Bioinformatics Analyses of the Transcriptome Reveal Ube3a-Dependent Effects on Mitochondrial-Related Pathways


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
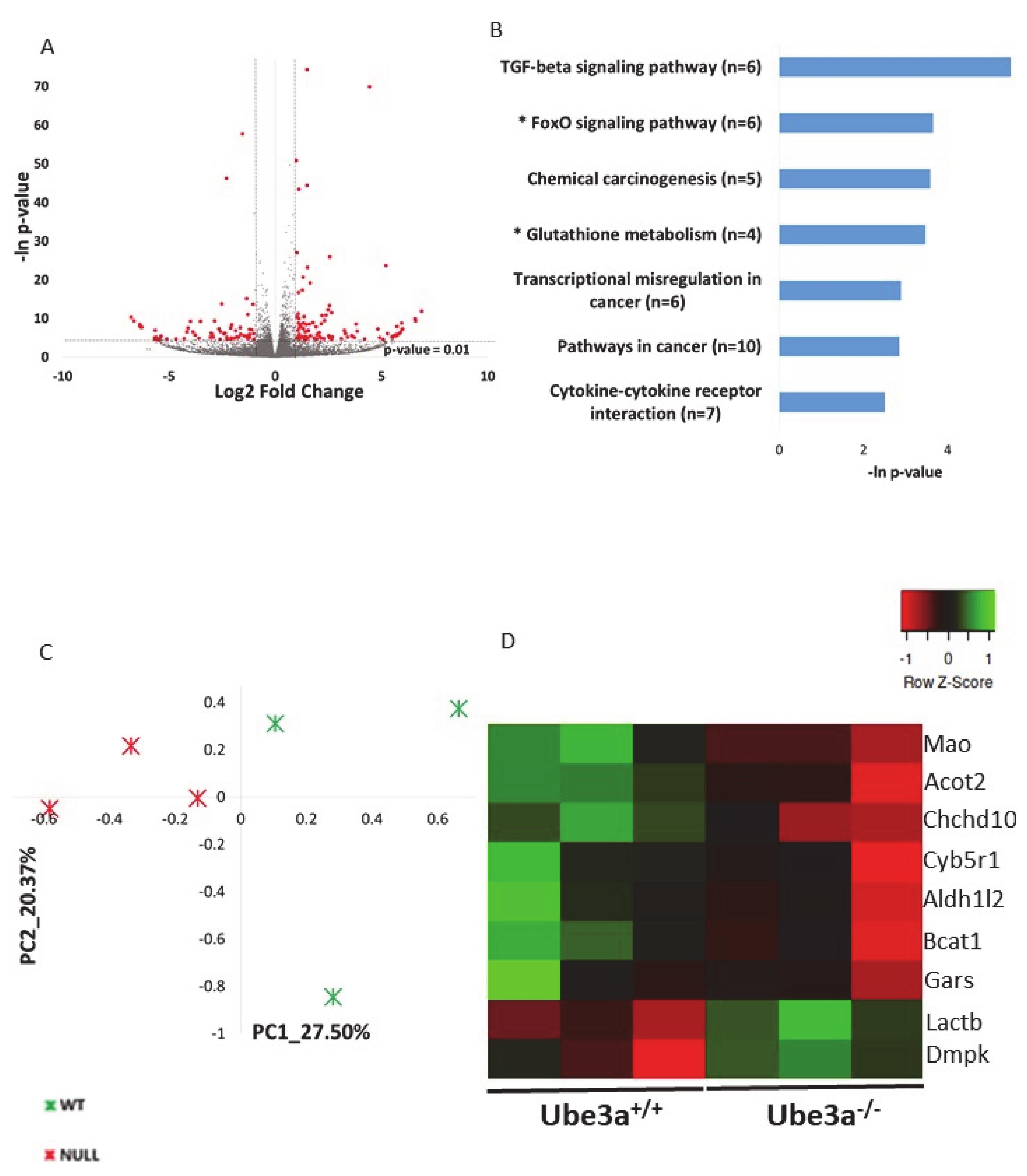
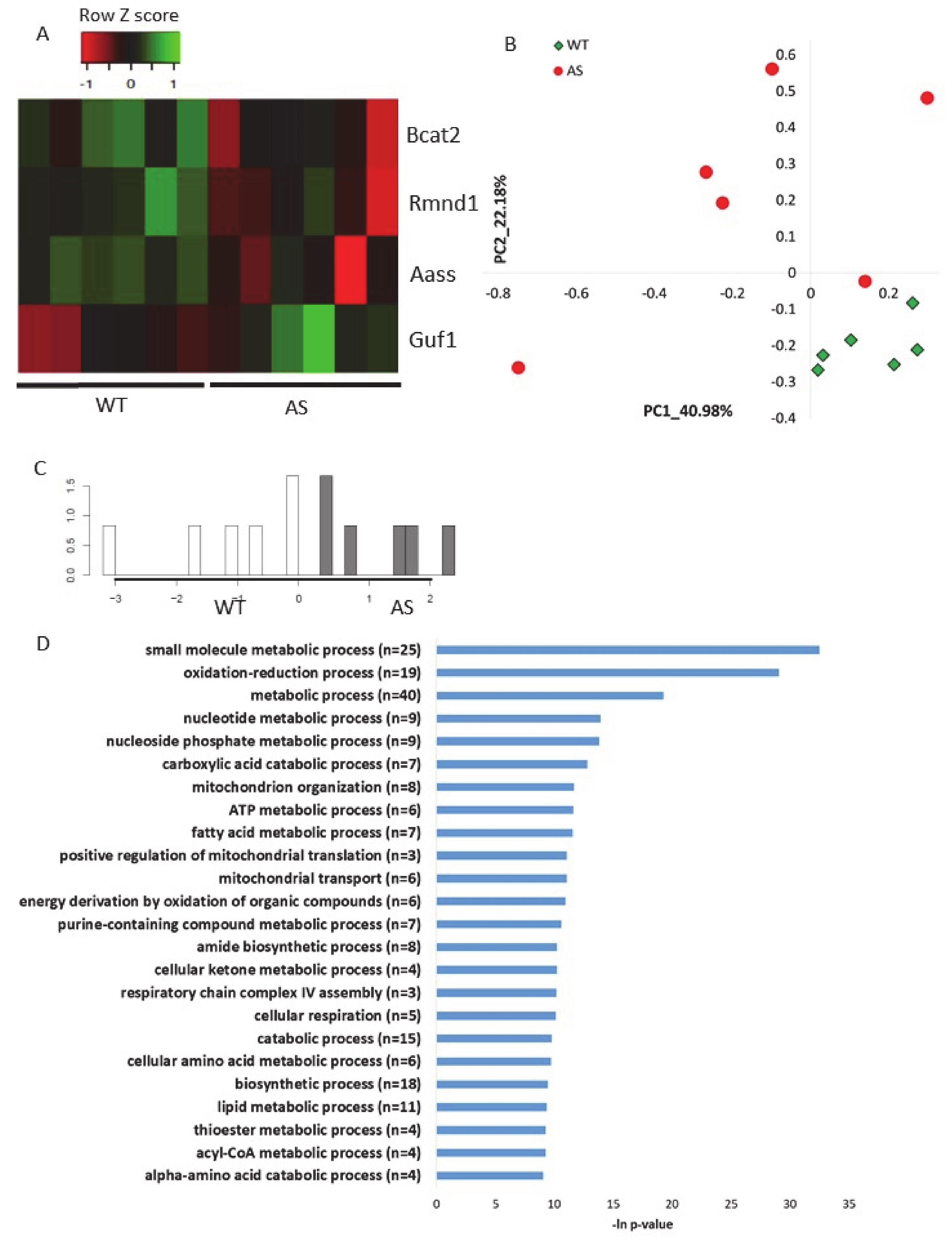
2.1. Ube3a Deletion Affects the Expression of Genes Involved in Mitochondrial Functioning
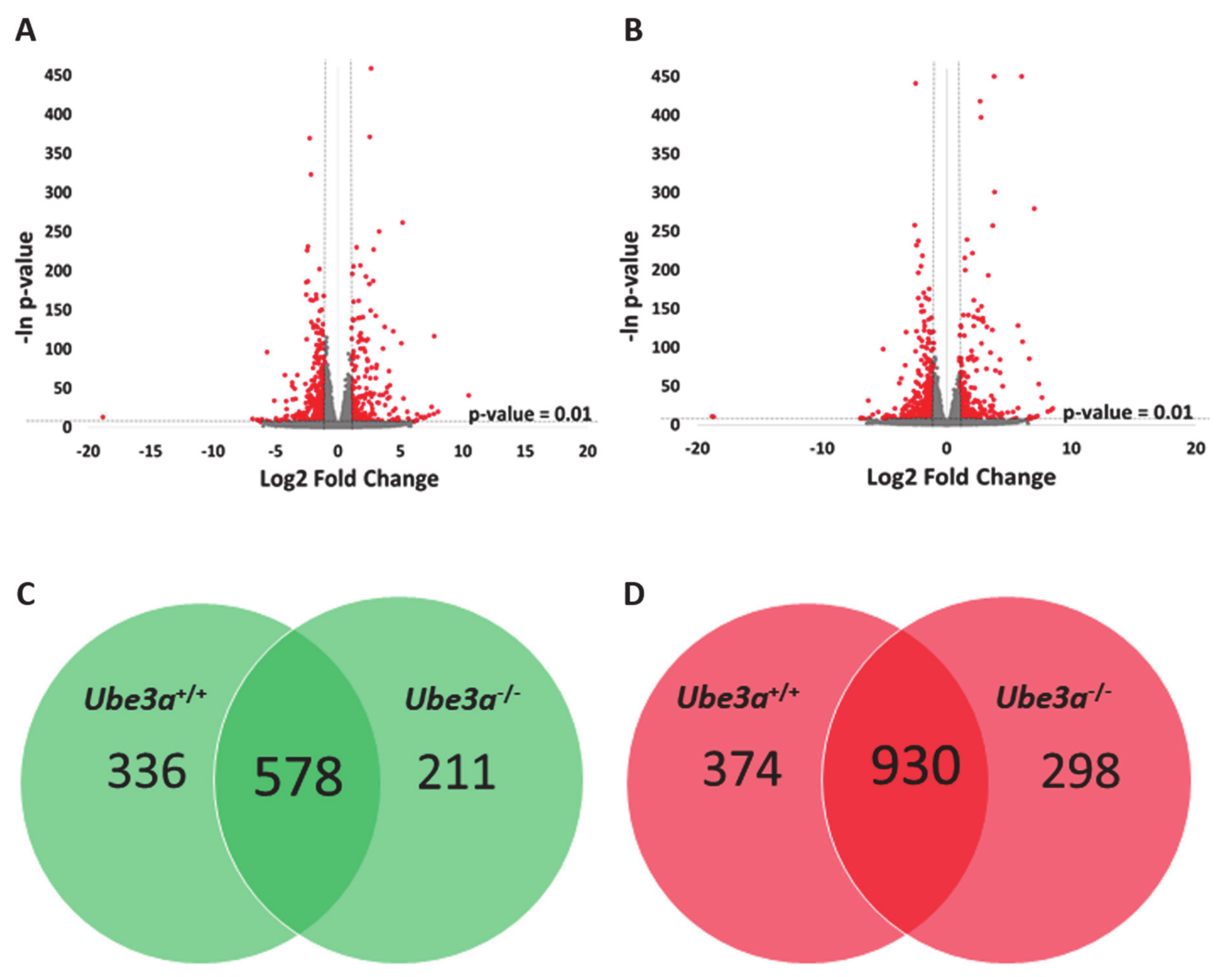
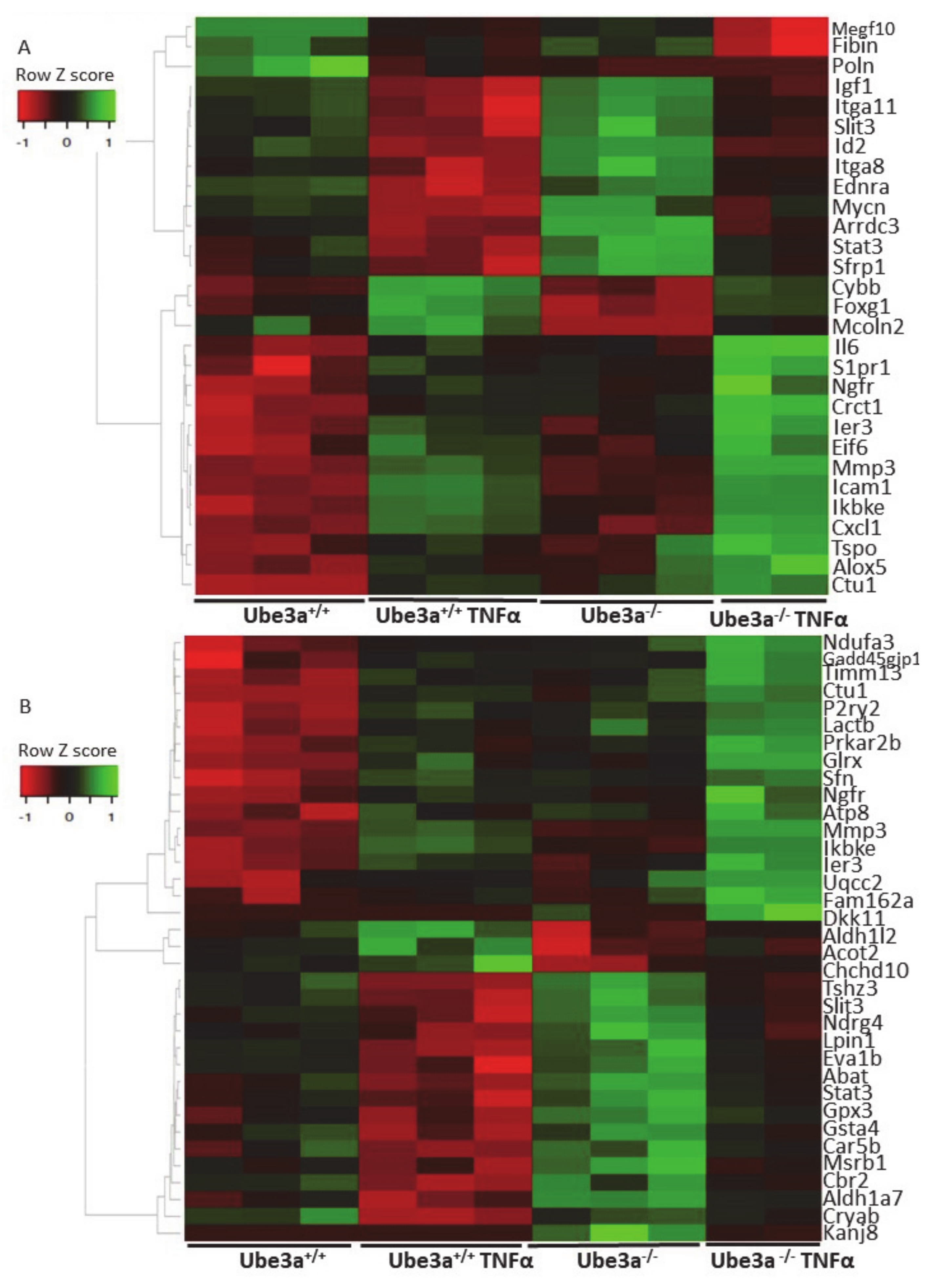
2.2. TNFα Induces Significant Changes in Ube3a+/+ and Ube3a−/− Cells
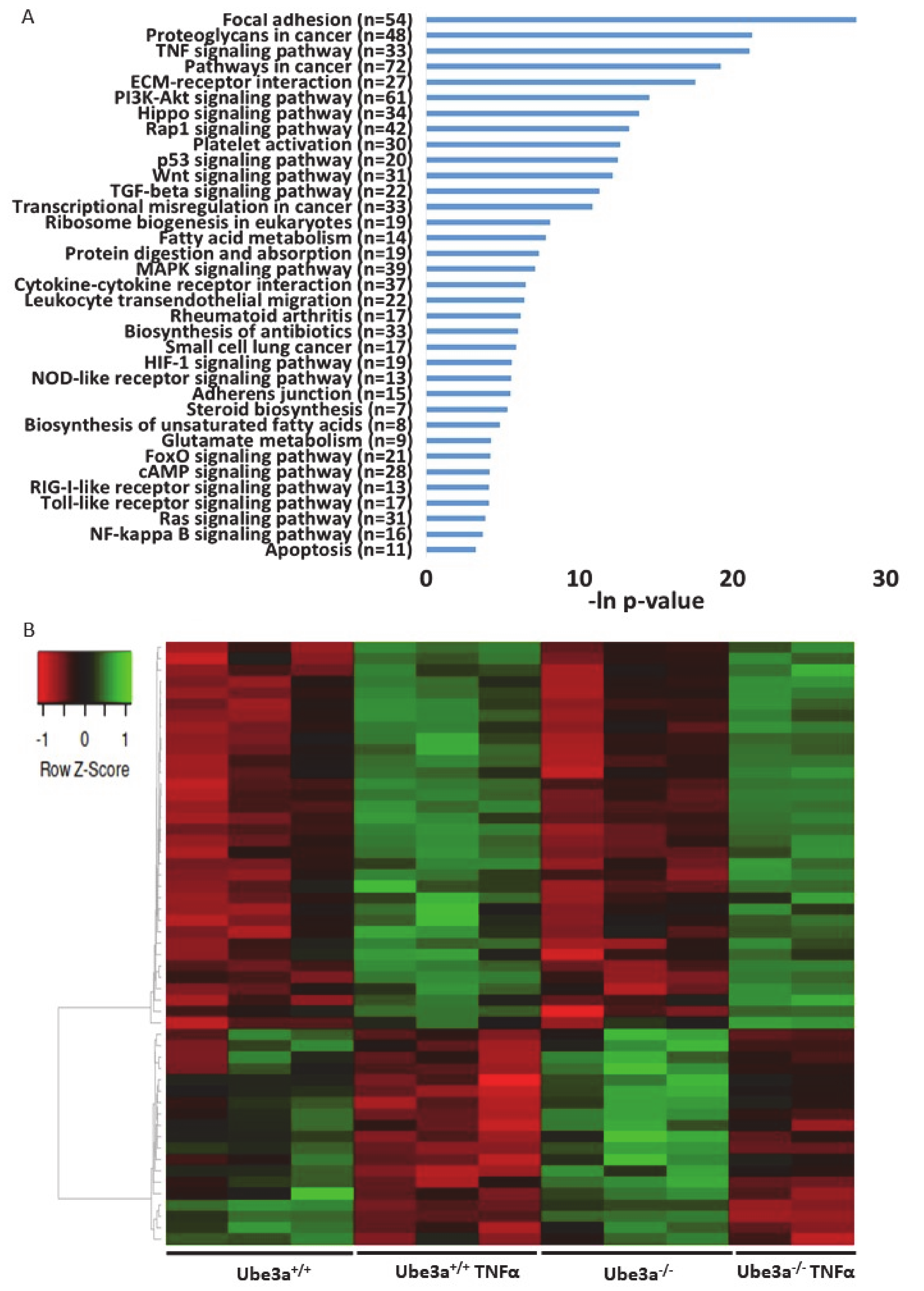
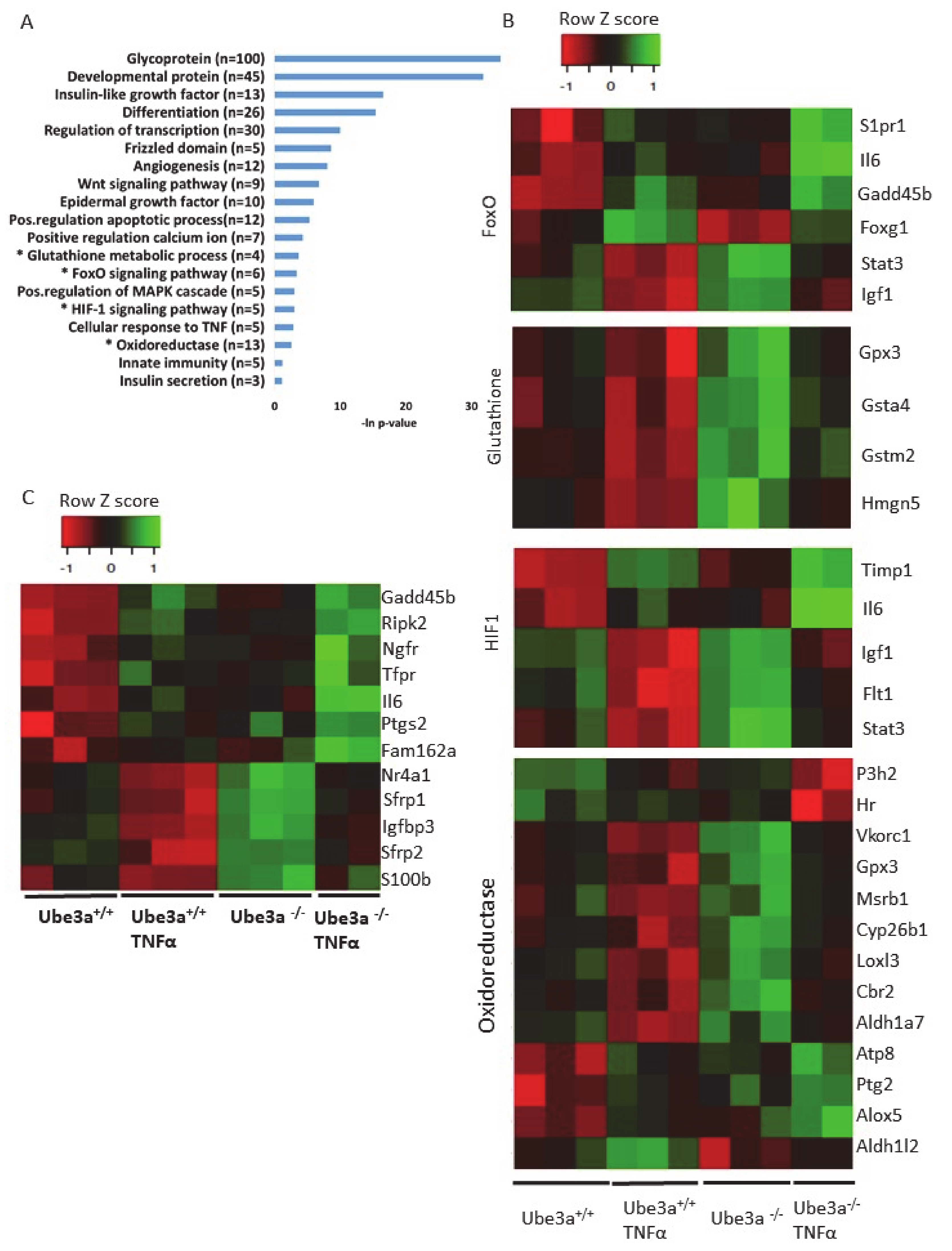
2.3. TNFα Differentially Affects Mitochondria-Associated Pathways in Ube3a Knockout MEFs
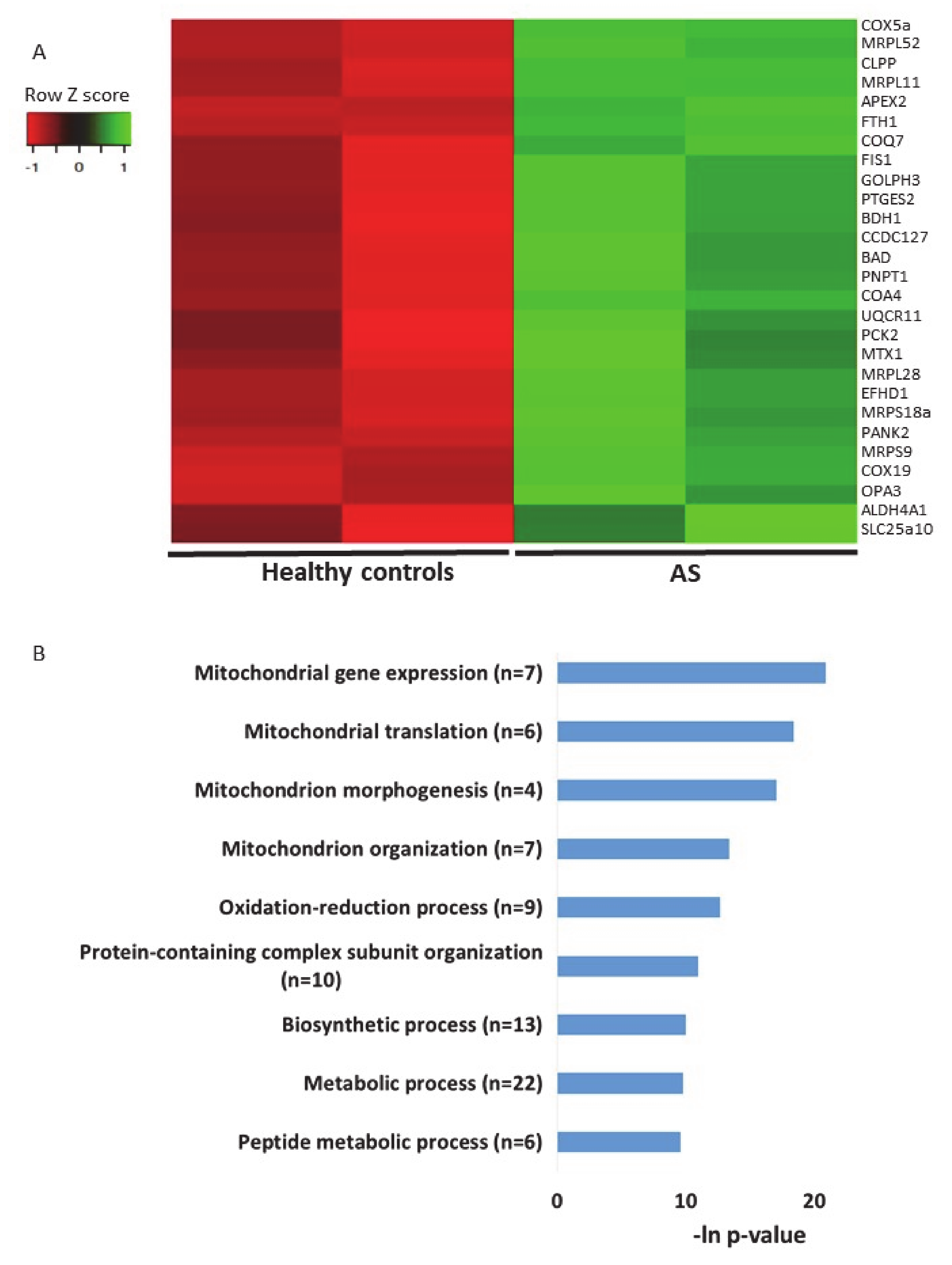
2.4. Additional Transcriptomics Datasets from other AS Models Show Differential Expression of Mitochondrial-Localized Genes
3. Discussion
4. Methods
4.1. MEFs Generation
4.2. TNFα Treatment
4.3. RNA-Seq Library Preparation
4.4. Bioinformatics Analysis
4.5. Factor Regression Analysis
4.6. Random Forest Analysis and LDA
4.7. Publicly Available RNA Sequencing Data
4.8. Data Availability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AS | Angelman Syndrome |
| ROS | Reactive Oxygen Species |
| TNFα | Tumor Necrosis Factor α |
| PCA | Principal Component Analysis |
| LDA | Linear Discriminant Analysis |
| RF | Random Forest |
References
- Ramamoorthy, S.; Nawaz, Z. E6-associated protein (E6-AP) is a dual function coactivator of steroid hormone receptors. Nucl. Recept. Signal. 2008, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sailer, C.; Offensperger, F.; Julier, A.; Kammer, K.-M.; Walker-Gray, R.; Gold, M.G.; Scheffner, M.; Stengel, F. Structural dynamics of the E6AP/UBE3A-E6-p53 enzyme-substrate complex. Nat. Commun. 2018, 9, 4441. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Li, Y.; Chang, W.-T.; Chen, Z.-C.; Cheng, J.-T.; Tsai, C.-C. Ubiquitin-protein ligase E3a (UBE3A) as a new biomarker of cardiac hypertrophy in cell models. J. Food Drug Anal. 2018, 27, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Gustin, R.M.; Bichell, T.J.V.; Bubser, M.; Daily, J.; Filonova, I.; Mrelashvili, D.; Deutch, A.Y.; Colbran, R.J.; Weeber, E.J.; Haas, K.F. Tissue-specific variation of Ube3a protein expression in rodents and in a mouse model of Angelman syndrome. Neurobiol. Dis. 2010, 39, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, Z.; Lonard, D.M.; Smith, C.L.; Lev-Lehman, E.; Tsai, S.Y.; Tsai, M.-J.; O’Malley, B.W. The Angelman Syndrome-Associated Protein, E6-AP, Is a Coactivator for the Nuclear Hormone Receptor Superfamily. Mol. Cell. Boil. 1999, 19, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Bernassola, F.; Karin, M.; Ciechanover, A.; Melino, G. The HECT Family of E3 Ubiquitin Ligases: Multiple Players in Cancer Development. Cancer Cell 2008, 14, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, Z.; Lonard, D.M.; Dennis, A.P.; Smith, C.L.; O’Malley, B.W. Proteasome-dependent degradation of the human estrogen receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 1858–1862. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, S.; Dhananjayan, S.C.; DeMayo, F.J.; Nawaz, Z. Isoform-Specific Degradation of PR-B by E6-AP Is Critical for Normal Mammary Gland Development. Mol. Endocrinol. 2010, 24, 2099–2113. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Huibregtse, J.M.; Howley, P. The HumanE6-APGene (UBE3A) Encodes Three Potential Protein Isoforms Generated by Differential Splicing. Genom. 1997, 41, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Trezza, R.A.; Sonzogni, M.; Bossuyt, S.N.V.; Zampeta, F.I.; Punt, A.M.; Berg, M.V.D.; Rotaru, D.; Koene, L.M.C.; Munshi, S.T.; Stedehouder, J.; et al. Loss of nuclear UBE3A causes electrophysiological and behavioral deficits in mice and is associated with Angelman syndrome. Nat. Neurosci. 2019, 22, 1235–1247. [Google Scholar] [CrossRef] [PubMed]
- LaSalle, J.M.; Reiter, L.T.; Chamberlain, S.J. Epigenetic regulation ofUBE3Aand roles in human neurodevelopmental disorders. Epigenomics 2015, 7, 1213–1228. [Google Scholar] [CrossRef] [PubMed]
- Miao, S.; Chen, R.; Ye, J.; Tan, G.-H.; Li, S.; Zhang, J.; Jiang, Y.-H.; Xiong, Z. The Angelman Syndrome Protein Ube3a Is Required for Polarized Dendrite Morphogenesis in Pyramidal Neurons. J. Neurosci. 2013, 33, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Arciuch, V.G.A.; Elguero, M.E.; Poderoso, J.J.; Carreras, M.C. Mitochondrial Regulation of Cell Cycle and Proliferation. Antioxidants Redox. Signal. 2012, 16, 1150–1180. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.; Green, D.R. Mitochondrial Regulation of Cell Death. Cold Spring Harb. Perspect. Boil. 2013, 5, a008706. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Levav-Cohen, Y.; Wolyniec, K.; Alsheich-Bartok, O.; Chan, A.-L.; Woods, S.J.; Jiang, Y.-H.; Haupt, S.; Haupt, Y. E6AP is required for replicative and oncogene-induced senescence in mouse embryo fibroblasts. Oncogene 2011, 31, 2199–2209. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Deng, S.; Liu, H.; Liu, Y.; Yang, Z.; Xing, T.; Jing, B.; Zhang, X. Knockdown of ubiquitin protein ligase E3A affects proliferation and invasion, and induces apoptosis of breast cancer cells through regulation of annexin A2. Mol. Med. Rep. 2012, 12, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Khan, O.Y.; Fu, G.; Ismail, A.; Srinivasan, S.; Cao, X.; Tu, Y.; Lu, S.; Nawaz, Z. Multifunction Steroid Receptor Coactivator, E6-Associated Protein, Is Involved in Development of the Prostate Gland. Mol. Endocrinol. 2006, 20, 544–559. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Simchi, L.; Panov, J.; Morsy, O.; Feuermann, Y.; Kaphzan, H. Novel Insights into the Role of UBE3A in Regulating Apoptosis and Proliferation. J. Clin. Med. 2020, 9, 1573. [Google Scholar] [CrossRef] [PubMed]
- Kishino, T.; Lalande, M.; Wagstaff, J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 1997, 15, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Urraca, N.; Cleary, J.; Brewer, V.; Pivnick, E.K.; McVicar, K.; Thibert, R.L.; Schanen, N.C.; Esmer, C.; Lamport, D.; Reiter, L.T. The Interstitial Duplication 15q11.2-q13 Syndrome Includes Autism, Mild Facial Anomalies and a Characteristic EEG Signature. Autism Res. 2013, 6, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Bolton, P.F.; Dennis, N.; Browne, C.; Thomas, N.; Veltman, M.; Thompson, R.; Jacobs, P. The phenotypic manifestations of interstitial duplications of proximal 15q with special reference to the autistic spectrum disorders. Am. J. Med Genet. 2001, 105, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Cook, E.H.; Lindgren, V.; Leventhal, B.L.; Courchesne, R.; Lincoln, A.; Shulman, C.; Lord, C.; Courchesne, E. Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am. J. Hum. Genet. 1997, 60, 928–934. [Google Scholar] [PubMed]
- Lopez, S.J.; Segal, D.J.; LaSalle, J.M. UBE3A: An E3 Ubiquitin Ligase With Genome-Wide Impact in Neurodevelopmental Disease. Front. Mol. Neurosci. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Isles, A.R.; Ingason, A.; Lowther, C.; Walters, J.T.R.; Gawlick, M.; Stöber, G.; Rees, E.; Martin, J.; Little, R.B.; Potter, H.; et al. Parental Origin of Interstitial Duplications at 15q11.2-q13.3 in Schizophrenia and Neurodevelopmental Disorders. PLoS Genet. 2016, 12, e1005993. [Google Scholar] [CrossRef] [PubMed]
- Shai, A.; Pitot, H.C.; Lambert, P.F. E6-associated protein is required for human papillomavirus type 16 E6 to cause cervical cancer in mice. Cancer Res. 2010, 70, 5064–5073. [Google Scholar] [CrossRef] [PubMed]
- Raghu, D.; Paul, P.J.; Gulati, T.; Deb, S.; Khoo, C.; Russo, A.; Gallo, E.; Blandino, G.; Chan, A.-L.; Takano, E.; et al. E6AP promotes prostate cancer by reducing p27 expression. Oncotarget 2017, 8, 42939–42948. [Google Scholar] [CrossRef] [PubMed]
- Mani, A.; Oh, A.S.; Bowden, E.T.; Lahusen, T.; Lorick, K.L.; Weissman, A.M.; Schlegel, R.; Wellstein, A.; Riegel, A.T. E6AP Mediates Regulated Proteasomal Degradation of the Nuclear Receptor Coactivator Amplified in Breast Cancer 1 in Immortalized Cells. Cancer Res. 2006, 66, 8680–8686. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Mohsin, S.K.; Gatalica, Z.; Fu, G.; Sharma, P.; Nawaz, Z. Decreased Expression of E6-Associated Protein in Breast and Prostate Carcinomas. Endocrinol. 2005, 146, 1707–1712. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Filipek, P.A.; Juranek, J.; Smith, M.; Mays, L.Z.; Ramos, E.R.; Bocian, M.; Ms, D.M.; Ma, T.M.L.; Modahl, C.; Spence, M.A.; et al. Mitochondrial dysfunction in autistic patients with 15q inverted duplication. Ann. Neurol. 2003, 53, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Fan, W.; Coskun, P.E.; Vesa, J.; Gold, J.-A.; Jiang, Y.-H.; Potluri, P.; Procaccio, V.; Acab, A.; Weiss, J.H.; et al. Mitochondrial dysfunction in CA1 hippocampal neurons of the UBE3A deficient mouse model for Angelman syndrome. Neurosci. Lett. 2009, 487, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Murphy, M.P. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann. New York Acad. Sci. 2010, 1201, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Santini, E.; Turner, K.L.; Ramaraj, A.B.; Murphy, M.P.; Klann, E.; Kaphzan, H. Mitochondrial Superoxide Contributes to Hippocampal Synaptic Dysfunction and Memory Deficits in Angelman Syndrome Model Mice. J. Neurosci. 2015, 35, 16213–16220. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn, K.J.; Nalbandian, A.; Gomez, A.; Wei, D.; Walker, N.; Kimonis, V. Administration of CoQ10 analogue ameliorates dysfunction of the mitochondrial respiratory chain in a mouse model of Angelman syndrome. Neurobiol. Dis. 2015, 76, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, B.A.; Lenning, J.; Khetarpal, N.; Tran, C.; Wu, J.Y.; Berri, A.M.; Dernay, K.; Haacke, E.M.; Shafie-Khorassani, F.; Podolsky, R.H.; et al. In vivo imaging of prodromal hippocampus CA1 subfield oxidative stress in models of Alzheimer disease and Angelman syndrome. FASEB J. 2017, 31, 4179–4186. [Google Scholar] [CrossRef] [PubMed]
- Burette, A.C.; Judson, M.C.; Li, A.N.; Chang, E.F.; Seeley, W.W.; Philpot, B.D.; Weinberg, R.J. Subcellular organization of UBE3A in human cerebral cortex. Mol. Autism 2018, 9, 54. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Zhao, G.; Li, H. Forward and Reverse Signaling Mediated by Transmembrane Tumor Necrosis Factor-Alpha and TNF Receptor 2: Potential Roles in an Immunosuppressive Tumor Microenvironment. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.R.; Wang, J.; Pacey, S.; Warren, A.Y.; Pober, J.S.; Al-Lamki, R.S. Tumor necrosis factor receptor-2 signaling pathways promote survival of cancer stem-like CD133+ cells in clear cell renal carcinoma. FASEB BioAdvances 2020, 2, 126–144. [Google Scholar] [CrossRef] [PubMed]
- De Vos, K.; Goossens, V.; Boone, E.; Vercammen, D.; Vancompernolle, K.; Vandenabeele, P.; Haegeman, G.; Fiers, W.; Grooten, J. The 55-kDa Tumor Necrosis Factor Receptor Induces Clustering of Mitochondria through Its Membrane-proximal Region. J. Boil. Chem. 1998, 273, 9673–9680. [Google Scholar] [CrossRef] [PubMed]
- Baregamian, N.; Song, J.; Bailey, C.E.; Papaconstantinou, J.; Evers, B.M.; Chung, D.H. Tumor Necrosis Factor-α and Apoptosis Signal-Regulating Kinase 1 Control Reactive Oxygen Species Release, Mitochondrial Autophagy and C-Jun N-Terminal Kinase/P38 Phosphorylation During Necrotizing Enterocolitis. Oxidative Med. Cell. Longev. 2009, 2, 297–306. [Google Scholar] [CrossRef]
- Corda, S.; LaPlace, C.; Vicaut, E.; Duranteau, J. Rapid Reactive Oxygen Species Production by Mitochondria in Endothelial Cells Exposed to Tumor Necrosis Factor- α Is Mediated by Ceramide. Am. J. Respir. Cell Mol. Boil. 2001, 24, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Doll, D.N.; Rellick, S.L.; Barr, T.L.; Ren, X.; Simpkins, J.W. Rapid mitochondrial dysfunction mediates TNF-alpha-induced neurotoxicity. J. Neurochem. 2015, 132, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Fernándezcheca, J.C.; Kaplowitz, N.; Garcia-Ruiz, C.; Colell, A.; Miranda, M.; Marí, M.; Ardite, E.; Morales, A. GSH transport in mitochondria: Defense against TNF-induced oxidative stress and alcohol-induced defect. Am. J. Physiol. Content 1997, 273, G7–G17. [Google Scholar] [CrossRef]
- Ardestani, S.; Deskins, D.L.; Young, P.P. Membrane TNF-alpha-activated programmed necrosis is mediated by Ceramide-induced reactive oxygen species. J. Mol. Signal. 2013, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Eeg-Olofsson, O.; Al-Zuhair, A.G.; Teebi, A.S.; Daoud, A.S.; Zaki, M.; Besisso, M.S.; Al-Essa, M.M. Rett Syndrome: A Mitochondrial Disease? J. Child Neurol. 1990, 5, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Kriaucionis, S.; Paterson, A.W.J.; Curtis, J.; Guy, J.; MacLeod, N.; Bird, A. Gene Expression Analysis Exposes Mitochondrial Abnormalities in a Mouse Model of Rett Syndrome. Mol. Cell. Boil. 2006, 26, 5033–5042. [Google Scholar] [CrossRef] [PubMed]
- Gold, W.; Williamson, S.; Kaur, S.; Hargreaves, I.; Land, J.; Pelka, G.; Tam, P.; Christodoulou, J. Mitochondrial dysfunction in the skeletal muscle of a mouse model of Rett syndrome (RTT): Implications for the disease phenotype. Mitochondrion 2014, 15, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Prabakaran, S.; Swatton, E.J.; Ryan, M.M.; Huffaker, S.J.; Huang, J.-J.; Griffin, J.L.; Wayland, M.; Freeman, T.; Dudbridge, F.; Lilley, K.S.; et al. Mitochondrial dysfunction in schizophrenia: Evidence for compromised brain metabolism and oxidative stress. Mol. Psychiatry 2004, 9, 684–697. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.F.; Elwell, C.; Johnson, M.H. Mitochondrial Dysfunction in Autism Spectrum Disorders. Autism-Open Access 2016, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Izzo, A.; Mollo, N.; Nitti, M.; Paladino, S.; Cali, G.; Genesio, R.; Bonfiglio, F.; Cicatiello, R.; Barbato, M.; Sarnataro, V.; et al. Mitochondrial dysfunction in down syndrome: Molecular mechanisms and therapeutic targets. Mol. Med. 2018, 24, 2. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.I.; Alleva, L.M.; Vissel, B. The roles of TNF in brain dysfunction and disease. Pharmacol. Ther. 2010, 128, 519–548. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Huang, L.; Li, X.; Li, H.; Zhou, Y.; Zhu, H.; Pan, T.; Kendrick, K.M.; Xu, W. Immunological cytokine profiling identifies TNF-α as a key molecule dysregulated in autistic children. Oncotarget 2017, 8, 82390–82398. [Google Scholar] [CrossRef] [PubMed]
- Hollis, F.; Kanellopoulos, A.K.; Bagni, C. Mitochondrial dysfunction in Autism Spectrum Disorder: Clinical features and perspectives. Curr. Opin. Neurobiol. 2017, 45, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2015, 44, D1251–D1257. [Google Scholar] [CrossRef] [PubMed]
- Koyavski, L.; Panov, J.; Simchi, L.; Rayi, P.R.; Sharvit, L.; Feuermann, Y.; Kaphzan, H. Sex-Dependent Sensory Phenotypes and Related Transcriptomic Expression Profiles Are Differentially Affected by Angelman Syndrome. Mol. Neurobiol. 2019, 56, 5998–6016. [Google Scholar] [CrossRef] [PubMed]
- Germain, N.D.; Chen, P.-F.; Plocik, A.M.; Glatt-Deeley, H.; Brown, J.D.; Fink, J.J.; Bolduc, A.K.; Robinson, T.M.; Levine, E.S.; Reiter, L.T.; et al. Gene expression analysis of human induced pluripotent stem cell-derived neurons carrying copy number variants of chromosome 15q11-q13.1. Mol. Autism 2014, 5, 44. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, J.; Wang, J.; Mao, L.; Tang, B.; Vanderklish, P.W.; Liao, X.; Xiong, Z.-Q.; Liao, L. HAP1 is an in vivo UBE3A target that augments autophagy in a mouse model of Angelman syndrome. Neurobiol. Dis. 2019, 132, 104585. [Google Scholar] [CrossRef] [PubMed]
- Parikshak, N.N.; Swarup, V.; Belgard, T.; Irimia, M.; Ramaswami, G.; Gandal, M.J.; Hartl, C.; Leppä, V.; Ubieta, L.D.L.T.; Huang, J.; et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nat. 2016, 540, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Boil. 2014, 15, 31. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Koh, H. Role of FOXO transcription factors in crosstalk between mitochondria and the nucleus. J. Bioenerg. Biomembr. 2017, 16, 18224–18341. [Google Scholar] [CrossRef] [PubMed]
- Ribas, V.; García-Ruiz, C.; Fernándezcheca, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Houten, S.M.; Wanders, R.J.A. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. [Google Scholar] [CrossRef] [PubMed]
- LaNoue, K.F.; E Tischler, M. Electrogenic characteristics of the mitochondrial glutamate-aspartate antiporter. J. Boil. Chem. 1974, 249. [Google Scholar]
- Liu, J.; Rone, M.B.; Papadopoulos, V. Protein-Protein Interactions Mediate Mitochondrial Cholesterol Transport and Steroid Biosynthesis. J. Boil. Chem. 2006, 281, 38879–38893. [Google Scholar] [CrossRef] [PubMed]
- Klimova, T.; Chandel, N.S. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008, 15, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Fernándezcheca, J.C. Mitochondrial Glutathione, a Key Survival Antioxidant. Antioxidants Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.D.; Grivennikova, V. Oxidation of NADH and ROS production by respiratory complex I. Biochim. et Biophys. Acta (BBA) - Gen. Subj. 2016, 1857, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Shulyakova, N.; Andreazza, A.C.; Mills, L.R.; Eubanks, J.H. Mitochondrial Dysfunction in the Pathogenesis of Rett Syndrome: Implications for Mitochondria-Targeted Therapies. Front. Cell. Neurosci. 2017, 11, 185. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Gavinio, R.; Asano, T.; Uemura, N.; Ito, H.; Taniguchi, Y.; Kobayashi, Y.; Maki, T.; Shen, J.; Takeda, S.; et al. PINK1 and Parkin complementarily protect dopaminergic neurons in vertebrates. Hum. Mol. Genet. 2013, 22, 2423–2434. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Jiang, S.; Zhang, L.; Yu, Z. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Colell, A.; García-Ruiz, C.; Miranda, M.; Ardite, E.; Marí, M.; Morales, A.; Corrales, F.; Kaplowitz, N.; Fernándezcheca, J.C. Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gastroenterol. 1998, 115, 1541–1551. [Google Scholar] [CrossRef]
- Qutub, A.A.; Popel, A.S. Reactive Oxygen Species Regulate Hypoxia-Inducible Factor 1α Differentially in Cancer and Ischemia. Mol. Cell. Boil. 2008, 28, 5106–5119. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Noël, G.; Galligan, J.T.; Sowa, M.E.; Arndt, V.; Overton, T.M.; Harper, J.W.; Howley, P. Identification and Proteomic Analysis of Distinct UBE3A/E6AP Protein Complexes. Mol. Cell. Boil. 2012, 32, 3095–3106. [Google Scholar] [CrossRef] [PubMed]
- Simon, H.-U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Wolyniec, K.; Levav-Cohen, Y.; Jiang, Y.-H.; Haupt, S.; Haupt, Y. The E6AP E3 ubiquitin ligase regulates the cellular response to oxidative stress. Oncogene 2012, 32, 3510–3519. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.-H.; Armstrong, D.; Albrecht, U.; Atkins, C.M.; Noebels, J.; Eichele, G.; Sweatt, J.D.; Beaudet, A.L. Mutation of the Angelman Ubiquitin Ligase in Mice Causes Increased Cytoplasmic p53 and Deficits of Contextual Learning and Long-Term Potentiation. Neuron 1998, 21, 799–811. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinform. 2014, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.M.; Chang, J.; Lucas, J.E.; Nevins, J.R.; Wang, Q.; West, M. High-Dimensional Sparse Factor Modeling: Applications in Gene Expression Genomics. J. Am. Stat. Assoc. 2008, 103, 1438–1456. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.M.; Baseler, M.; Lane, H.C.; Lempicki, R. The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Boil. 2007, 8, R183. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Bult, C.J.; Blake, A.J.; Smith, C.L.; Kadin, A.J.; Richardson, E.J.; Anagnostopoulos, A.; Asabor, R.; Baldarelli, R.M.; Beal, J.S.; Bello, S.M.; et al. Mouse Genome Database (MGD) 2019. Nucleic Acids Res. 2018, 47, D801–D806. [Google Scholar] [CrossRef] [PubMed]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef] [PubMed]
- Wenric, S.; Shemirani, R. Using Supervised Learning Methods for Gene Selection in RNA-Seq Case-Control Studies. Front. Genet. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Ram, M.; Najafi, A.; Shakeri, M. Classification and Biomarker Genes Selection for Cancer Gene Expression Data Using Random Forest. Iran. J. Pathol. 2017, 12, 339–347. [Google Scholar] [PubMed]
- Liaw, A.; Wiener, M. Classification and Regression with Random Forest. R. News. 2002, 2, 18–22. [Google Scholar]
- Venables, W.N.; Ripley, B.D. MASS package. Mod. Appl. Stat. with S; Springer: New York, NY, USA, 2002. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinform. 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panov, J.; Simchi, L.; Feuermann, Y.; Kaphzan, H. Bioinformatics Analyses of the Transcriptome Reveal Ube3a-Dependent Effects on Mitochondrial-Related Pathways. Int. J. Mol. Sci. 2020, 21, 4156. https://doi.org/10.3390/ijms21114156
Panov J, Simchi L, Feuermann Y, Kaphzan H. Bioinformatics Analyses of the Transcriptome Reveal Ube3a-Dependent Effects on Mitochondrial-Related Pathways. International Journal of Molecular Sciences. 2020; 21(11):4156. https://doi.org/10.3390/ijms21114156
Chicago/Turabian StylePanov, Julia, Lilach Simchi, Yonatan Feuermann, and Hanoch Kaphzan. 2020. "Bioinformatics Analyses of the Transcriptome Reveal Ube3a-Dependent Effects on Mitochondrial-Related Pathways" International Journal of Molecular Sciences 21, no. 11: 4156. https://doi.org/10.3390/ijms21114156
APA StylePanov, J., Simchi, L., Feuermann, Y., & Kaphzan, H. (2020). Bioinformatics Analyses of the Transcriptome Reveal Ube3a-Dependent Effects on Mitochondrial-Related Pathways. International Journal of Molecular Sciences, 21(11), 4156. https://doi.org/10.3390/ijms21114156