,110]. Shortly following brain ischemia-reperfusion injury, neurons and neuroglial cells trigger molecular reactions that lead to the activation of astrocytes up to 28 days after ischemia. Activated astrocytes multiply quickly and change their shape and function [110]. After activation, astrocytes secrete proinflammatory cytokines, metalloproteinases and chemokines [110]. Substances released from astrocytes, e.g., interleukin-1β and matrix metalloproteinases, increase the permeability of the blood-brain barrier and enhance the transfer of leukocytes from the blood to the brain parenchyma [110]. The influx of these cells leads to further progressive ischemic tissue injury [22,23]. Microglia, like astrocytes, also belong to the first line of defense and are activated within a few minutes following brain ischemia [110]. Increased activation is noted 2–3 days after the onset of ischemia, persisting for several weeks postischemia [110]. Activated microglial cells change their shape to amoeboid and thus acquire phagocytic capacity [110]. Microglia secrete proinflammatory substances such as matrix metalloproteinase-9, tumor necrosis factor α and interleukin-1, which are involved in blood-brain barrier injury. Intensified influx of monocytes into ischemic brain parenchyma is observed during 24 h postischemia as a result of additional damage to the blood-brain barrier by astrocytic and microglial inflammatory mediators. An increased number of monocytes in brain parenchyma is observed up to 7 days after ischemia [110]. Over time, anti-inflammatory macrophages begin to dominate damaged brain parenchyma because they are necessary for parenchyma regeneration processes [110]. Other cells involved in the innate immune response triggered by brain ischemia are neutrophils which appear in injured brain tissue immediately after ischemia [110]. They concentrate close to the ischemic region and release oxygen free radicals, proinflammatory cytokines and proteolytic enzymes which cause additional destruction of brain tissue [110]. The number of neutrophils appearing in the brain after ischemia directly corresponds to the size of the ischemic injury region [110]. Furthermore, T and B lymphocytes infiltrate brain tissue postischemia. Brain ischemia also triggers penetration of brain parenchyma by natural killer cells [110]. Dendritic cells are involved in the immune response following brain ischemia as well [110]. Mast cells present in brain vessels and meninges also participate in the neuroinflammatory response following brain ischemia. Mast cells secrete cytoplasmic granules containing heparin, histamine, TNF-α and proteases, e.g., tryptase, chymase, matrix metalloproteinase-2 and matrix metalloproteinase-9, contributing to additional injury to the blood-brain barrier, brain edema and neutrophil infiltration in damaged brain parenchyma [110].
Lesions in white matter and neuroglial cell proliferation were noted in the brains of animals and humans following an episode of brain ischemia with reperfusion [18,19,22,23,63,64,67,68,111,112]. Ischemic injury of the brain in animals causes severe damage of the corpus callosum and subcortical white matter [19,63,64,113]. These observations are consistent with proliferation of neuroglial cells in both regions following ischemia-reperfusion brain injury [114]. Ischemia with reperfusion of the brain increases the permeability of the blood-brain barrier, which allows inflammatory cells with inflammatory mediators and amyloid to penetrate from the blood to the brain parenchyma, which in turn causes massive lesions in white matter [26,66,88,105,106,109,115].
Evidence suggests that transient brain ischemia with reperfusion in animals and humans causes massive loss of neuronal cells in the regions either belonging, or not, to areas of the brain selectively sensitive to an ischemic episode [14,18,19]. Progressive processes that continue during reperfusion after an ischemic episode are involved in ischemic brain neurodegeneration [14,19]. These processes develop not only in the early stages following brain ischemia, but also in the late periods after the resumption of brain circulation [14]. Over the years postischemia, developing neuropathological processes cause generalized brain atrophy [14,17,18,19,21]. A general brain examination performed following brain injury due to ischemia-reperfusion with survival up to 2 years showed hallmarks of brain hydrocephalus [17,18,19,21]. Dilatation of the subarachnoid space around the brain hemispheres has also been noted [18]. Complete hippocampal and striatal atrophy was reported [18,19]. The brain cortex after ischemia in animals and humans was narrow, showing artificially increased neuronal density [14,18,19]. In addition, features of late brain parenchyma atrophy, such as diffuse changes in white matter in the form of cavitations and rarefaction, revealing advanced spongiosis, have also been observed [18,19]. This phenomenon can be explained by a massive loss of neuronal cells, which is accompanied by an increased permeability of the blood-brain barrier occurring both in the early and late stages following ischemia-reperfusion brain injury [20,69,70,103].
9. The Development of Dementia Postischemia
After experimental brain ischemia injury, changes in behavior have been observed [9,10,11,12,73]. Locomotor hyperactivity was noted following ischemic brain damage, as in Alzheimer’s disease subjects, and is correlated with neuronal disappearance and development of neuroinflammation [20,22,23]. Postischemic brain damage causes the loss of reference and working memory with progress of a spatial memory deficit [9]. The progression of cognitive deficit develops slowly together with increase in length of recirculation time [9]. Long-lasting motor hyperactivity with cognitive deficits and decreased anxiety were also documented following repetitive transient postischemic brain injury in animals. The behavioral abnormality was associated with huge brain atrophy [17,18,19,21,67,68,74,75]. Learning and memory insufficiency following brain ischemia with recirculation move forward irreversibly and persevere forever [9].
Soon after outcome of ischemic neuropathology in humans, the slow and progressive development of dementia was found [15,116]. The occurrence of dementia following the first ischemic, and the recurrent, stroke is evaluated around 10% and 33–41%, respectively [116]. For the duration of a 25-year follow-up, the occurrence of dementia was estimated around 48% [116]. Worldwide, dementia following stroke occurs in between 5% and 50% of survivors, depending on diagnostic criteria, population demographics and geographical location [13]. In fact, it is certain that dementia following brain ischemia has many risk factors in common with the development of Alzheimer’s disease-type dementia. Among other things, reduced glucose metabolism in the brain is an early event in the pathology of Alzheimer’s disease and cerebral ischemia and may precede the neuropathological accumulation of amyloid in both disease entities [117,118]. In the progression of pathology, amyloid accumulation appears to play a key role, among other things, in the vessels of the brain, which again leads to a gradual decrease in cerebral blood flow postischemia, closing the vicious circle [118]. It is highly likely that after brain ischemia, alterations may precede Alzheimer’s disease dementia and cause all the consequences associated with the development of dementia in this disease entity.
10. Discussion
First of all, we present the reaction of the amyloid protein precursor gene and its product to transient brain ischemia with reperfusion. Evidence revealed postischemic overexpression of the amyloid protein precursor gene which correlated with the substantial rise of amyloid in the intra- and extracellular spaces of the brain [24,60] and serum [26,88,109] with generation of diffuse and senile amyloid plaques (Figure 1) [24,72].
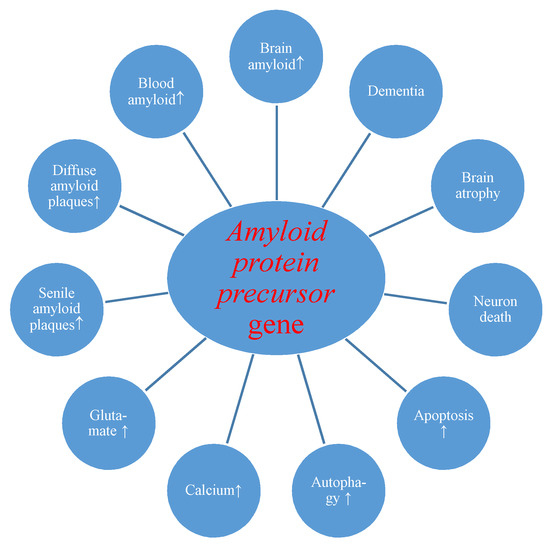
Figure 1.
Potential role of amyloid protein precursor gene changes during brain injury due to ischemia-reperfusion. ↑-increase.
The studies also made known postischemic overexpression of the tau protein gene in the brain tissue which correlated with the increase of tau protein in the intra- and extracellular spaces [36] and plasma [29,30] with development of neurofibrillary tangles (Figure 2) [27].
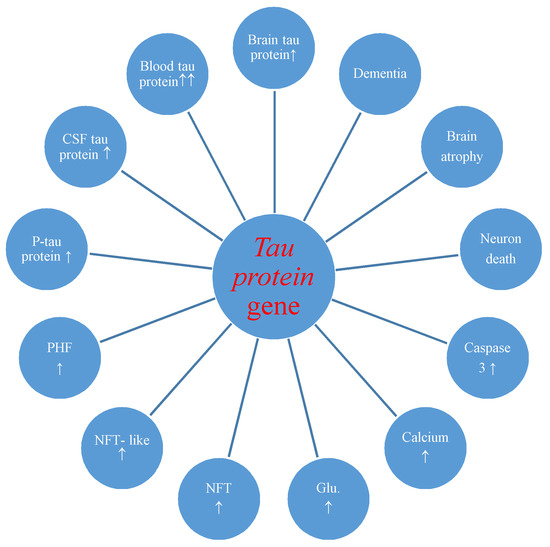
Figure 2.
Potential role of tau protein gene changes during brain injury due to ischemia-reperfusion. CSF-cerebrospinal fluid, P-tau-phosphorylated tau protein, PHF-paired helical filaments, NFT-like-neurofibrillary tangle-like, NFT-neurofibrillary tangle, Glu.-glutamate, ↑-increase.
Overexpression of the amyloid and tau protein genes begins at the same times as neuronal death and neurodegeneration postischemia (Figure 1 and Figure 2) [18,19]. Increased amyloid in brain parenchyma and blood [24,26,60,88,109] was correlated with a parallel growth of tau protein in brain tissue and plasma postischemia [29,30,36], and these alterations forecast a poorer clinical outcome. Postischemic tau protein gene overexpression also paralleled overexpression of the caspase 3 gene, which plays a significant role in apoptosis of neurons [91,93,94]. Additionally, it was noted that stimulated caspase shows a relationship with the occurrence of a neurofibrillary tangle (Figure 2) [36]. Also, postischemic neurodegeneration and dementia showed a negative relationship with the amount of amyloid and tau protein (Figure 1 and Figure 2) [19,36]. Presented facts indicate that neuronal injury and death in the postischemic brain need amyloid and tau protein. Therefore a new manner to control neuronal survival or death is presented (Figure 1 and Figure 2). Triggered neuropathological alterations, such as excitotoxicity, oxidative stress, autophagy, mitophagy, apoptosis and neuroinflammation through amyloid and tau protein clarify their probable neuropathological machinery in postischemic neurodegeneration (Figure 1 and Figure 2). Thus, it is highly likely that amyloid and tau protein, in addition, increase postischemic injury or neuronal death (Figure 1 and Figure 2).
11. Conclusions
Evidence points to proteomic and genomic alterations of amyloid and tau protein in the postischemic brain (Figure 1 and Figure 2). As a consequence, bilateral injury to the brain triggers postischemic neurodegeneration with development of dementia of an Alzheimer’s disease phenotype. Even so, a considerable move forward has, in recent times, been completed in research of the neuropathogenecity of amyloid and tau protein postischemia. However, strategic processes engaged in irreparable ischemic neurodegeneration produced through both proteins (Figure 1 and Figure 2) are, in spite of everything, unknown. In this way, animal reversible models of brain ischemia seem to be a helpful approach for clarifying the role of genes and their proteins straightforwardly connected with Alzheimer’s disease. With detailed study, the genomic and proteomic processes can speed up the existing knowledge about the neuropathogenesis of the postischemic brain, and stimulate upcoming exploration on brain ischemia with innovative trends.
Author Contributions
Conceptualization, R.P. and M.U.-K.; methodology, R.P. and M.U.-K.; software, S.J.; validation, R.P., S.J.C. and M.U.-K.; formal analysis, R.P.; investigation, M.U.-K. and S.J.; resources, M.U.-K. and S.J.; data curation, R.P.; writing—original draft preparation, R.P. and M.U.-K.; writing—review and editing, R.P. and S.J.C.; visualization, R.P.; supervision, R.P.; project administration, R.P. All authors have read and agreed to the published version of the manuscript.
Funding
The authors acknowledge the financial support from the following institutions: the Mossakowski Medical Research Centre, Polish Academy of Sciences, Warsaw, Poland (T3-RP) and the Medical University of Lublin, Lublin, Poland (DS 475/19-SJC).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pluta, R.; Furmaga-Jabłońska, W.; Maciejewski, R.; Ułamek-Kozioł, M.; Jabłoński, M. Brain ischemia activates β- and γ-secretase cleavage of amyloid precursor protein: Significance in sporadic Alzheimer’s disease. Mol. Neurobiol. 2013, 47, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Jabłoński, M.; Ułamek-Kozioł, M.; Kocki, J.; Brzozowska, J.; Januszewski, S.; Furmaga-Jabłońska, W.; Bogucka-Kocka, A.; Maciejewski, R.; Czuczwar, S.J. Sporadic Alzheimer’s disease begins as episodes of brain ischemia and ischemically dysregulated Alzheimer’s disease genes. Mol. Neurobiol. 2013, 48, 500–515. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R. Brain Ischemia: Alzheimer’s Disease Mechanisms; Nova Science Publishers: New York, NY, USA, 2019; p. 311. [Google Scholar]
- Pluta, R.; Ułamek-Kozioł, M. The role of degenerative pathways in the development of irreversible consequences after brain ischemia. Neural Regen. Res. 2019, 14, 982–983. [Google Scholar] [CrossRef]
- Pluta, R.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J. Common proteomic and genomic contribution to ischemic brain damage and Alzheimer’s disease. In Alzheimer’s Disease; Wisniewski, T., Ed.; Codon Publications: Brisbane, Australia, 2019; pp. 53–68. [Google Scholar]
- Pluta, R.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S. Amyloid pathology in the brain after ischemia. Folia Neuropathol. 2019, 57, 220–226. [Google Scholar] [CrossRef]
- Ułamek-Kozioł, M.; Czuczwar, S.J.; Januszewski, S.; Pluta, R. Substantiation for the use of curcumin during the development of neurodegeneration after brain ischemia. Int. J. Mol. Sci. 2020, 21, 517. [Google Scholar] [CrossRef]
- Ułamek-Kozioł, M.; Czuczwar, S.J.; Januszewski, S.; Pluta, R. Proteomic and genomic changes in tau protein; which are associated with Alzheimer’s disease after ischemia-reperfusion brain injury. Int. J. Mol. Sci. 2020, 21, 892. [Google Scholar] [CrossRef]
- Kiryk, A.; Pluta, R.; Figiel, I.; Mikosz, M.; Ułamek, M.; Niewiadomska, G.; Jabłoński, M.; Kaczmarek, L. Transient brain ischemia due to cardiac arrest causes irreversible long-lasting cognitive injury. Behav. Brain Res. 2011, 219, 1–7. [Google Scholar] [CrossRef]
- De la Tremblaye, P.B.; Plamondon, H. Impaired conditioned emotional response and object recognition are concomitant to neuronal damage in the amygdale and perirhinal cortex in middle-aged ischemic rats. Behav. Brain Res. 2011, 219, 227–233. [Google Scholar] [CrossRef]
- Li, J.; Wang, Y.J.; Zhang, M.; Fang, C.Q.; Zhou, H.D. Cerebral ischemia aggravates cognitive impairment in a rat model of Alzheimer’s disease. Life Sci. 2011, 89, 86–92. [Google Scholar] [CrossRef]
- Cohan, C.H.; Neumann, J.T.; Dave, K.R.; Alekseyenko, A.; Binkert, M.; Stransky, K.; Lin, H.W.; Barnes, C.A.; Wright, C.B.; Perez-Pinzon, M.A. Effect of cardiac arrest on cognitive impairment and hippocampal plasticity in middle-aged rats. PLoS ONE 2015, 10, e0124918. [Google Scholar] [CrossRef] [PubMed]
- Surawan, J.; Areemit, S.; Tiamkao, S.; Sirithanawuthichai, T.; Saensak, S. Risk factors associated with post-stroke dementia: A systematic review and meta-analysis. Neurol. Int. 2017, 9, 7216. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bivard, A.; Lillicrap, T.; Maréchal, B.; Garcia-Esperon, C.; Holliday, E.; Krishnamurthy, V.; Levi, C.R.; Parsons, M. Transient ischemic attack results in delayed brain atrophy and cognitive decline. Stroke 2018, 49, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, Y. Dementia and death after stroke in older adults during a 10-year follow-up: Results from a competing risk model. J. Nutr. Health Aging 2018, 22, 297–301. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Hypoxia/ischemia activate processing of amyloid precursor protein: Impact of vascular dysfunction in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2017, 140, 536–549. [Google Scholar] [CrossRef] [PubMed]
- Hossmann, K.A.; Schmidt-Kastner, R.; Ophoff, B.G. Recovery of integrative central nervous function after one hour global cerebro-circulatory arrest in normothermic cat. J. Neurol. Sci. 1987, 77, 305–320. [Google Scholar] [CrossRef]
- Pluta, R. The role of apolipoprotein E in the deposition of β-amyloid peptide during ischemia–reperfusion brain injury. A model of early Alzheimer’s disease. Ann. NY Acad. Sci. 2000, 903, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Ułamek, M.; Jabłoński, M. Alzheimer’s mechanisms in ischemic brain degeneration. Anat. Rec. 2009, 292, 1863–1881. [Google Scholar] [CrossRef]
- Pluta, R.; Januszewski, S.; Jabłoński, M.; Ułamek, M. Factors in creepy delayed neuronal death in hippocampus following brain ischemia-reperfusion injury with long-term survival. Acta Neurochir. 2010, 106, 37–41. [Google Scholar]
- Jabłoński, M.; Maciejewski, R.; Januszewski, S.; Ułamek, M.; Pluta, R. One year follow up in ischemic brain injury and the role of Alzheimer factors. Physiol. Res. 2011, 60 (Suppl. 1), 113–119. [Google Scholar]
- Sekeljic, V.; Bataveljic, D.; Stamenkovic, S.; Ułamek, M.; Jabłoński, M.; Radenovic, L.; Pluta, R.; Andjus, P.R. Cellular markers of neuroinflammation and neurogenesis after ischemic brain injury in the long-term survival rat model. Brain Struct. Funct. 2012, 217, 411–420. [Google Scholar] [CrossRef]
- Radenovic, L.; Nenadic, M.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J.; Andjus, P.R.; Pluta, R. Heterogeneity in brain distribution of activated microglia and astrocytes in a rat ischemic model of Alzheimer’s disease after 2 years of survival. Aging (Albany NY) 2020, 12. [Google Scholar] [CrossRef]
- Pluta, R.; Kida, E.; Lossinsky, A.S.; Golabek, A.A.; Mossakowski, M.J.; Wisniewski, H.M. Complete cerebral ischemia with short-term survival in rats induced by cardiac arrest. I. Extracellular accumulation of Alzheimer’s β-amyloid protein precursor in the brain. Brain Res. 1994, 649, 323–328. [Google Scholar] [CrossRef]
- Qi, J.; Wu, H.; Yang, Y.; Wand, D.; Chen, Y.; Gu, Y.; Liu, T. Cerebral ischemia and Alzheimer’s disease: The expression of amyloid-β and apolipoprotein E in human hippocampus. J. Alzheimers Dis. 2007, 12, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H.; Mörtberg, E.; Song, L.; Chang, L.; Provuncher, G.K.; Patel, P.P.; Ferrell, E.; Fournier, D.R.; Kan, C.W.; Campbell, T.G.; et al. Hypoxia due to cardiac arrest induces a time-dependent increase in serum amyloid β levels in humans. PLoS ONE 2011, 6, e28263. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Hirano, A.; Katagiri, T.; Sasaki, H.; Yamada, S. Neurofibrillary tangle formation in the nucleus basalis of Meynert ipsilateral to a massive cerebral infarct. Ann. Neurol. 1988, 23, 620–623. [Google Scholar] [CrossRef]
- Wen, Y.; Yang, S.H.; Liu, R.; Perez, E.J.; Brun-Ziukemagel, A.M.; Koulen, P.; Simpkins, J.W. Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats. Biochim. Biophys. Acta 2007, 1772, 473–483. [Google Scholar] [CrossRef]
- Mörtberg, E.; Zetterberg, H.; Nordmark, J.; Blennow, K.; Catry, C.; Decraemer, H.; Vanmechelen, E.; Rubertsson, S. Plasma tau protein in comatose patients after cardiac arrest treated with therapeutic hypothermia. Acta Anaesthesiol. Scand. 2011, 55, 1132–1138. [Google Scholar] [CrossRef] [PubMed]
- Randall, J.; Mörtberg, E.; Provuncher, G.K.; Fournier, D.R.; Duffy, D.C.; Rubertsson, S.; Blennow, K.; Zetterberg, H.; Wilson, D.H. Tau proteins in serum predict neurological outcome after hypoxic brain injury from cardiac arrest: Results of a pilot study. Resuscitation 2013, 84, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Majd, S.; Power, J.H.; Koblar, S.A.; Grantham, H.J. Early glycogen synthase kinase-3 and protein phosphatase 2A independent tau dephosphorylation during global brain ischaemia and reperfusion following cardiac arrest and the role of the adenosine monophosphate kinase pathway. Eur. J. Neurosci. 2016, 44, 1987–1997. [Google Scholar] [PubMed]
- Bi, M.; Gladbach, A.; van Eersel, J.; Ittner, A.; Przybyla, M.; van Hummel, A.; Chua, S.W.; van der Hoven, J.; Lee, W.S.; Müller, J.; et al. Tau exacerbates excitotoxic brain damage in an animal model of stroke. Nat. Commun. 2017, 8, 473. [Google Scholar] [CrossRef]
- Fujii, H.; Takahashi, T.; Mukai, T.; Tanaka, S.; Hosomi, N.; Maruyama, H.; Sakai, N.; Matsumoto, M. Modifications of tau protein after cerebral ischemia and reperfusion in rats are similar to those occurring in Alzheimer’s disease–Hyperphosphorylation and cleavage of 4- and 3-repeat tau. J. Cereb. Blood Flow Metab. 2017, 37, 2441–2457. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Yuldasheva, N.Y.; Batten, T.F.C.; Pickles, A.R.; Kellett, K.A.B.; Saha, S. Tau pathology and neurochemical changes associated with memory dysfunction in an optimized murine model of global cerebral ischaemia–A potential model for vascular dementia? Neurochem. Int. 2018, 118, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Kovalska, M.; Tothova, B.; Kovalska, L.; Tatarkova, Z.; Kalenska, D.; Tomascova, A.; Adamkov, M.; Lehotsky, J. Association of induced hyperhomocysteinemia with Alzheimer’s disease-like neurodegeneration in rat cortical neurons after global ischemia-reperfusion injury. Neurochem. Res. 2018, 43, 1766–1778. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J. Tau protein dysfunction after brain ischemia. J. Alzheimers Dis. 2018, 66, 429–437. [Google Scholar] [CrossRef]
- Pluta, R.; Bogucka-Kocka, A.; Ułamek-Kozioł, M.; Bogucki, J.; Czuczwar, S.J. Ischemic tau protein gene induction as an additional key factor driving development of Alzheimer’s phenotype changes in CA1 area of hippocampus in an ischemic model of Alzheimer’s disease. Pharmacol. Rep. 2018, 70, 881–884. [Google Scholar] [CrossRef]
- Pluta, R.; Ułamek-Kozioł, M.; Kocki, J.; Bogucki, J.; Januszewski, S.; Bogucka-Kocka, A.; Czuczwar, S.J. Expression of the tau protein and amyloid protein precursor processing genes in the CA3 area of the hippocampus in the ischemic model of Alzheimer’s disease in the rat. Mol. Neurobiol. 2020, 57, 1281–1290. [Google Scholar] [CrossRef]
- Kocki, J.; Ułamek-Kozioł, M.; Bogucka-Kocka, A.; Januszewski, S.; Jabłoński, M.; Gil-Kulik, P.; Brzozowska, J.; Petniak, A.; Furmaga-Jabłońska, W.; Bogucki, J.; et al. Dysregulation of amyloid precursor protein; β-secretase; presenilin 1 and 2 genes in the rat selectively vulnerable CA1 subfield of hippocampus following transient global brain ischemia. J. Alzheimers Dis. 2015, 47, 1047–1056. [Google Scholar] [CrossRef]
- Pluta, R.; Kocki, J.; Ułamek-Kozioł, M.; Petniak, A.; Gil-Kulik, P.; Januszewski, S.; Bogucki, J.; Jabłoński, M.; Brzozowska, J.; Furmaga-Jabłońska, W.; et al. Discrepancy in expression of β-secretase and amyloid-β protein precursor in Alzheimer-related genes in the rat medial temporal lobe cortex following transient global brain ischemia. J. Alzheimers Dis. 2016, 51, 1023–1031. [Google Scholar] [CrossRef]
- Pluta, R.; Kocki, J.; Ułamek-Kozioł, M.; Bogucka-Kocka, A.; Gil-Kulik, P.; Januszewski, S.; Jabłoński, M.; Petniak, A.; Brzozowska, J.; Bogucki, J.; et al. Alzheimer-associated presenilin 2 gene is dysregulated in rat medial temporal lobe cortex after complete brain ischemia due to cardiac arrest. Pharmacol. Rep. 2016, 68, 155–161. [Google Scholar] [CrossRef]
- Hall, E.D.; Oostveen, J.A.; Dunn, E.; Carter, D.B. Increased amyloid protein precursor and apolipoprotein E immunoreactivity in the selectively vulnerable hippocampus following transient forebrain ischemia in gerbils. Exp. Neurol. 1995, 135, 17–27. [Google Scholar] [CrossRef]
- Tomimoto, H.; Akiguchi, I.; Wakita, H.; Nakamura, S.; Kimura, J. Ultrastructural localization of amyloid protein precursor in the normal and postischemic gerbil brain. Brain Res. 1995, 672, 187–195. [Google Scholar] [CrossRef]
- Ishimaru, H.; Ishikawa, K.; Haga, S.; Shoji, M.; Ohe, Y.; Haga, C.; Sasaki, A.; Takashashi, A.; Maruyama, Y. Accumulation of apolipoprotein E and β-amyloid-like protein in a trace of the hippocampal CA1 pyramidal cell layer after ischaemic delayed neuronal death. Neuroreport 1996, 7, 3063–3067. [Google Scholar] [CrossRef] [PubMed]
- Yokota, M.; Saido, T.C.; Tani, E.; Yamaura, I.; Minami, N. Cytotoxic fragment of amyloid precursor protein accumulates in hippocampus after global forebrain ischemia. J. Cereb. Blood Flow Metab. 1996, 16, 1219–1223. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R. Experimental model of neuropathological changes characteristic for Alzheimer’s disease. Folia Neuropathol. 1997, 35, 94–98. [Google Scholar]
- Pluta, R.; Barcikowska, M.; Dębicki, G.; Ryba, M.; Januszewski, S. Changes in amyloid precursor protein and apolipoprotein E immunoreactivity following ischemic brain injury in rat with long-term survival: Influence of idebenone treatment. Neurosci. Lett. 1997, 232, 95–98. [Google Scholar] [CrossRef]
- Pluta, R.; Barcikowska, M.; Mossakowski, M.J.; Zelman, I. Cerebral accumulation of beta-amyloid following ischemic brain injury with long-term survival. Acta Neurochir. 1998, 71, 206–208. [Google Scholar]
- Lin, B.; Schmidt-Kastner, R.; Busto, R.; Ginsberg, M.D. Progressive parenchymal deposition of β-amyloid precursor protein in rat brain following global cerebral ischemia. Acta Neuropathol. 1999, 97, 359–368. [Google Scholar] [CrossRef]
- Pluta, R. No effect of anti-oxidative therapy on cerebral amyloidosis following ischemia–reperfusion brain injury. Folia Neuropathol. 2000, 38, 188–190. [Google Scholar]
- Lin, B.; Ginsberg, M.D.; Busto, R. Hyperglycemic but not normoglycemic global ischemia induces marked early intraneuronal expression of β-amyloid precursor protein. Brain Res. 2001, 888, 107–116. [Google Scholar] [CrossRef]
- Sinigaglia-Coimbra, R.; Cavalheiro, E.A.; Coimbra, C.G. Postischemic hypertermia induces Alzheimer-like pathology in the rat brain. Acta Neuropathol. 2002, 103, 444–452. [Google Scholar] [CrossRef]
- Fujioka, M.; Taoka, T.; Matsuo, Y.; Mishima, K.; Ogoshi, K.; Kondo, Y.; Isuda, M.; Fujiwara, M.; Asano, T.; Sakaki, T.; et al. Magnetic resonance imaging shows delayed ischemic striatal neurodegeneration. Ann. Neurol. 2003, 54, 732–747. [Google Scholar] [CrossRef]
- Pluta, R.; Jabłoński, M. Alzheimer’s factors in ischemic brain injury. In Brain Injury; Pathogenesis; Monitoring; Recovery and Management; Agrawal, A., Ed.; InTech Open Book: Rjeka, Croatia, 2012; pp. 97–138. [Google Scholar]
- Pluta, R.; Ułamek-Kozioł, M.; Januszewski, S.; Ściślewska, M.; Bogucka-Kocka, A.; Kocki, J. Alzheimer’s factors in postischemic dementia. Rom. J. Morphol. Embryol. 2012, 53, 461–466. [Google Scholar] [PubMed]
- Banati, R.B.; Gehrmann, J.; Wießner, C.; Hossmann, K.A.; Kreutzberg, G.W. Glial expression of the β-amyloid precursor protein (APP) in global ischemia. J. Cereb. Blood Flow Metab. 1995, 15, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Palacios, G.; Mengod, G.; Tortosa, A.; Ferrer, I.; Palacios, J.M. Increased β-amyloid precursor protein expression in astrocytes in the gerbil hippocampus following ischaemia: Association with proliferation of astrocytes. Eur. J. Neurosci. 1995, 7, 501–510. [Google Scholar] [CrossRef]
- Nihashi, T.; Inao, S.; Kajita, Y.; Kawai, T.; Sugimoto, T.; Niwa, M.; Kabeya, R.; Hata, N.; Hayashi, S.; Yoshida, J. Expression and distribution of beta amyloid precursor protein and beta amyloid peptide in reactive astrocytes after transient middle cerebral artery occlusion. Acta Neurochir. 2001, 143, 287–295. [Google Scholar] [CrossRef]
- Badan, I.; Platt, D.; Kessler, C.; Popa-Wagner, A. Temporal dynamics of degenerative and regenerative events associated with cerebral ischemia in aged rats. Gerontology 2003, 49, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Badan, I.; Dinca, I.; Buchhold, B.; Suofu, Y.; Walker, L.; Gratz, M.; Platt, D.; Kessler, C.H.; Popa-Wagner, A. Accelerated accumulation of N- and C-terminal beta APP fragments and delayed recovery of microtubule-associated protein 1B expression following stroke in aged rats. Eur. J. Neurosci. 2004, 19, 2270–2280. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Loike, J.D.; Brionne, T.C.; Lu, E.; Anankov, R.; Yan, F.; Silverstein, S.C.; Husemann, J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat. Med. 2003, 9, 453–457. [Google Scholar] [CrossRef]
- Takuma, K.; Baba, A.; Matsuda, T. Astrocyte apoptosis: Implications for neuroprotection. Prog. Neurobiol. 2004, 72, 111–127. [Google Scholar] [CrossRef]
- Pluta, R.; Ułamek, M.; Januszewski, S. Micro-blood–brain barrier openings and cytotoxic fragments of amyloid precursor protein accumulation in white matter after ischemic brain injury in long-lived rats. Acta Neurochir. 2006, 96, 267–271. [Google Scholar]
- Pluta, R.; Januszewski, S.; Ułamek, M. Ischemic blood–brain barrier and amyloid in white matter as etiological factors in leukoaraiosis. Acta Neurochir. 2008, 102, 353–356. [Google Scholar]
- Yam, P.S.; Takasago, T.; Dewar, D.; Graham, D.I.; McCulloch, J. Amyloid precursor protein accumulates in white matter at the margin of a focal ischaemic lesion. Brain Res. 1997, 760, 150–157. [Google Scholar] [CrossRef]
- Pluta, R.; Misicka, A.; Barcikowska, M.; Spisacka, S.; Lipkowski, A.W.; Januszewski, S. Possible reverse transport of β-amyloid peptide across the blood-brain barrier. Acta Neurochir. 2000, 76, 73–77. [Google Scholar]
- Pluta, R. Glial expression of the β-amyloid peptide in cardiac arrest. J. Neurol. Sci. 2002, 203–204, 277–280. [Google Scholar] [CrossRef]
- Pluta, R. Astroglial expression of the beta-amyloid in ischemia-reperfusion brain injury. Ann. NY Acad. Sci. 2002, 977, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R. Blood–brain barrier dysfunction and amyloid precursor protein accumulation in microvascular compartment following ischemia–reperfusion brain injury with 1-year survival. Acta Neurochir. 2003, 86, 117–122. [Google Scholar]
- Pluta, R. Pathological opening of the blood–brain barrier to horseradish peroxidase and amyloid precursor protein following ischemia–reperfusion brain injury. Chemotherapy 2005, 51, 223–226. [Google Scholar] [CrossRef]
- Pluta, R. Role of ischemic blood–brain barrier on amyloid plaques development in Alzheimer’s disease brain. Curr. Neurovasc. Res. 2007, 4, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Van Groen, T.; Puurunen, K.; Maki, H.M.; Sivenius, J.; Jolkkonen, J. Transformation of diffuse beta-amyloid precursor protein and beta-amyloid deposits to plaques in the thalamus after transient occlusion of the middle cerebral artery in rats. Stroke 2005, 36, 1551–1556. [Google Scholar] [CrossRef]
- Pluta, R.; Jolkkonen, J.; Cuzzocrea, S.; Pedata, F.; Cechetto, D.; Popa-Wagner, A. Cognitive impairment with vascular impairment and degeneration. Curr. Neurovasc. Res. 2011, 8, 342–350. [Google Scholar] [CrossRef]
- Pluta, R.; Kocki, J.; Maciejewski, R.; Ułamek-Kozioł, M.; Jabłoński, M.; Bogucka-Kocka, A.; Czuczwar, S.J. Ischemia signaling to Alzheimer-related genes. Folia Neuropathol. 2012, 50, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Jabłoński, M.; Czuczwar, S.J. Postischemic dementia with Alzheimer phenotype: Selectively vulnerable versus resistant areas of the brain and neurodegeneration versus β-amyloid peptide. Folia Neuropathol. 2012, 50, 101–109. [Google Scholar] [PubMed]
- Giulian, D.; Haverkamp, L.J.; Li, J.; Karshin, W.L.; Yu, J.; Tom, D.; Li, X.; Kirkpatrick, J.B. Senile plaques stimulate microglia to release a neurotoxin found in Alzheimer brain. Neurochem. Int. 1995, 27, 119–137. [Google Scholar] [CrossRef]
- Dewar, D.; Graham, D.I.; Teasdale, G.M.; McCulloch, J. Alz-50 and ubiquitin immunoreactivity is induced by permanent focal cerebral ischaemia in the cat. Acta Neuropathol. 1993, 86, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Dewar, D.; Graham, D.I.; Teasdale, G.M.; McCulloch, J. Cerebral ischemia induces alterations in tau and ubiquitin proteins. Dementia 1994, 5, 168–173. [Google Scholar] [CrossRef]
- Dewar, D.; Dawson, D. Tau protein is altered by focal cerebral ischaemia in the rat: An immunohistochemical and immunoblotting study. Brain Res. 1995, 684, 70–78. [Google Scholar] [CrossRef]
- Irving, E.A.; Yatsushiro, K.; McCulloch, J.; Dewar, D. Rapid alteration of tau in oligodendrocytes after focal ischemic injury in the rat: Involvement of free radicals. J. Cereb. Blood Flow Metab. 1997, 17, 612–622. [Google Scholar] [CrossRef]
- Uchihara, T.; Nakamura, A.; Arai, T.; Ikeda, K.; Tsuchiya, K. Microglial tau undergoes phosphorylation-independent modification after ischemia. Glia 2004, 45, 180–187. [Google Scholar] [CrossRef]
- Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Tau blocks traffic of organelles; neurofilaments; and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 2002, 156, 1051–1063. [Google Scholar] [CrossRef]
- Wen, Y.; Yang, S.; Liu, R.; Simpkins, J.W. Transient cerebral ischemia induces site-specific hyperphosphorylation of tau protein. Brain Res. 2004, 1022, 30–38. [Google Scholar] [CrossRef]
- Wen, Y.; Yang, S.; Liu, R.; Brun-Zinkernagel, A.M.; Koulen, P.; Simpkins, J.W. Transient cerebral ischemia induces aberrant neuronal cell cycle re-entry and Alzheimer’s disease-like tauopathy in female rats. J. Biol. Chem. 2004, 279, 22684–22692. [Google Scholar] [CrossRef] [PubMed]
- Jendroska, K.; Poewe, W.; Daniel, S.E.; Pluess, J.; Iwerssen-Schmidt, H.; Paulsen, J.; Barthel, S.; Schelosky, L.; Cervos-Navarr, J.; DeArmond, S.J. Ischemic stress induces deposition of amyloid beta immunoreactivity in human brain. Acta Neuropathol. 1995, 90, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, H.M.; Maslinska, D. Beta-protein immunoreactivity in the human brain after cardiac arrest. Folia Neuropathol. 1996, 34, 65–71. [Google Scholar] [PubMed]
- Jendroska, K.; Hoffmann, O.M.; Patt, S. Amyloid β peptide and precursor protein (APP) in mild and severe brain ischemia. Ann. NY Acad Sci. 1997, 826, 401–405. [Google Scholar] [CrossRef]
- Lee, P.H.; Bang, O.Y.; Hwang, E.M.; Lee, J.S.; Joo, U.S.; Mook-Jung, I.; Huh, K. Circulating beta amyloid protein is elevated in patients with acute ischemic stroke. J. Neural. Transm. 2005, 112, 1371–1379. [Google Scholar] [CrossRef]
- Maślińska, D.; Laure-Kamionowska, M.; Taraszewska, A.; Deręgowski, K.; Maśliński, S. Immunodistribution of amyloid beta protein (Aβ) and advanced glycation end-product receptors (RAGE) in choroid plexus and ependyma of resuscitated patients. Folia Neuropathol. 2011, 49, 295–300. [Google Scholar] [PubMed]
- Pluta, R.; Salińska, E.; Puka, M.; Stafiej, A.; Łazarewicz, J.W. Early changes in extracellular amino acids and calcium concentrations in rabbit hippocampus following complete 15-min cerebral ischemia. Resuscitation 1988, 16, 193–210. [Google Scholar] [CrossRef]
- Ułamek-Kozioł, M.; Kocki, J.; Bogucka-Kocka, A.; Petniak, A.; Gil-Kulik, P.; Januszewski, S.; Bogucki, J.; Jabłoński, M.; Furmaga-Jabłońska, W.; Brzozowska, J.; et al. Dysregulation of autophagy; mitophagy and apoptotic genes in the medial temporal lobe cortex in an ischemic model of Alzheimer’s disease. J. Alzheimers Dis. 2016, 54, 113–121. [Google Scholar] [CrossRef]
- Pluta, R.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J. Dysregulation of Alzheimer’s disease-related genes and proteins following cardiac arrest. Folia Neuropathol. 2017, 55, 283–288. [Google Scholar] [CrossRef]
- Ułamek-Kozioł, M.; Kocki, J.; Bogucka-Kocka, A.; Januszewski, S.; Bogucki, J.; Czuczwar, S.J.; Pluta, R. Autophagy; mitophagy and apoptotic gene changes in the hippocampal CA1 area in a rat ischemic model of Alzheimer’s disease. Pharmacol. Rep. 2017, 69, 1289–1294. [Google Scholar] [CrossRef]
- Ułamek-Kozioł, M.; Czuczwar, S.J.; Kocki, J.; Januszewski, S.; Bogucki, J.; Bogucka-Kocka, A.; Pluta, R. Dysregulation of autophagy; mitophagy; and apoptosis genes in the CA3 region of the hippocampus in the ischemic model of Alzheimer’s disease in the rat. J. Alzheimers Dis. 2019, 72, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Orzyłowska, O.; Oderfeld-Nowak, B.; Zaremba, M.; Januszewski, S.; Mossakowski, M.J. Prolonged and concomitant induction of astroglial immunoreactivity of interleukin-1 beta and interleukin-6 in the rat hippocampus after transient global ischemia. Neurosci. Lett. 1999, 263, 72–76. [Google Scholar] [CrossRef]
- Hofmeijer, J.; van Putten, M.J. Ischemic cerebral damage: An appraisal of synaptic failure. Stroke 2012, 43, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.T.; Cohan, C.H.; Dave, K.R.; Wright, C.B.; Perez-Pinzon, M.A. Global cerebral ischemia: Synaptic and cognitive dysfunction. Curr. Drug Targets. 2013, 14, 20–35. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.W.; Han, X.J.; Shi, Z.S.; Lei, Z.G.; Xu, Z.C. Remodeling of synapses in the CA1 area of the hippocampus after transient global ischemia. Neuroscience 2012, 218, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Ułamek-Kozioł, M.; Furmaga-Jabłońska, W.; Januszewski, S.; Brzozowska, J.; Sciślewska, M.; Jabłoński, M.; Pluta, R. Neuronal autophagy: Self-eating or self-cannibalism in Alzheimer’s disease. Neurochem. Res. 2013, 38, 1769–1773. [Google Scholar] [CrossRef] [PubMed]
- Curcio, M.; Salazar, I.L.; Mele, M.; Canzoniero, L.M.; Duarte, C.B. Calpains and neuronal damage in the ischemic brain: The swiss knife in synaptic injury. Prog. Neurobiol. 2016, 143, 1–35. [Google Scholar] [CrossRef]
- Mossakowski, M.J.; Lossinsky, A.S.; Pluta, R.; Wisniewski, H.M. Changes in cerebral microcirculation system following experimentally induced cardiac arrest: A SEM and TEM study. In Microcirculatory Stasis in the Brain; Tomita, M., Ed.; Elsevier Science Publishers, B.V.: Amsterdam, The Netherlands, 1993; pp. 99–106. [Google Scholar]
- Mossakowski, M.J.; Lossinsky, A.S.; Pluta, R.; Wisniewski, H.M. Abnormalities of the blood-brain barrier in global cerebral ischemia in rats due to experimental cardiac arrest. Acta Neurochir. 1994, 60, 274–276. [Google Scholar]
- Pluta, R.; Lossinsky, A.S.; Wiśniewski, H.M.; Mossakowski, M.J. Early blood–brain barrier changes in the rat following transient complete cerebral ischemia induced by cardiac arrest. Brain Res. 1994, 633, 41–52. [Google Scholar] [CrossRef]
- Wisniewski, H.M.; Pluta, R.; Lossinsky, A.S.; Mossakowski, M.J. Ultrastructural studies of cerebral vascular spasm after cardiac arrest-related global cerebral ischemia in rats. Acta Neuropathol. 1995, 90, 432–440. [Google Scholar] [CrossRef]
- Pluta, R.; Barcikowska, M.; Januszewski, S.; Misicka, A.; Lipkowski, A.W. Evidence of blood–brain barrier permeability/leakage for circulating human Alzheimer’s β-amyloid-(1–42)-peptide. Neuroreport 1996, 7, 1261–1265. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Misicka, A.; Januszewski, J.; Barcikowska, M.; Lipkowski, A.W. Transport of human β-amyloid peptide through the rat blood-brain barrier after global cerebral ischemia. Acta Neurochir. 1997, 70, 247–249. [Google Scholar]
- Pluta, R.; Lossinsky, A.S.; Walski, M.; Wiśniewski, H.M.; Mossakowski, M.J. Platelet occlusion phenomenon after short- and long-term survival following complete cerebral ischemia in rats produced by cardiac arrest. J. Hirnforsch. 1994, 35, 463–471. [Google Scholar] [PubMed]
- Pluta, R.; Barcikowska, M.; Misicka, A.; Lipkowski, A.W.; Spisacka, S.; Januszewski, S. Ischemic rats as a model in the study of the neurobiological role of human β-amyloid peptide. Time-dependent disappearing diffuse amyloid plaques in brain. Neuroreport 1999, 10, 3615–3619. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H.; Cao, H.Y.; Wang, Y.R.; Jiao, S.S.; Bu, X.L.; Zeng, F.; Wang, Q.H.; Li, J.; Deng, J.; Zhou, H.D.; et al. Serum Aβ is predictive for short-term neurological deficits after acute ischemic stroke. Neurotox. Res. 2015, 27, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Dabrowska, S.; Andrzejewska, A.; Lukomska, B.; Jankowski, M. Neuroinflammation as a target for treatment of stroke using mesenchymal stem cells and extracellular vesicles. J. Neuroinflamation 2019, 16, 178. [Google Scholar] [CrossRef]
- Fernando, M.S.; Simpson, J.E.; Matthews, F.; Brayne, C.; Lewis, C.E.; Barber, R.; Kalaria, R.N.; Forster, G.; Esteves, F.; Wharton, S.B.; et al. White matter lesions in an unselected cohort of the elderly: Molecular pathology suggests origin from chronic hypoperfusion injury. Stroke 2006, 37, 1391–1398. [Google Scholar] [CrossRef]
- Scherr, M.; Trinka, E.; Mc Coy, M.; Krenn, Y.; Staffen, W.; Kirschner, M.; Bergmann, H.J.; Mutzenbach, J.S. Cerebral hypoperfusion during carotid artery stenosis can lead to cognitive deficits that may be independent of white matter lesion load. Curr. Neurovasc. Res. 2012, 9, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Wakita, H.; Tomimoto, H.; Akiguchi, I.; Kimura, J. Glial activation and white matter changes in the rat brain induced by chronic cerebral hypoperfusion: An immunohistochemical study. Acta Neuropathol. 1994, 87, 484–492. [Google Scholar] [CrossRef]
- Yoshizaki, K.; Adachi, K.; Kataoka, S.; Watanabe, A.; Tabira, T.; Takahashi, K.; Wakita, H. Chronic cerebral hypoperfusion induced by right unilateral common carotid artery occlusion causes delayed white matter lesions and cognitive impairment in adult mice. Exp. Neurol. 2008, 210, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Anfuso, C.D.; Assero, G.; Lupo, G.; Nicota, A.; Cannavo, G.; Strosznajder, R.P.; Rapisarda, P.; Pluta, R.; Alberghia, M. Amyloid beta(1-42) and its beta(25-35) fragment induce activation and membrane translocation of cytosolic phospholipase A(2) in bovine retina capillary pericytes. Biochim. Biophys. Acta 2004, 1686, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R. Ischemia-reperfusion Pathways in Alzheimer’s Disease; Nova Science Publishers Inc: New York, NY, USA, 2007. [Google Scholar]
- Pluta, R. Unresolved questions concerning etiology of Alzheimer’s disease: Hypometabolism. Nutrition 2011, 27, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Popa-Wagner, A.; Buga, A.M.; Popescu, B.; Muresanu, D. Vascular cognitive impairment; dementia; aging and energy demand. A vicious cycle. J. Neural Transm. 2015, 122, 47–54. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).