Iron and Sphingolipids as Common Players of (Mal)Adaptation to Hypoxia in Pulmonary Diseases


Abstract
1. Introduction
2. Maladaptation to Hypoxia: The Case of HAPE and Respiratory Pathologies
2.1. High-Altitude Pulmonary Edema
2.2. Acute Respiratory Distress Syndrome
2.3. Chronic Obstructive Pulmonary Disease
2.4. Cystic Fibrosis
3. The Variegate Pathways Involved in Hypoxia Adaptation
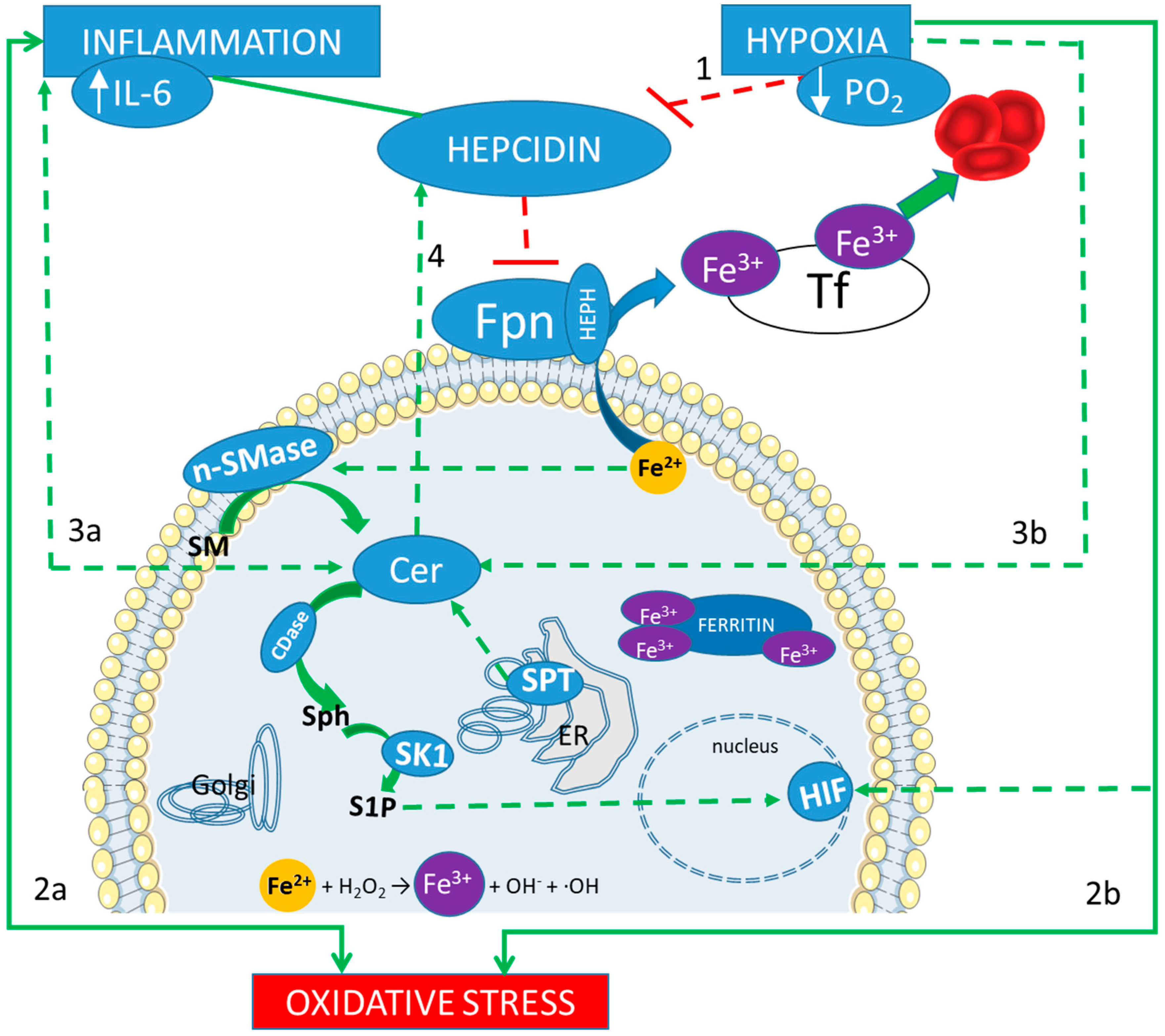
3.1. Hypoxia and Iron Metabolism: Role of Hepcidin
3.1.1. Iron Metabolism and HAPE
3.1.2. Iron Metabolism and ARDS
3.1.3. Iron Metabolism and COPD
3.1.4. Iron Metabolism and CF
3.2. Hypoxia and Sphingolipid Metabolism
3.2.1. Role of Ceramide
3.2.2. Role of Sphingosine 1 Phosphate
3.2.3. SPL Metabolism and HAPE
3.2.4. SPL Metabolism and ARDS
3.2.5. SPL Metabolism and COPD
3.2.6. SPL Metabolism and CF
4. Perspectives: Sphingolipid and Iron Metabolism in the Prognosis and Treatment of Chronic Respiratory Diseases
4.1. Sphingolipids and Iron Interplay
4.2. The Potential Prognostic Factors
4.3. Enhancing Hypoxia Adaptive Mechanism for the Treatment of Inflammatory Anemia
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CDase | ceramidase |
| Cer | ceramide |
| CF | Cystic Fibrosis |
| CFTR | Cystic Fibrosis Transmembrane conductance Regulator |
| COPD | Chronic Obstructive Pulmonary Disease |
| Fpn | ferroportin |
| HAPE | High altitude pulmonary edema |
| Hb | hemoglobin |
| HIF | Hypoxia-inducible factor |
| HPV | Hypoxic Pulmonary Vasocostriction |
| nSMase | neutral sphingomyelinase |
| O2 | oxygen |
| S1P | sphingosine 1 phosphate |
| Sph | sphingosine |
| SPL | sphingolipids |
References
- Nanduri, J.; Yuan, G.; Kumar, G.K.; Semenza, G.L.; Prabhakar, N.R. Transcriptional responses to intermittent hypoxia. Respir Physiol Neurobiol. Transcriptional responses to intermittent hypoxia. Respir. Physiol. Neurobiol. 2008, 164, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Koskenkorva-Frank, T.S.; Weiss, G.; Koppenol, W.H.; Burckhardt, S. The complex interplay of iron metabolism, reactive oxygen species, and reactive nitrogen species: Insights into the potential of various iron therapies to induce oxidative and nitrosative stress. Free Radic. Biol. Med. 2013, 65, 1174–1194. [Google Scholar] [CrossRef] [PubMed]
- Ademowo, O.S.; Dias, H.K.I.; Burton, D.G.A.; Griffiths, H.R. Lipid (per) oxidation in mitochondria: An emerging target in the ageing process? Biogerontology 2017, 18, 859–879. [Google Scholar] [CrossRef] [PubMed]
- Diab, K.J.; Adamowicz, J.J.; Kamocki, K.; Rush, N.I.; Garrison, J.; Gu, Y.; Schweitzer, K.S.; Skobeleva, A.; Rajashekhar, G.; Hubbard, W.C.; et al. Stimulation of sphingosine 1-phosphate signaling as an alveolar cell survival strategy in emphysema. Am. J. Respir. Crit. Care Med. 2010, 181, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Suresh, M.V.; Balijepalli, S.; Zhang, B.; Singh, V.V.; Swamy, S.; Panicker, S.; Dolgachev, V.A.; Subramanian, C.; Ramakrishnan, S.K.; Thomas, B.; et al. Hypoxia-Inducible Factor (HIF)-1alpha Promotes Inflammation and Injury Following Aspiration-Induced Lung Injury in Mice. Shock 2019, 52, 612–621. [Google Scholar] [CrossRef]
- Hackett, P.H.; Roach, R.C. High-altitude illness. N. Engl. J. Med. 2001, 345, 107–114. [Google Scholar] [CrossRef]
- Montgomery, S.T.; Mall, M.A.; Kicic, A.; Stick, S.M.; AREST, C.F. Hypoxia and sterile inflammation in cystic fibrosis airways: Mechanisms and potential therapies. Eur. Respir. J. 2017, 49. [Google Scholar] [CrossRef]
- Dunham-Snary, K.J.; Wu, D.; Sykes, E.A.; Thakrar, A.; Parlow, L.R.G.; Mewburn, J.D.; Parlow, J.L.; Archer, S.L. Hypoxic Pulmonary Vasoconstriction: From Molecular Mechanisms to Medicine. Chest 2017, 151, 181–192. [Google Scholar] [CrossRef]
- Force, A.D.T.; Ranieri, V.M.; Rubenfeld, G.D. Acute Respiratory Distress Syndrome. JAMA 2012, 307, 2526–2533. [Google Scholar]
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, Patterns of Care, and Mortality for Patients with Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef]
- Umbrello, M.; Formenti, P.; Bolgiaghi, L.; Chiumello, D. Current Concepts of ARDS: A Narrative Review. Int. J. Mol. Sci. 2016, 18, 64. [Google Scholar] [CrossRef] [PubMed]
- Jenq, C.C.; Tsai, F.C.; Tsai, T.Y.; Hsieh, S.Y.; Lai, Y.W.; Tian, Y.C.; Chang, M.Y.; Lin, C.Y.; Fang, J.T.; Yang, C.W.; et al. Effect of Anemia on Prognosis in Patients on Extracorporeal Membrane Oxygenation. Artif. Organs 2018, 42, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Silver, M.R. Anemia in the long-term ventilator-dependent patient with respiratory failure. Chest 2005, 128, 568S–575S. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.V.; Bota, D.P.; Mélot, C.; Vincent, J.L. Time course of hemoglobin concentrations in nonbleeding intensive care unit patients. Crit. Care Med. 2003, 31, 406–410. [Google Scholar] [PubMed]
- Barnes, J.P.; Celli, B.R. Systemic manifestations and comorbidities of COPD. Eur. Respir. J. 2009, 33, 1165–1185. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, C.; Peto, R. The natural history of chronic airflow obstruction. Br. Med. J. 1977, 1, 1645–1648. [Google Scholar] [CrossRef] [PubMed]
- Rovina, N.; Koutsoukou, A.; Koulouris, N.G. Inflammation and immune response in COPD: Where do we stand? Mediat. Inflamm. 2013, 2013, 413735. [Google Scholar] [CrossRef]
- Burgel, P.R. The role of small airways in obstructive airway diseases. Eur. Respir. Rev. 2011, 20, 23–33. [Google Scholar] [CrossRef]
- Burgel, P.R.; Bourdin, A.; Chanez, P.; Chabot, F.; Chaouat, A.; Chinet, T.; de Blic, J.; Devillier, P.; Deschildre, A.; Didier, A.; et al. Update on the roles of distal airways in COPD. Eur. Respir. Rev. 2011, 20, 7–22. [Google Scholar] [CrossRef]
- Seimetz, M.; Parajuli, N.; Pichl, A.; Veit, F.; Kwapiszewska, G.; Weisel, F.C.; Milger, K.; Egemnazarov, B.; Turowska, A.; Fuchs, B.; et al. Inducible NOS Inhibition Reverses Tobacco-Smoke-Induced Emphysema and Pulmonary Hypertension in Mice. Cell 2011, 147, 293–305. [Google Scholar] [CrossRef]
- Barr, R.G. The epidemiology of vascular dysfunction relating to chronic obstructive pulmonary disease and emphysema. Proc. Am. Thorac. Soc. 2011, 8, 522–527. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Peinado, V.I.; Barbera, J.A.; Ramirez, J.; Gomez, F.P.; Roca, J.; Jover, L.; Gimferrer, J.M.; Rodriguez-Roisin, R. Endothelial dysfunction in pulmonary arteries of patients with mild COPD. Am. J. Physiol. 1998, 274, L908–L913. [Google Scholar] [CrossRef] [PubMed]
- Peinado, V.I.; Pizarro, S.; Barbera, J.A. Pulmonary vascular involvement in COPD. Chest 2008, 134, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factor 1 and cardiovascular disease. Annu. Rev. Physiol. 2014, 76, 39–56. [Google Scholar] [CrossRef]
- Sheikh, A.Q.; Saddouk, F.Z.; Ntokou, A.; Mazurek, R.; Greif, D.M. Cell Autonomous and Non-cell Autonomous Regulation of SMC Progenitors in Pulmonary Hypertension. Cell Rep. 2018, 23, 1152–1165. [Google Scholar] [CrossRef]
- Youn, B.S.; Mantel, C.; Broxmeyer, H.E. Chemokines, chemokine receptors and hematopoiesis. Immunol. Rev. 2000, 177, 150–174. [Google Scholar] [CrossRef]
- Matsui, H.; Grubb, B.R.; Tarran, R.; Randell, S.H.; Gatzy, J.T.; Davis, C.W.; Boucher, R.C. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 1998, 95, 1005–1015. [Google Scholar] [CrossRef]
- Worlitzsch, D.; Tarran, R.; Ulrich, M.; Schwab, U.; Cekici, A.; Meyer, K.C.; Birrer, P.; Bellon, G.; Berger, J.; Weiss, T.; et al. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J. Clin. Investig. 2002, 109, 317–325. [Google Scholar] [CrossRef]
- Sly, P.D.; Gangell, C.L.; Chen, L.; Ware, R.S.; Ranganathan, S.; Mott, L.S.; Murray, C.P.; Stick, S.M.; AREST CF Investigators. Risk factors for bronchiectasis in children with cystic fibrosis. N. Engl. J. Med. 2013, 368, 1963–1970. [Google Scholar] [CrossRef]
- Fritzsching, B.; Zhou-Suckow, Z.; Trojanek, J.B.; Schubert, S.C.; Schatterny, J.; Hirtz, S.; Agrawal, R.; Muley, T.; Kahn, N.; Sticht, C.; et al. Hypoxic epithelial necrosis triggers neutrophilic inflammation via IL-1 receptor signaling in cystic fibrosis lung disease. Am. J. Respir. Crit. Care Med. 2015, 191, 902–913. [Google Scholar] [CrossRef]
- Wang, Z.; Jin, N.; Ganguli, S.; Swartz, D.R.; Li, L.; Rhoades, R.A. Rho-kinase activation is involved in hypoxia-induced pulmonary vasoconstriction. Am. J. Respir. Cell Mol. Biol. 2001, 25, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Young, J.M.; Williams, D.R.; Thompson, A.A.R. Thin Air, Thick Vessels: Historical and Current Perspectives on Hypoxic Pulmonary Hypertension. Front. Med. 2019, 6, 93. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1: Mediator of physiological and pathophysiological responses to hypoxia. J. Appl. Physiol. 2000, 88, 1474–1480. [Google Scholar] [CrossRef]
- Rankin, E.B.; Biju, M.P.; Liu, Q.; Unger, T.L.; Rha, J.; Johnson, R.S.; Simon, M.C.; Keith, B.; Haase, V.H. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J. Clin. Investig. 2007, 117, 1068–1077. [Google Scholar] [CrossRef]
- Wang, L.G.; Semenza, G.L. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc. Natl. Acad. Sci. USA 1993, 90, 4304–4308. [Google Scholar] [CrossRef]
- Moore, L.G. Measuring high-altitude adaptation. J. Appl. Physiol. 2017, 123, 1371–1385. [Google Scholar] [CrossRef] [PubMed]
- Cheong, H.I.; Janocha, A.J.; Monocello, L.T.; Garchar, A.C.; Gebremedhin, A.; Erzurum, S.C.; Beall, C.M. Alternative hematological and vascular adaptive responses to high-altitude hypoxia in East African highlanders. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L172–L177. [Google Scholar] [CrossRef] [PubMed]
- Corante, N.; Anza-Ramírez, C.; Figueroa-Mujíca, R.; Macarlupú, J.L.; Vizcardo-Galindo, G.; Bilo, G.; Parati, G.; Gamboa, J.L.; León-Velarde, F.; Villafuerte, F.C. Excessive Erythrocytosis and Cardiovascular Risk in Andean Highlanders. High Alt. Med. Biol. 2018, 19, 221–231. [Google Scholar] [CrossRef]
- Ganz, T. Anemia of Inflammation. N. Engl. J. Med. 2019, 381, 1148–1157. [Google Scholar] [CrossRef]
- Tabeling, C.; Yu, H.; Wang, L.; Ranke, H.; Goldenberg, N.M.; Zabini, D.; Noe, E.; Krauszman, A.; Gutbier, B.; Yin, J.; et al. CFTR and sphingolipids mediate hypoxic pulmonary vasoconstriction. Proc. Natl. Acad. Sci. USA 2015, 112, E1614–E1623. [Google Scholar] [CrossRef]
- Ghidoni, R.; Caretti, A.; Signorelli, P. Role of Sphingolipids in the Pathobiology of Lung Inflammation. Mediat. Inflamm. 2015, 2015, 487508. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.G.; Talbot, N.P.; Privat, C.; Rivera-Ch, M.; Nickol, A.H.; Ratcliffe, P.J.; Dorrington, K.L.; León-Velarde, F.; Robbins, P.A. Effects of iron supplementation and depletion on hypoxic pulmonary hypertension: Two randomized controlled trials. JAMA 2009, 302, 1444–1450. [Google Scholar] [CrossRef] [PubMed]
- Bart, N.K.; Curtis, M.K.; Cheng, H.Y.; Hungerford, S.L.; McLaren, R.; Petousi, N.; Dorrington, K.L.; Robbins, P.A. Elevation of iron storage in humans attenuates the pulmonary vascular response to hypoxia. J. Appl. Physiol. 2016, 121, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Gassmann, M.; Muckenthaler, M.U. Adaptation of iron requirement to hypoxic conditions at high altitude. J. Appl. Physiol. 2015, 119, 1432–1440. [Google Scholar] [CrossRef]
- Goetze, O.; Schmitt, J.; Spliethoff, K.; Theurl, I.; Weiss, G.; Swinkelsu, D.W.; Tjalsma, H.; Maggiorini, M.; Krayenbühl, P.; Rau, M.; et al. Adaptation of iron transport and metabolism to acute high-altitude hypoxia in mountaineers. Hepatology 2013, 58, 2153–2162. [Google Scholar] [CrossRef]
- Piperno, A.; Galimberti, S.; Mariani, R.; Pelucchi, S.; Ravasi, G.; Lombardi, C.; Bilo, G.; Revera, M.; Giuliano, A.; Faini, A.; et al. Modulation of hepcidin production during hypoxia-induced erythropoiesis in humans in vivo: Data from the HIGHCARE project. Blood 2011, 117, 2953–2959. [Google Scholar] [CrossRef]
- Lasocki, S.; Puy, H.; Mercier, G.; Lehmann, S.; Hepcidane study group. Impact of iron deficiency diagnosis using hepcidin mass spectrometry dosage methods on hospital stay and costs after a prolonged ICU stay: Study protocol for a multicentre, randomised, single-blinded medico-economic trial. Anaesth. Crit. Care Pain Med. 2017, 36, 391–396. [Google Scholar] [CrossRef]
- Galesloot, T.E.; Vermeulen, S.H.; Geurts-Moespot, A.J.; Klaver, S.M.; Kroot, J.J.; van Tienoven, D.; Wetzels, J.F.; Kiemeney, L.A.; Sweep, F.C.; den Heijer, M.; et al. Serum hepcidin: Reference ranges and biochemical correlates in the general population. Blood 2011, 117, e218–e225. [Google Scholar] [CrossRef]
- Lasocki, S.; Baron, G.; Driss, F.; Westerman, M.; Puy, H.; Boutron, I.; Beaumont, C.; Montravers, P. Diagnostic accuracy of serum hepcidin for iron deficiency in critically ill patients with anemia. Intensive Care Med. 2010, 36, 1044–1048. [Google Scholar] [CrossRef]
- Fein, E.; Merle, U.; Ehehalt, R.; Herrmann, T.; Kulaksiz, H. Regulation of hepcidin in HepG2 and RINm5F cells. Peptides 2007, 28, 951–957. [Google Scholar] [CrossRef]
- Vrtacnik, P.; Marc, J.; Ostanek, B. Hypoxia mimetic deferoxamine influences the expression of histone acetylation- and DNA methylation-associated genes in osteoblasts. Connect. Tissue Res. 2015, 56, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.J.; Das, N.K.; Ramakrishnan, S.K.; Jain, C.; Jurkovic, M.T.; Wu, J.; Nemeth, E.; Lakhal-Littleton, S.; Colacino, J.A.; Shah, Y.M. Hepatic hepcidin/intestinal HIF-2α axis maintains iron absorption during iron deficiency and overload. J. Clin. Investig. 2018, 129, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Renassia, C.; Peyssonnaux, C. New insights into the links between hypoxia and iron homeostasis. Curr. Opin. Hematol. 2019, 26, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Altamura, S.; Bärtsch, P.; Dehnert, C.; Maggiorini, M.; Weiss, G.; Theurl, I.; Muckenthaler, M.U.; Mairbäurl, H. Increased hepcidin levels in high-altitude pulmonary edema. J. Appl. Physiol. 2015, 118, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Connelly, K.G.; Moss, M.; Parsons, P.E.; Moore, E.E.; Moore, F.A.; Giclas, P.C.; Seligman, P.A.; Repine, J.E. Serum ferritin as a predictor of the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1997, 155, 21–25. [Google Scholar] [CrossRef]
- Nickol, A.H.; Frise, M.C.; Cheng, H.Y.; McGahey, A.; McFadyen, B.M.; Harris-Wright, T.; Bart, N.K.; Curtis, M.K.; Khandwala, S.; O’Neill, D.P.; et al. A cross-sectional study of the prevalence and associations of iron deficiency in a cohort of patients with chronic obstructive pulmonary disease. BMJ Open 2015, 5, e007911. [Google Scholar] [CrossRef]
- Cloonan, S.M.; Mumby, S.; Adcock, I.M.; Choi, A.M.K.; Chung, K.F.; Quinlan, G.J. The “Iron”-y of Iron Overload and Iron Deficiency in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2017, 196, 1103–1112. [Google Scholar] [CrossRef]
- Tassiopoulos, S.; Kontos, A.; Konstantopoulos, K.; Hadzistavrou, C.; Vaiopoulos, G.; Aessopos, A.; Tassiopoulos, T. Erythropoietic response to hypoxaemia in diffuse idiopathic pulmonary fibrosis, as opposed to chronic obstructive pulmonary disease. Respir. Med. 2001, 95, 471–475. [Google Scholar] [CrossRef][Green Version]
- Pavlisa, G.; Vrbanic, V.; Kusec, V.; Jaksic, B. Erythropoietin response after correction of severe hypoxaemia due to acute respiratory failure in chronic obstructive pulmonary disease patients. Clin. Sci. 2004, 106, 43–51. [Google Scholar] [CrossRef]
- John, M.; Hoernig, S.; Doehner, W.; Okonko, D.D.; Witt, C.; Anker, S.D. Anemia and inflammation in COPD. Chest 2005, 127, 825–829. [Google Scholar] [CrossRef]
- Fischer, R.; Simmerlein, R.; Huber, R.M.; Schiffl, H.; Lang, S.M. Lung disease severity, chronic inflammation, iron deficiency, and erythropoietin response in adults with cystic fibrosis. Pediatr. Pulmonol. 2007, 42, 1193–1197. [Google Scholar] [CrossRef] [PubMed]
- Chillappagari, S.; Venkatesan, S.; Garapati, V.; Mahavadi, P.; Munder, A.; Seubert, A.; Sarode, G.; Guenther, A.; Schmeck, B.T.; Tümmler, B.; et al. Impaired TLR4 and HIF expression in cystic fibrosis bronchial epithelial cells downregulates hemeoxygenase-1 and alters iron homeostasis in vitro. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L791–L799. [Google Scholar] [CrossRef] [PubMed]
- O’connor, T.M.; McGrath, D.S.; Short, C.; O’donnell, M.J.; Sheehy, M.; Bredin, C.P. Subclinical anaemia of chronic disease in adult patients with cystic fibrosis. J. Cyst. Fibros. 2002, 1, 31–34. [Google Scholar] [CrossRef]
- Feizi, T. Demonstration by monoclonal antibodies that carbohydrate structures of glycoproteins and glycolipids are onco-developmental antigens. Nature 1985, 314, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Hannun, A.Y.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell. Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Chiricozzi, E.; Loberto, N.; Schiumarini, D.; Samarani, M.; Mancini, G.; Tamanini, A.; Lippi, G.; Dechecchi, M.C.; Bassi, R.; Giussani, P.; et al. Sphingolipids role in the regulation of inflammatory response: From leukocyte biology to bacterial infection. J. Leukoc. Biol. 2018, 103, 445–456. [Google Scholar] [CrossRef]
- Gault, R.C.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar]
- Tettamanti, G.; Bassi, R.; Viani, P.; Riboni, L. Salvage pathways in glycosphingolipid metabolism. Biochimie 2003, 85, 423–437. [Google Scholar] [CrossRef]
- Ueda, N.; Kaushal, G.P.; Hong, X.; Shah, S.V. Role of enhanced ceramide generation in DNA damage and cell death in chemical hypoxic injury to LLC-PK1 cells. Kidney Int. 1998, 54, 399–406. [Google Scholar] [CrossRef]
- Yoshimura, S.; Banno, Y.; Nakashima, S.; Hayashi, K.; Yamakawa, H.; Sawada, M.; Sakai, N.; Nozawa, Y. Inhibition of neutral sphingomyelinase activation and ceramide formation by glutathione in hypoxic PC12 cell death. J. Neurochem. 1999, 73, 675–683. [Google Scholar] [CrossRef]
- Basnakian, A.G.; Ueda, N.; Hong, X.; Galitovsky, V.E.; Yin, X.; Shah, S.V. Ceramide synthase is essential for endonuclease-mediated death of renal tubular epithelial cells induced by hypoxia-reoxygenation. Am. J. Physiol. Ren. Physiol. 2005, 288, F308–F314. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.S.; Ahn, K.H.; Kim, S.K.; Jeon, H.J.; Ji, J.E.; Choi, J.M.; Jung, K.M.; Jung, S.Y.; Kim, D.K. Hypoxia-induced neuronal apoptosis is mediated by de novo synthesis of ceramide through activation of serine palmitoyltransferase. Cell. Signal. 2010, 22, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Noureddine, L.; Azzam, R.; Nemer, G.; Bielawski, J.; Nasser, M.; Bitar, F.; Dbaibo, G.S. Modulation of total ceramide and constituent ceramide species in the acutely and chronically hypoxic mouse heart at different ages. Prostaglandins Other Lipid Mediat. 2008, 86, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Azzam, R.; Hariri, F.; El-Hachem, N.; Kamar, A.; Dbaibo, G.; Nemer, G.; Bitar, F. Regulation of de novo ceramide synthesis: The role of dihydroceramide desaturase and transcriptional factors NFATC and Hand2 in the hypoxic mouse heart. DNA Cell Biol. 2013, 32, 310–319. [Google Scholar] [CrossRef]
- Devlin, C.M.; Lahm, T.; Hubbard, W.C.; Van Demark, M.; Wang, K.C.; Wu, X.; Bielawska, A.; Obeid, L.M.; Ivan, M.; Petrache, I. Dihydroceramide-based response to hypoxia. J. Biol. Chem. 2011, 286, 38069–38078. [Google Scholar] [CrossRef]
- Klevstig, M.; Ståhlman, M.; Lundqvist, A.; Scharin Täng, M.; Fogelstrand, P.; Adiels, M.; Andersson, L.; Kolesnick, R.; Jeppsson, A.; Borén, J.; et al. Targeting acid sphingomyelinase reduces cardiac ceramide accumulation in the post-ischemic heart. J. Mol. Cell Cardiol. 2016, 93, 69–72. [Google Scholar] [CrossRef]
- Cogolludo, A.; Villamor, E.; Perez-Vizcaino, F.; Moreno, L. Ceramide and Regulation of Vascular Tone. Int. J. Mol. Sci. 2019, 20, 411. [Google Scholar] [CrossRef]
- Ebenezer, L.D.; Fu, P.; Natarajan, V. Targeting sphingosine-1-phosphate signaling in lung diseases. Pharmacol. Ther. 2016, 168, 143–157. [Google Scholar] [CrossRef]
- Yang, Y.; Uhlig, S. The role of sphingolipids in respiratory disease. Ther. Adv. Respir. Dis. 2011, 5, 325–344. [Google Scholar] [CrossRef]
- Takuwa, Y.; Takuwa, N.; Sugimoto, N. The Edg family G protein-coupled receptors for lysophospholipids: Their signaling properties and biological activities. J. Biochem. 2002, 131, 767–771. [Google Scholar] [CrossRef]
- Yasuo, M.; Mizuno, S.; Allegood, J.; Kraskauskas, D.; Bogaard, H.J.; Spiegel, S.; Voelkel, N.F. Fenretinide causes emphysema, which is prevented by sphingosine 1-phoshate. PLoS ONE 2013, 8, e53927. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Berka, V.; Song, A.; Sun, K.; Wang, W.; Zhang, W.; Ning, C.; Li, C.; Zhang, Q.; Bogdanov, M.; et al. Elevated sphingosine-1-phosphate promotes sickling and sickle cell disease progression. J. Clin. Investig. 2014, 124, 2750–2761. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Zhang, Y.; D’Alessandro, A.; Nemkov, T.; Song, A.; Wu, H.; Liu, H.; Adebiyi, M.; Huang, A.; Wen, Y.E.; et al. Sphingosine-1-phosphate promotes erythrocyte glycolysis and oxygen release for adaptation to high-altitude hypoxia. Nat. Commun. 2016, 7, 12086. [Google Scholar] [CrossRef] [PubMed]
- Takuwa, Y.; Okamoto, Y.; Yoshioka, K.; Takuwa, N. Sphingosine-1-phosphate signaling in physiology and diseases. Biofactors 2012, 38, 329–337. [Google Scholar] [CrossRef]
- Chawla, S.; Rahar, B.; Singh, M.; Bansal, A.; Saraswat, D.; Saxena, S. Exogenous sphingosine-1-phosphate boosts acclimatization in rats exposed to acute hypobaric hypoxia: Assessment of haematological and metabolic effects. PLoS ONE 2014, 9, e98025. [Google Scholar] [CrossRef]
- Barbacini, P.; Casas, J.; Torretta, E.; Capitanio, D.; Maccallini, G.; Hirschler, V.; Gelfi, C. Regulation of Serum Sphingolipids in Andean Children Born and Living at High Altitude (3775 m). Int. J. Mol. Sci. 2019, 20, 2835. [Google Scholar] [CrossRef]
- Guo, L.; Tan, G.; Liu, P.; Li, H.; Tang, L.; Huang, L.; Ren, Q. Three plasma metabolite signatures for diagnosing high altitude pulmonary edema. Sci. Rep. 2015, 5, 15126. [Google Scholar] [CrossRef] [PubMed]
- Petrache, I.; Natarajan, V.; Zhen, L.; Medler, T.R.; Richter, A.T.; Cho, C.; Hubbard, W.C.; Berdyshev, E.V.; Tuder, R.M. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat. Med. 2005, 11, 491–498. [Google Scholar] [CrossRef]
- Singleton, P.A.; Dudek, S.M.; Chiang, E.T.; Garcia, J.G. Regulation of sphingosine 1-phosphate-induced endothelial cytoskeletal rearrangement and barrier enhancement by S1P1 receptor, PI3 kinase, Tiam1/Rac1, and alpha-actinin. FASEB J. 2005, 19, 1646–1656. [Google Scholar] [CrossRef]
- Kolliputi, N.; Galam, L.; Parthasarathy, P.T.; Tipparaju, S.M.; Lockey, R.F. NALP-3 inflammasome silencing attenuates ceramide-induced transepithelial permeability. J. Cell. Physiol. 2012, 227, 3310–3316. [Google Scholar] [CrossRef]
- Spengler, D.; Winoto-Morbach, S.; Kupsch, S.; Vock, C.; Blöchle, K.; Frank, S.; Rintz, N.; Diekötter, M.; Janga, H.; Weckmann, M.; et al. Novel therapeutic roles for surfactant-inositols and -phosphatidylglycerols in a neonatal piglet ARDS model: A translational study. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L32–L53. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, R.; Barreira, B.; Moreno, E.; Lara-Acedo, V.; Morales-Cano, D.; Martínez-Ramas, A.; de Olaiz Navarro, B.; Herrero, R.; Lorente, J.Á.; Cogolludo, Á.; et al. Role of acid sphingomyelinase and IL-6 as mediators of endotoxin-induced pulmonary vascular dysfunction. Thorax 2017, 72, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Liu, H.; Zhang, X.; Wen, G.; Zhu, C.; Zhao, Y.; Niu, W.; Qin, Y.; Chen, H.; Bai, C.; et al. Transcriptomic analysis of lung tissues after hUC-MSCs and FTY720 treatment of lipopolysaccharide-induced acute lung injury in mouse models. Int. Immunopharmacol. 2018, 63, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Camp, S.M.; Chiang, E.T.; Sun, C.; Usatyuk, P.V.; Bittman, R.; Natarajan, V.; Garcia, J.G.; Dudek, S.M. Pulmonary Endothelial Cell Barrier Enhancement by Novel FTY720 Analogs: Methoxy-FTY720, Fluoro-FTY720, and beta-Glucuronide-FTY720. Chem. Phys. Lipids 2016, 194, 85–93. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, W.; Heng, Z.; Zheng, J.; Li, P.; Yuan, X.; Niu, W.; Bai, C.; Liu, H. Combination therapy of human umbilical cord mesenchymal stem cells and FTY720 attenuates acute lung injury induced by lipopolysaccharide in a murine model. Oncotarget 2017, 8, 77407–77414. [Google Scholar] [CrossRef][Green Version]
- Jamalkandi, S.A.; Mirzaie, M.; Jafari, M.; Mehrani, H.; Shariati, P.; Khodabandeh, M. Signaling network of lipids as a comprehensive scaffold for omics data integration in sputum of COPD patients. Biochim. Biophys. Acta 2015, 1851, 1383–1393. [Google Scholar]
- Scarpa, M.C.; Baraldo, S.; Marian, E.; Turato, G.; Calabrese, F.; Saetta, M.; Maestrelli, P. Ceramide expression and cell homeostasis in chronic obstructive pulmonary disease. Respiration 2013, 85, 342–349. [Google Scholar] [CrossRef]
- Bodas, M.; Pehote, G.; Silverberg, D.; Gulbins, E.; Vij, N. Autophagy augmentation alleviates cigarette smoke-induced CFTR-dysfunction, ceramide-accumulation and COPD-emphysema pathogenesis. Free Radic. Biol. Med. 2019, 131, 81–97. [Google Scholar] [CrossRef]
- Arana, L.; Gangoiti, P.; Ouro, A.; Trueba, M.; Gómez-Muñoz, A. Ceramide and ceramide 1-phosphate in health and disease. Lipids Health Dis. 2010, 9, 15. [Google Scholar] [CrossRef]
- Filosto, S.; Castillo, S.; Danielson, A.; Franzi, L.; Khan, E.; Kenyon, N.; Last, J.; Pinkerton, K.; Tuder, R.; Goldkorn, T. Neutral sphingomyelinase 2: A novel target in cigarette smoke-induced apoptosis and lung injury. Am. J. Respir. Cell. Mol. Biol. 2011, 44, 350–360. [Google Scholar] [CrossRef]
- Zulueta, A.; Caretti, A.; Campisi, G.M.; Brizzolari, A.; Abad, J.L.; Paroni, R.; Signorelli, P.; Ghidoni, R. Inhibitors of ceramide de novo biosynthesis rescue damages induced by cigarette smoke in airways epithelia. Naunyn Schmiedebergs Arch. Pharmacol. 2017, 390, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Demedts, I.K.; Demoor, T.; Bracke, K.R.; Joos, G.F.; Brusselle, G.G. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir. Res. 2006, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Karandashova, S.; Kummarapurugu, A.B.; Zheng, S.; Chalfant, C.E.; Voynow, J.A. Neutrophil elastase increases airway ceramide levels via upregulation of serine palmitoyltransferase. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L206–L214. [Google Scholar] [CrossRef] [PubMed]
- De Cunto, G.; Brancaleone, V.; Riemma, M.A.; Cerqua, I.; Vellecco, V.; Spaziano, G.; Cavarra, E.; Bartalesi, B.; D’Agostino, B.; Lungarella, G.; et al. Functional contribution of sphingosine-1-phosphate to airway pathology in cigarette smoke-exposed mice. Br. J. Pharmacol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Barnawi, J.; Jersmann, H.; Haberberger, R.; Hodge, S.; Meech, R. Reduced DNA methylation of sphingosine-1 phosphate receptor 5 in alveolar macrophages in COPD: A potential link to failed efferocytosis. Respirology 2017, 22, 315–321. [Google Scholar] [CrossRef]
- Cordts, F.; Pitson, S.; Tabeling, C.; Gibbins, I.; Moffat, D.F.; Jersmann, H.; Hodge, S.; Haberberger, R.V. Expression profile of the sphingosine kinase signalling system in the lung of patients with chronic obstructive pulmonary disease. Life Sci. 2011, 89, 806–811. [Google Scholar] [CrossRef]
- Josipovic, I.; Pflüger, B.; Fork, C.; Vasconez, A.E.; Oo, J.A.; Hitzel, J.; Seredinski, S.; Gamen, E.; Heringdorf, D.M.Z.; Chen, W.; et al. Long noncoding RNA LISPR1 is required for S1P signaling and endothelial cell function. J. Mol. Cell. Cardiol. 2018, 116, 57–68. [Google Scholar] [CrossRef]
- Aureli, M.; Schiumarini, D.; Loberto, N.; Bassi, R.; Tamanini, A.; Mancini, G.; Tironi, M.; Munari, S.; Cabrini, G.; Dechecchi, M.C.; et al. Unravelling the role of sphingolipids in cystic fibrosis lung disease. Chem. Phys. Lipids 2016, 200, 94–103. [Google Scholar] [CrossRef]
- Rockfield, S.; Chhabra, R.; Robertson, M.; Rehman, N.; Bisht, R.; Nanjundan, M. Links Between Iron and Lipids: Implications in Some Major Human Diseases. Pharm. (Basel) 2018, 11, 113. [Google Scholar] [CrossRef]
- Chen, K.; Ho, T.S.; Lin, G.; Tan, K.L.; Rasband, M.N.; Bellen, H.J. Loss of Frataxin activates the iron/sphingolipid/PDK1/Mef2 pathway in mammals. Elife 2016, 5, e20732. [Google Scholar] [CrossRef]
- Bettencourt, C.; Forabosco, P.; Wiethoff, S.; Heidari, M.; Johnstone, D.M.; Botía, J.A.; Collingwood, J.F.; Hardy, J.; UK Brain Expression Consortium (UKBEC); Milward, E.A.; et al. Gene co-expression networks shed light into diseases of brain iron accumulation. Neurobiol. Dis. 2016, 87, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Natarajan, S.K.; Mott, J.L.; Kharbanda, K.K.; Harrison-Findik, D.D. Ceramide Induces Human Hepcidin Gene Transcription through JAK/STAT3 Pathway. PLoS ONE 2016, 11, e0147474. [Google Scholar] [CrossRef] [PubMed]
- Shakoury-Elizeh, M.; Protchenko, O.; Berger, A.; Cox, J.; Gable, K.; Dunn, T.M.; Prinz, W.A.; Bard, M.; Philpott, C.C. Metabolic response to iron deficiency in Saccharomyces cerevisiae. J. Biol. Chem. 2010, 285, 14823–14833. [Google Scholar] [CrossRef] [PubMed]
- Boshuizen, M.; van Hezel, M.E.; van Manen, L.; Straat, M.; Somsen, Y.B.O.; Spoelstra-de Man, A.M.E.; Blumberg, N.; van Bruggen, R.; Juffermans, N.P. The effect of red blood cell transfusion on iron metabolism in critically ill patients. Transfusion 2019, 59, 1196–1201. [Google Scholar] [CrossRef] [PubMed]
- Maceyka, M.; Spiegel, S. Sphingolipid metabolites in inflammatory disease. Nature 2014, 510, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Teichgräber, V.; Ulrich, M.; Endlich, N.; Riethmüller, J.; Wilker, B.; De Oliveira-Munding, C.C.; van Heeckeren, A.M.; Barr, M.L.; von Kürthy, G.; Schmid, K.W.; et al. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat. Med. 2008, 14, 382–391. [Google Scholar] [CrossRef]
- Michaud, M.D.; Robitaille, G.A.; Gratton, J.P.; Richard, D.E. Sphingosine-1-phosphate: A novel nonhypoxic activator of hypoxia-inducible factor-1 in vascular cells. Arter. Thromb. Vasc. Biol. 2009, 29, 902–908. [Google Scholar] [CrossRef]
- Glaser, U.G.; Fandrey, J. Sphingolipids in inflammatory hypoxia. Biol. Chem. 2018, 399, 1169–1174. [Google Scholar] [CrossRef]
- Dubois, N.; Rio, E.; Ripoche, N.; Ferchaud-Roucher, V.; Gaugler, M.H.; Campion, L.; Krempf, M.; Carrie, C.; Mahé, M.; Mirabel, X.; et al. Plasma ceramide, a real-time predictive marker of pulmonary and hepatic metastases response to stereotactic body radiation therapy combined with irinotecan. Radiother. Oncol. 2016, 119, 229–235. [Google Scholar] [CrossRef]
- Bowler, R.P.; Jacobson, S.; Cruickshank, C.; Hughes, G.J.; Siska, C.; Ory, D.S.; Petrache, I.; Schaffer, J.E.; Reisdorph, N.; Kechris, K. Plasma sphingolipids associated with chronic obstructive pulmonary disease phenotypes. Am. J. Respir. Crit. Care Med. 2015, 191, 275–284. [Google Scholar] [CrossRef]
- Brodlie, M.; McKean, M.C.; Johnson, G.E.; Gray, J.; Fisher, A.J.; Corris, P.A.; Lordan, J.L.; Ward, C. Ceramide is increased in the lower airway epithelium of people with advanced cystic fibrosis lung disease. Am. J. Respir. Crit. Care Med. 2010, 182, 369–375. [Google Scholar] [CrossRef]
- Grassme, H.; Riethmuller, J.; Gulbins, E. Ceramide in cystic fibrosis. Handb. Exp. Pharmacol. 2013, 216, 265–274. [Google Scholar]
- Christofidou-Solomidou, M.; Pietrofesa, R.A.; Arguiri, E.; Schweitzer, K.S.; Berdyshev, E.V.; McCarthy, M.; Corbitt, A.; Alwood, J.S.; Yu, Y.; Globus, R.K.; et al. Space radiation-associated lung injury in a murine model. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L416–L428. [Google Scholar] [CrossRef] [PubMed]
- Duru, S.; Bilgin, E.; Ardic, S. Hepcidin: A useful marker in chronic obstructive pulmonary disease. Ann. Thorac. Med. 2012, 7, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Tandara, L.; Grubisic, T.Z.; Ivan, G.; Jurisic, Z.; Tandara, M.; Gugo, K.; Mladinov, S.; Salamunic, I. Systemic inflammation up-regulates serum hepcidin in exacerbations and stabile chronic obstructive pulmonary disease. Clin. Biochem. 2015, 48, 1252–1257. [Google Scholar] [CrossRef] [PubMed]
- Uijterschout, L.; Swinkels, D.W.; Akkermans, M.D.; Zandstra, T.; Nuijsink, M.; Hendriks, D.; Hudig, C.; Tjalsma, H.; Vos, R.; van Goudoever, J.B.; et al. The value of soluble transferrin receptor and hepcidin in the assessment of iron status in children with cystic fibrosis. J. Cyst. Fibros. 2014, 13, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Basu, M.; Malhotra, A.S.; Pal, K.; Prasad, R.; Kumar, R.; Prasad, B.A.; Sawhney, R.C. Erythropoietin levels in lowlanders and high-altitude natives at 3450 m. Aviat. Space Environ. Med. 2007, 78, 963–967. [Google Scholar] [CrossRef]
- Markoulaki, D.; Kostikas, K.; Papatheodorou, G.; Koutsokera, A.; Alchanatis, M.; Bakakos, P.; Gourgoulianis, K.I.; Roussos, C.; Koulouris, N.G.; Loukides, S. Hemoglobin, erythropoietin and systemic inflammation in exacerbations of chronic obstructive pulmonary disease. Eur. J. Intern. Med. 2011, 22, 103–107. [Google Scholar] [CrossRef]
- Sun, X.; Ma, S.F.; Wade, M.S.; Acosta-Herrera, M.; Villar, J.; Pino-Yanes, M.; Zhou, T.; Liu, B.; Belvitch, P.; Moitra, J.; et al. Functional promoter variants in sphingosine 1-phosphate receptor 3 associate with susceptibility to sepsis-associated acute respiratory distress syndrome. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 305, L467–L477. [Google Scholar] [CrossRef]
- Lea, S.R.; Metcalfe, H.J.; Plumb, J.; Beerli, C.; Poll, C.; Singh, D.; Abbott-Banner, K.H. Neutral sphingomyelinase-2, acid sphingomyelinase, and ceramide levels in COPD patients compared to controls. Int. J. Chron. Obs. Pulmon. Dis. 2016, 11, 2139–2147. [Google Scholar] [CrossRef]
- Chen, N.; Hao, C.; Peng, X.; Lin, H.; Yin, A.; Hao, L.; Tao, Y.; Liang, X.; Liu, Z.; Xing, C.; et al. Roxadustat for Anemia in Patients with Kidney Disease Not Receiving Dialysis. N. Eng. J. Med. 2019, 381, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; Warrington, S.; Cortezi, B.; Zöllner, S.; Vauléon, S.; Swinkels, D.W.; Summo, L.; Schwoebel, F.; Riecke, K. Safety, Pharmacokinetics and pharmacodynamics of the anti-hepcidin Spiegelmer lexaptepid pegol in healthy subjects. Br. J. Pharmacol. 2016, 173, 1580–1588. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.; Harikumar, K.B. Sphingosine 1-Phosphate: A Novel Target for Lung Disorders. Front. Immunol. 2017, 8, 296. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.A.; Riethmüller, J.; Seitz, A.P.; Gardner, A.; Boudreau, R.; Kamler, M.; Kleuser, B.; Schuchman, E.; Caldwell, C.C.; Edwards, M.J.; et al. Sphingolipids as targets for inhalation treatment of cystic fibrosis. Adv. Drug Deliv. Rev. 2018, 133, 66–75. [Google Scholar] [CrossRef]


{kind=link}
| Parameter | High Altitude Good Adaptation | High Altitude Bad Adaptation (HAPE) | ARDS | COPD | Cystic Fibrosis |
|---|---|---|---|---|---|
| hepcidin | Low [44,45,46] | High [54] | High (anemic ICU patients [49]) | Low in stable [57,124] high in exacerbations [125] | Low in stable kids [126] |
| ferritin | Low [44,45] | Normal/high [54] | High [55] | Normal/high [56] | Normal/high [61] |
| Erythropoietin (EPO) | High [44,45] | High [127] | ? | high [56] low in exacerbations [128] | Normal/high [61] |
| hemoglobin | High [35] | Very high/low [44,45] | Low (ICU patients) [14] | high [128] Low in worse prognosis [128] | Normal/Low [61] |
| SPL Metabolites | S1P high [86] | Cer High [87] | S1PR3 high [129] and SMase high [92] in worse prognosis | Cer High [98,130] | Cer high [121] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ottolenghi, S.; Zulueta, A.; Caretti, A. Iron and Sphingolipids as Common Players of (Mal)Adaptation to Hypoxia in Pulmonary Diseases. Int. J. Mol. Sci. 2020, 21, 307. https://doi.org/10.3390/ijms21010307
Ottolenghi S, Zulueta A, Caretti A. Iron and Sphingolipids as Common Players of (Mal)Adaptation to Hypoxia in Pulmonary Diseases. International Journal of Molecular Sciences. 2020; 21(1):307. https://doi.org/10.3390/ijms21010307
Chicago/Turabian StyleOttolenghi, Sara, Aida Zulueta, and Anna Caretti. 2020. "Iron and Sphingolipids as Common Players of (Mal)Adaptation to Hypoxia in Pulmonary Diseases" International Journal of Molecular Sciences 21, no. 1: 307. https://doi.org/10.3390/ijms21010307
APA StyleOttolenghi, S., Zulueta, A., & Caretti, A. (2020). Iron and Sphingolipids as Common Players of (Mal)Adaptation to Hypoxia in Pulmonary Diseases. International Journal of Molecular Sciences, 21(1), 307. https://doi.org/10.3390/ijms21010307