mACPpred: A Support Vector Machine-Based Meta-Predictor for Identification of Anticancer Peptides

 ,
, 
Abstract
1. Introduction
2. Results
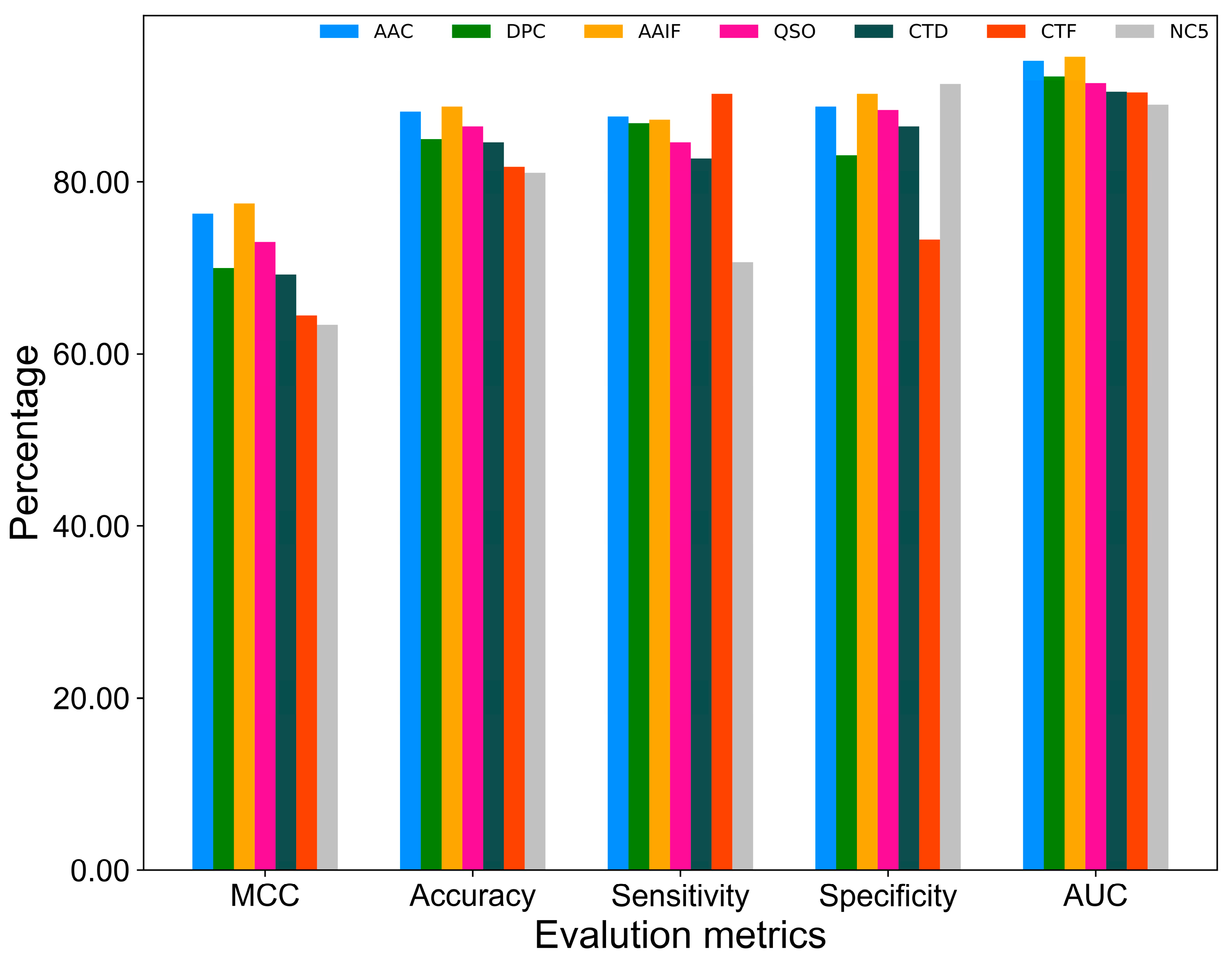
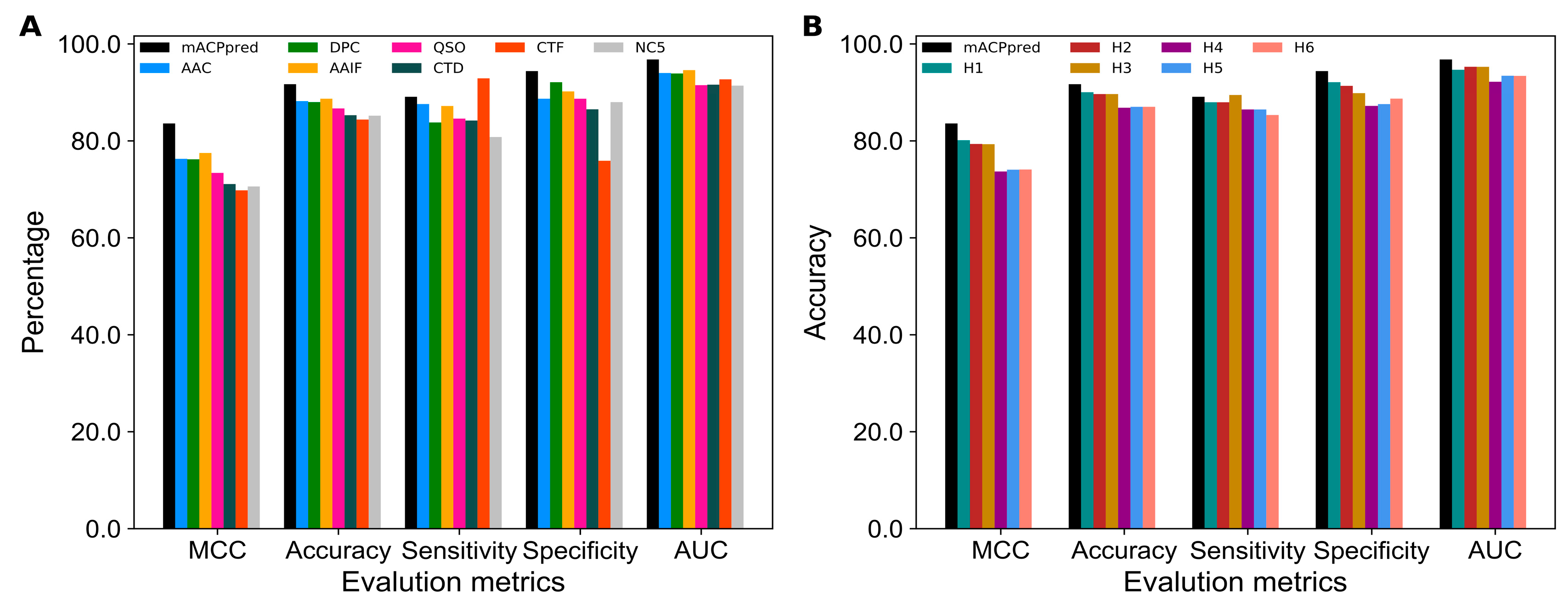
2.1. Performance of Various Feature Encodings
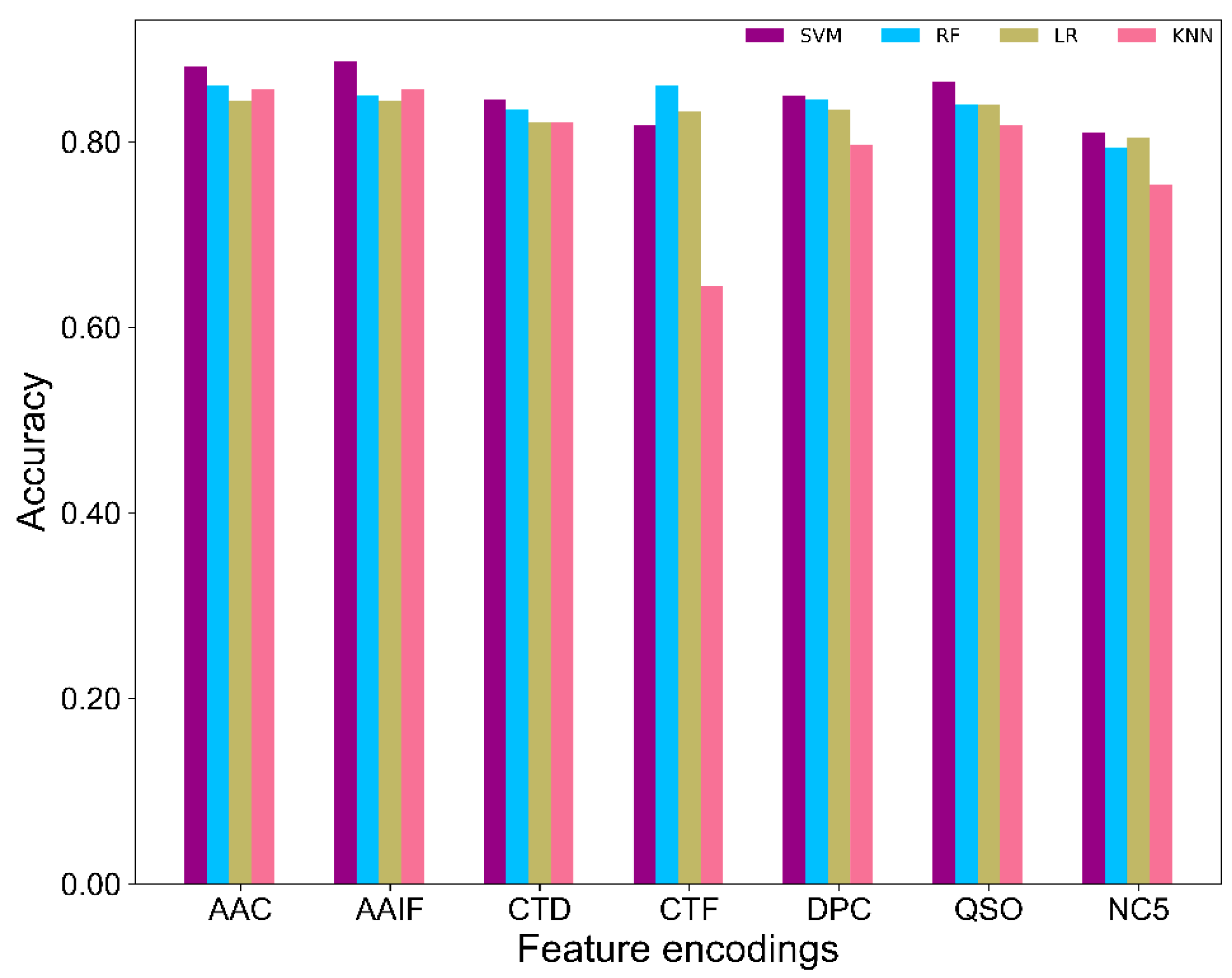
2.2. Comparison of SVM and Other Classifiers
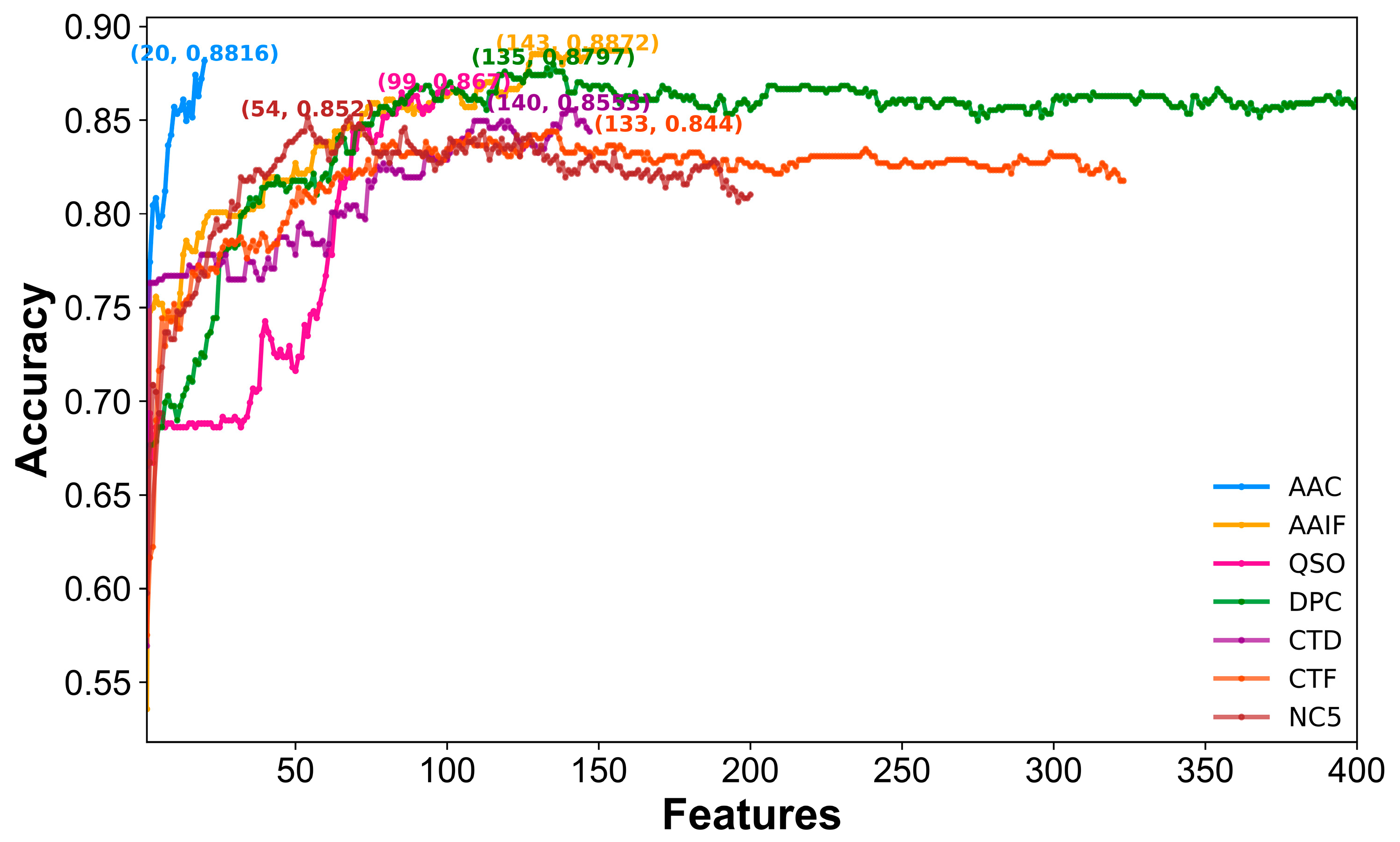
2.3. Selection of the Optimal Features for Each Encoding
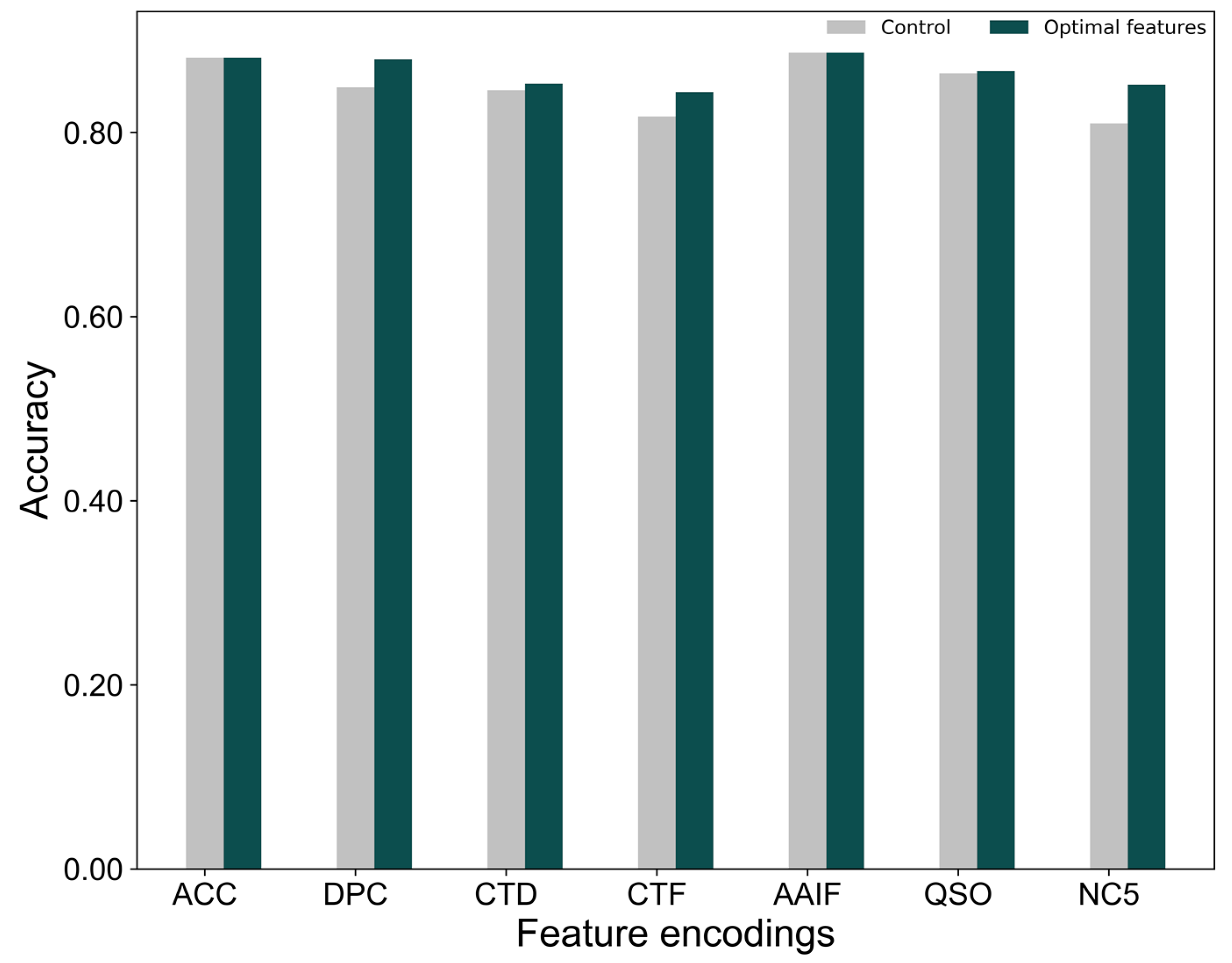
2.4. Construction of the Final Predictor
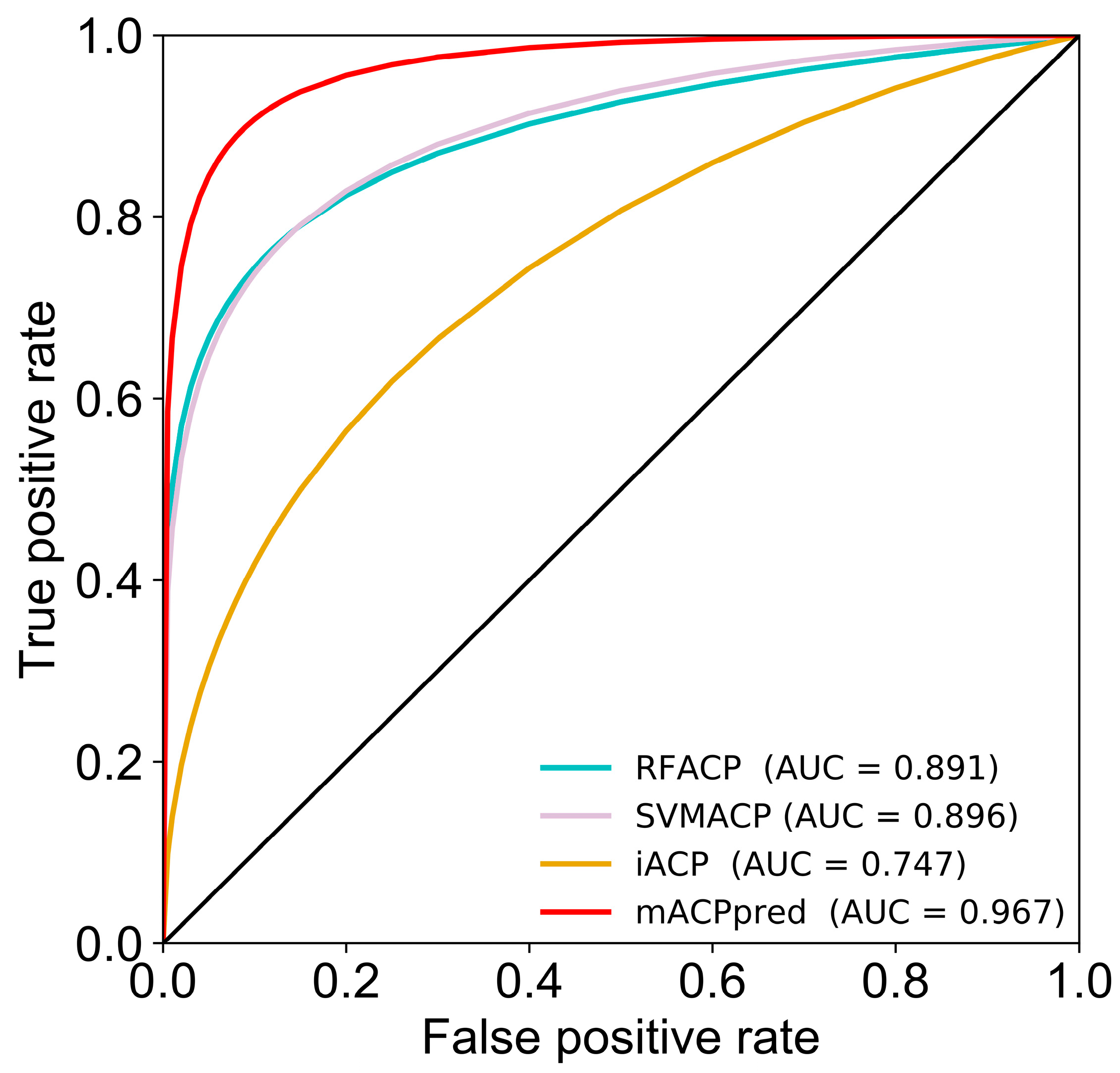
2.5. Performance Comparison on the Independent Dataset
2.6. Webserver Implementation
3. Discussion
4. Materials and Methods
4.1. Dataset Collection and Processing
4.2. Feature Extraction
4.2.1. Sequence-Based Features
- AAC has been widely used in numerous sequence-based prediction tasks [46], which represents the occurrence frequencies of 20 standard amino acids in a given peptide sequence that generates a 20-dimensional vector.
- DPC encoding of the given peptide sequence results in a fixed length of a 400-dimensional feature vector that summarizes amino acids fraction, the sequence-order, and fragment information.
- QSO encoding of the given peptide sequence results in a fixed length of a 100-dimensional feature vector, by measuring the physicochemical distance between the amino acids within the sequence. A detailed description of QSO feature encoding along with a set of equations has been provided in previous studies [47,48,49].
- In NC5, each amino acid is encoded as a 20-dimensional 0/1 vector. For example, the amino acid of type A and type C are encoded as (1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0) and (0,1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0), respectively. BPF(w) dimension is 20 × w, where w is sequence window length. Since the minimal length of the peptide in our dataset is 5, we fixed at this value and considered both N-and C- terminals (NC5) that generate a 200-dimensional vector.
4.2.2. Physicochemical Properties-Based Features
- In AAIF, we used only eight high-quality amino acid indices as reported in a previous study [57], which are LIFS790101 [58], CEDJ970104 [59], MIYS990104 [60], NAKH920108 [61], TSAJ990101 [62], MAXF760101 [63], BIOV880101 [64], BLAM930101 [65]. AAIF generates a 160 (=20 amino acids * 8 properties) dimensional vector, which has been successfully applied in numerous sequence-based prediction tasks [66,67,68].
- We used seven different types of physicochemical properties listed in Table 3 where 20 standard amino acids are classified into 3 different classes according to their attributes. In CTD, composition, transition, and distribution are respectively encoded as a 21, 21, 105-dimensional feature vector. A detailed description of CTD with a set of equations has been provided in previous studies [69,70].
4.3. Support Vector Machine
4.4. Ten-Fold Cross-Validation
4.5. Feature Selection
4.6. Performance Evaluation of ACPs Prediction
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Salehi, B.; Zucca, P.; Sharifi-Rad, M.; Pezzani, R.; Rajabi, S.; Setzer, W.N.; Varoni, E.M.; Iriti, M.; Kobarfard, F.; Sharifi-Rad, J. Phytotherapeutics in cancer invasion and metastasis. Phytother. Res. 2018, 32, 1425–1449. [Google Scholar] [CrossRef]
- Rahman, N. Realizing the promise of cancer predisposition genes. Nature 2014, 505, 302–308. [Google Scholar] [CrossRef]
- Wild, C.P.; Scalbert, A.; Herceg, Z. Measuring the exposome: A powerful basis for evaluating environmental exposures and cancer risk. Environ. Mol. Mutagen 2013, 54, 480–499. [Google Scholar] [CrossRef] [PubMed]
- Gillies, R.J.; Gatenby, R.A. Metabolism and its sequelae in cancer evolution and therapy. Cancer J. 2015, 21, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Storey, K.; Ryser, M.D.; Leder, K.; Foo, J. Spatial Measures of Genetic Heterogeneity During Carcinogenesis. Bull. Math. Biol. 2017, 79, 237–276. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Boyle, P.; Levin, B. World Cancer Report 2008; IARC Press, International Agency for Research on Cancer: Lyon, France, 2008. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Kakde, D.; Jain, D.; Shrivastava, V.; Kakde, R.; Patil, A. Cancer therapeutics-opportunities, challenges and advances in drug delivery. J. Appl. Pharm. Sci. 2011, 1, 1–10. [Google Scholar]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714. [Google Scholar] [CrossRef]
- Harris, F.; Dennison, S.R.; Singh, J.; Phoenix, D.A. On the selectivity and efficacy of defense peptides with respect to cancer cells. Med. Res. Rev. 2013, 33, 190–234. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.; Singh, H.; Andrabi, M.; Husain, S.A.; Ahmad, S. Databases and QSAR for cancer research. Cancer Inform. 2006, 2, 99–111. [Google Scholar] [CrossRef]
- Thundimadathil, J. Cancer treatment using peptides: Current therapies and future prospects. J. Amino Acids 2012, 2012, 967347. [Google Scholar] [CrossRef]
- Otvos, L., Jr. Peptide-based drug design: Here and now. Methods Mol. Biol. 2008, 494, 1–8. [Google Scholar] [PubMed]
- Wei, L.; Zhou, C.; Chen, H.; Song, J.; Su, R. ACPred-FL: A sequence-based predictor based on effective feature representation to improve the prediction of anti-cancer peptides. Bioinformatics 2018, 34, 4007–4016. [Google Scholar] [CrossRef]
- Gaspar, D.; Veiga, A.S.; Castanho, M.A. From antimicrobial to anticancer peptides. A review. Front. Microbiol. 2013, 4, 294. [Google Scholar] [CrossRef]
- Tyagi, A.; Tuknait, A.; Anand, P.; Gupta, S.; Sharma, M.; Mathur, D.; Joshi, A.; Singh, S.; Gautam, A.; Raghava, G.P. CancerPPD: A database of anticancer peptides and proteins. Nucleic Acids Res. 2015, 43, D837–D843. [Google Scholar] [CrossRef]
- Stephenson, N.; Shane, E.; Chase, J.; Rowland, J.; Ries, D.; Justice, N.; Zhang, J.; Chan, L.; Cao, R. Survey of machine learning techniques in drug discovery. Curr. Drug Metab. 2018. [Google Scholar] [CrossRef]
- Tan, J.-X.; Dao, F.-Y.; Lv, H.; Feng, P.-M.; Ding, H. Identifying Phage Virion Proteins by Using Two-Step Feature Selection Methods. Molecules 2018, 23, 2000. [Google Scholar] [CrossRef]
- Cascio, D.; Taormina, V.; Raso, G. An Automatic HEp-2 Specimen Analysis System Based on an Active Contours Model and an SVM Classification. Appl. Sci. 2019, 9, 307. [Google Scholar] [CrossRef]
- Manavalan, B.; Lee, J. SVMQA: Support-vector-machine-based protein single-model quality assessment. Bioinformatics 2017, 33, 2496–2503. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Shin, T.H.; Lee, G. PVP-SVM: Sequence-Based Prediction of Phage Virion Proteins Using a Support Vector Machine. Front. Microbiol. 2018, 9, 476. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Shin, T.H.; Lee, G. DHSpred: Support-vector-machine-based human DNase I hypersensitive sites prediction using the optimal features selected by random forest. Oncotarget 2018, 9, 1944–1956. [Google Scholar] [CrossRef] [PubMed]
- Basith, S.; Manavalan, B.; Shin, T.H.; Lee, G. iGHBP: Computational identification of growth hormone binding proteins from sequences using extremely randomised tree. Comput. Struct. Biotechnol. J. 2018, 16, 412–420. [Google Scholar] [CrossRef]
- Qiang, X.; Chen, H.; Ye, X.; Su, R.; Wei, L. M6AMRFS: Robust Prediction of N6-Methyladenosine Sites With Sequence-Based Features in Multiple Species. Front. Genet. 2018, 9, 495. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Li, F.; Marquez-Lago, T.T.; Leier, A.; Fan, C.; Kwoh, C.K.; Chou, K.C.; Song, J.; Jia, C. MULTiPly: A novel multi-layer predictor for discovering general and specific types of promoters. Bioinformatics 2019. [Google Scholar] [CrossRef]
- Manavalan, B.; Basith, S.; Shin, T.H.; Choi, S.; Kim, M.O.; Lee, G. MLACP: Machine-learning-based prediction of anticancer peptides. Oncotarget 2017, 8, 77121–77136. [Google Scholar] [CrossRef] [PubMed]
- Gabere, M.N.; Noble, W.S. Empirical comparison of web-based antimicrobial peptide prediction tools. Bioinformatics 2017, 33, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- Hanley, J.A.; McNeil, B.J. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 1982, 143, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Ding, H.; Feng, P.; Lin, H.; Chou, K.C. iACP: A sequence-based tool for identifying anticancer peptides. Oncotarget 2016, 7, 16895–16909. [Google Scholar] [CrossRef]
- Manavalan, B.; Lee, J.; Lee, J. Random forest-based protein model quality assessment (RFMQA) using structural features and potential energy terms. PLoS ONE 2014, 9, e106542. [Google Scholar] [CrossRef]
- Su, R.; Liu, X.; Wei, L.; Zou, Q. Deep-Resp-Forest: A deep forest model to predict anti-cancer drug response. Methods 2019. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Cao, R.-Z.; Wang, W.; Liu, T.-S.; Wang, L.-M.; He, C.-M. A two-step discriminated method to identify thermophilic proteins. Int. J. Biomath. 2017, 10, 1750050. [Google Scholar] [CrossRef]
- Conover, M.; Staples, M.; Si, D.; Sun, M.; Cao, R. AngularQA: Protein Model Quality Assessment with LSTM Networks. bioRxiv 2019, 560995. [Google Scholar] [CrossRef]
- Hou, J.; Wu, T.; Cao, R.; Cheng, J. Protein tertiary structure modeling driven by deep learning and contact distance prediction in CASP13. bioRxiv 2019, 552422. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Basith, S.; Shin, T.H.; Wei, L.; Lee, G.; Hancock, J. mAHTPred: A sequence-based meta-predictor for improving the prediction of anti-hypertensive peptides using effective feature representation. Bioinformatics 2018. [Google Scholar] [CrossRef] [PubMed]
- Qiang, X.; Zhou, C.; Ye, X.; Du, P.F.; Su, R.; Wei, L. CPPred-FL: A sequence-based predictor for large-scale identification of cell-penetrating peptides by feature representation learning. Brief Bioinform. 2018. [Google Scholar] [CrossRef]
- Wei, L.; Luan, S.; Nagai, L.A.E.; Su, R.; Zou, Q. Exploring sequence-based features for the improved prediction of DNA N4-methylcytosine sites in multiple species. Bioinformatics 2018. [Google Scholar] [CrossRef]
- Cao, R.; Adhikari, B.; Bhattacharya, D.; Sun, M.; Hou, J.; Cheng, J. QAcon: Single model quality assessment using protein structural and contact information with machine learning techniques. Bioinformatics 2017, 33, 586–588. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.; Kapoor, P.; Kumar, R.; Chaudhary, K.; Gautam, A.; Raghava, G.P. In silico models for designing and discovering novel anticancer peptides. Sci. Rep. 2013, 3, 2984. [Google Scholar] [CrossRef]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef]
- Novkovic, M.; Simunic, J.; Bojovic, V.; Tossi, A.; Juretic, D. DADP: The database of anuran defense peptides. Bioinformatics 2012, 28, 1406–1407. [Google Scholar] [CrossRef]
- Pirtskhalava, M.; Gabrielian, A.; Cruz, P.; Griggs, H.L.; Squires, R.B.; Hurt, D.E.; Grigolava, M.; Chubinidze, M.; Gogoladze, G.; Vishnepolsky, B.; et al. DBAASP v.2: An enhanced database of structure and antimicrobial/cytotoxic activity of natural and synthetic peptides. Nucleic Acids Res. 2016, 44, D1104–D1112. [Google Scholar] [CrossRef]
- Fan, L.; Sun, J.; Zhou, M.; Zhou, J.; Lao, X.; Zheng, H.; Xu, H. DRAMP: A comprehensive data repository of antimicrobial peptides. Sci. Rep. 2016, 6, 24482. [Google Scholar] [CrossRef]
- Zhao, X.; Wu, H.; Lu, H.; Li, G.; Huang, Q. LAMP: A Database Linking Antimicrobial Peptides. PLoS ONE 2013, 8, e66557. [Google Scholar] [CrossRef]
- Usmani, S.S.; Kumar, R.; Bhalla, S.; Kumar, V.; Raghava, G.P.S. In Silico Tools and Databases for Designing Peptide-Based Vaccine and Drugs. Adv. Protein Chem. Struct. Biol. 2018, 112, 221–263. [Google Scholar]
- Chou, K.C. Prediction of protein subcellular locations by incorporating quasi-sequence-order effect. Biochem. Biophys. Res. Commun. 2000, 278, 477–483. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Yang, B.; Xie, R.; Marquez-Lago, T.T.; Leier, A.; Hayashida, M.; Akutsu, T.; Zhang, Y.; Chou, K.C.; et al. Bastion3: A two-layer ensemble predictor of type III secreted effectors. Bioinformatics 2018. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, B.; Leier, A.; Marquez-Lago, T.T.; Hayashida, M.; Rocker, A.; Zhang, Y.; Akutsu, T.; Chou, K.C.; Strugnell, R.A.; et al. Bastion6: A bioinformatics approach for accurate prediction of type VI secreted effectors. Bioinformatics 2018, 34, 2546–2555. [Google Scholar] [CrossRef]
- Lin, T.W.; Wu, J.W.; Chang, D.T. Combining phylogenetic profiling-based and machine learning-based techniques to predict functional related proteins. PLoS ONE 2013, 8, e75940. [Google Scholar] [CrossRef]
- Shen, J.; Zhang, J.; Luo, X.; Zhu, W.; Yu, K.; Chen, K.; Li, Y.; Jiang, H. Predicting protein-protein interactions based only on sequences information. Proc. Natl. Acad. Sci. USA 2007, 104, 4337–4341. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, L.; Jia, L.; Ren, Y.; Yu, G. Protein-Protein Interactions Prediction Using a Novel Local Conjoint Triad Descriptor of Amino Acid Sequences. Int. J. Mol. Sci. 2017, 18, 2373. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Chen, H.; Su, R. M6APred-EL: A Sequence-Based Predictor for Identifying N6-methyladenosine Sites Using Ensemble Learning. Mol. Ther. Nucleic Acids 2018, 12, 635–644. [Google Scholar] [CrossRef]
- Zou, Q., Sr.; Xing, P.; Wei, L.; Liu, B. Gene2vec: Gene Subsequence Embedding for Prediction of Mammalian N6-Methyladenosine Sites from mRNA. RNA 2018, 25, 205–218. [Google Scholar] [CrossRef]
- Chen, W.; Lv, H.; Nie, F.; Lin, H. i6mA-Pred: Identifying DNA N6-methyladenine sites in the rice genome. Bioinformatics 2019. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yang, H.; Feng, P.; Ding, H.; Lin, H. iDNA4mC: Identifying DNA N4-methylcytosine sites based on nucleotide chemical properties. Bioinformatics 2017, 33, 3518–3523. [Google Scholar] [CrossRef] [PubMed]
- Saha, I.; Maulik, U.; Bandyopadhyay, S.; Plewczynski, D. Fuzzy clustering of physicochemical and biochemical properties of amino acids. Amino Acids 2012, 43, 583–594. [Google Scholar] [CrossRef]
- Lifson, S.; Sander, C. Antiparallel and parallel beta-strands differ in amino acid residue preferences. Nature 1979, 282, 109–111. [Google Scholar] [CrossRef]
- Cedano, J.; Aloy, P.; Perez-Pons, J.A.; Querol, E. Relation between amino acid composition and cellular location of proteins1. J. Mol. Biol. 1997, 266, 594–600. [Google Scholar] [CrossRef]
- Miyazawa, S.; Jernigan, R.L. Self-consistent estimation of inter-residue protein contact energies based on an equilibrium mixture approximation of residues. Proteins 1999, 34, 49–68. [Google Scholar] [CrossRef]
- Sipos, L.; von Heijne, G. Predicting the topology of eukaryotic membrane proteins. Eur. J. Biochem. 1993, 213, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.; Taylor, R.; Chothia, C.; Gerstein, M. The packing density in proteins: Standard radii and volumes. J. Mol. Biol. 1999, 290, 253–266. [Google Scholar] [CrossRef]
- Maxfield, F.R.; Scheraga, H.A. Status of empirical methods for the prediction of protein backbone topography. Biochemistry 1976, 15, 5138–5153. [Google Scholar] [CrossRef]
- Biou, V.; Gibrat, J.F.; Levin, J.M.; Robson, B.; Garnier, J. Secondary structure prediction: Combination of three different methods. Protein Eng. 1988, 2, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Blaber, M.; Zhang, X.J.; Matthews, B.W. Structural basis of amino acid alpha helix propensity. Science 1993, 260, 1637–1640. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Govindaraj, R.G.; Shin, T.H.; Kim, M.O.; Lee, G. iBCE-EL: A New Ensemble Learning Framework for Improved Linear B-Cell Epitope Prediction. Front. Immunol. 2018, 9, 1695. [Google Scholar] [CrossRef]
- Manavalan, B.; Shin, T.H.; Kim, M.O.; Lee, G. PIP-EL: A New Ensemble Learning Method for Improved Proinflammatory Peptide Predictions. Front. Immunol. 2018, 9, 1783. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yan, R.; Li, J.; Song, J. SOHPRED: A new bioinformatics tool for the characterization and prediction of human S-sulfenylation sites. Mol. Biosyst. 2016, 12, 2849–2858. [Google Scholar] [CrossRef]
- Bhadra, P.; Yan, J.; Li, J.; Fong, S.; Siu, S.W.I. AmPEP: Sequence-based prediction of antimicrobial peptides using distribution patterns of amino acid properties and random forest. Sci. Rep. 2018, 8, 1697. [Google Scholar] [CrossRef]
- Zhang, P.; Tao, L.; Zeng, X.; Qin, C.; Chen, S.Y.; Zhu, F.; Yang, S.Y.; Li, Z.R.; Chen, W.P.; Chen, Y.Z. PROFEAT Update: A Protein Features Web Server with Added Facility to Compute Network Descriptors for Studying Omics-Derived Networks. J. Mol. Biol. 2017, 429, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Shin, T.H.; Kim, M.O.; Lee, G. AIPpred: Sequence-Based Prediction of Anti-inflammatory Peptides Using Random Forest. Front. Pharmacol. 2018, 9, 276. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Subramaniyam, S.; Shin, T.H.; Kim, M.O.; Lee, G. Machine-Learning-Based Prediction of Cell-Penetrating Peptides and Their Uptake Efficiency with Improved Accuracy. J. Proteome Res. 2018, 17, 2715–2726. [Google Scholar] [CrossRef]
- Dao, F.Y.; Lv, H.; Wang, F.; Feng, C.Q.; Ding, H.; Chen, W.; Lin, H. Identify origin of replication in Saccharomyces cerevisiae using two-step feature selection technique. Bioinformatics 2018. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Li, Y.; Niu, M.; Zou, Q. ELM-MHC: An Improved MHC Identification Method with Extreme Learning Machine Algorithm. J. Proteome Res. 2019, 18, 1392–1401. [Google Scholar] [CrossRef]
- Peng, H.; Long, F.; Ding, C. Feature selection based on mutual information: Criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005, 27, 1226–1238. [Google Scholar] [CrossRef]
- Cao, R.; Bhattacharya, D.; Hou, J.; Cheng, J. DeepQA: Improving the estimation of single protein model quality with deep belief networks. BMC Bioinform. 2016, 17, 495. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Freitas, C.; Chan, L.; Sun, M.; Jiang, H.; Chen, Z. ProLanGO: Protein Function Prediction Using Neural Machine Translation Based on a Recurrent Neural Network. Molecules 2017, 22, 1732. [Google Scholar] [CrossRef]
- Wei, L.; Su, R.; Wang, B.; Li, X.; Zou, Q.; Gao, X. Integration of deep feature representations and handcrafted features to improve the prediction of N6-methyladenosine sites. Neurocomputing 2018, 324, 3–9. [Google Scholar] [CrossRef]
- Malik, A.; Ahmad, S. Sequence and structural features of carbohydrate binding in proteins and assessment of predictability using a neural network. BMC Struct. Biol. 2007, 7, 1. [Google Scholar] [CrossRef]
- Malik, A.; Firoz, A.; Jha, V.; Ahmad, S. PROCARB: A Database of Known and Modelled Carbohydrate-Binding Protein Structures with Sequence-Based Prediction Tools. Adv. Bioinform. 2010, 436036. [Google Scholar] [CrossRef]
- Su, R.; Hu, J.; Zou, Q.; Manavalan, B.; Wei, L. Empirical comparison and analysis of web-based cell-penetrating peptide prediction tools. Brief Bioinform. 2019. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature Encoding | Dimension | MCC | Accuracy | Sensitivity | Specificity |
|---|---|---|---|---|---|
| AAC | 20 | 0.763 | 0.882 | 0.876 | 0.887 |
| DPC | 135 | 0.762 | 0.880 | 0.838 | 0.921 |
| CTD | 140 | 0.711 | 0.853 | 0.842 | 0.865 |
| AAIF | 143 | 0.775 | 0.887 | 0.872 | 0.902 |
| QSO | 99 | 0.734 | 0.867 | 0.846 | 0.887 |
| CTF | 133 | 0.698 | 0.844 | 0.929 | 0.759 |
| NC5 | 54 | 0.706 | 0.852 | 0.808 | 0.880 |
| Methods | MCC | Accuracy | Sensitivity | Specificity | AUC | p Value |
|---|---|---|---|---|---|---|
| mACPpred | 0.829 | 0.914 | 0.885 | 0.943 | 0.967 | – |
| SVMACP [27] | 0.592 | 0.768 | 0.554 | 0.981 | 0.896 | 0.000382 |
| RFACP [27] | 0.511 | 0.707 | 0.414 | 1.000 | 0.891 | 0.000401 |
| iACP [30] | 0.338 | 0.667 | 0.580 | 0.753 | 0.747 | <0.00001 |
| Properties | Class1 | Class2 | Class3 |
|---|---|---|---|
| Hydrophobicity | Polar E, D, K, N, Q, R | Neutral A, G, H, P, S, T, Y | Hydrophobicity C, L, V, I, M, F, W |
| Normalized Van der Waals volume | 0–2.78 A, C, D, G, P, S, T | 2.95–4.0 E, I, L, N, V, Q | 4.03–8.08 M, H, K, F, R, Y, W |
| Polarity | 4.9–6.2 L, I, F, W, C, M, V, Y | 8.0–9.2 A, G, P, S, T | 10.4–13.0 H, Q, R, K, N, E, D |
| Polarizability | 0–0.108 A, D, G, S, T | 0.128–0.186 C, E, I, L, P, Q, V, N | 0.219–0.409 K, M, H, F, R, Y, W |
| Charge | Positive K, R | Neutral A, N, C, Q, G, H, I, L, M, F, P, S, T, W, Y, V | Negative D, E |
| Secondary Structure | Helix A, E, H, K, L, M, Q, R | Strand V, I, Y, C, W, F, T | Coil D, G, N, P, S |
| Solvent Accessibility | Buried A, C, F, G, I, L, V, W | Exposed D, E, K, N, Q, R | Intermediate M, S, P, T, H, Y |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boopathi, V.; Subramaniyam, S.; Malik, A.; Lee, G.; Manavalan, B.; Yang, D.-C. mACPpred: A Support Vector Machine-Based Meta-Predictor for Identification of Anticancer Peptides. Int. J. Mol. Sci. 2019, 20, 1964. https://doi.org/10.3390/ijms20081964
Boopathi V, Subramaniyam S, Malik A, Lee G, Manavalan B, Yang D-C. mACPpred: A Support Vector Machine-Based Meta-Predictor for Identification of Anticancer Peptides. International Journal of Molecular Sciences. 2019; 20(8):1964. https://doi.org/10.3390/ijms20081964
Chicago/Turabian StyleBoopathi, Vinothini, Sathiyamoorthy Subramaniyam, Adeel Malik, Gwang Lee, Balachandran Manavalan, and Deok-Chun Yang. 2019. "mACPpred: A Support Vector Machine-Based Meta-Predictor for Identification of Anticancer Peptides" International Journal of Molecular Sciences 20, no. 8: 1964. https://doi.org/10.3390/ijms20081964
APA StyleBoopathi, V., Subramaniyam, S., Malik, A., Lee, G., Manavalan, B., & Yang, D.-C. (2019). mACPpred: A Support Vector Machine-Based Meta-Predictor for Identification of Anticancer Peptides. International Journal of Molecular Sciences, 20(8), 1964. https://doi.org/10.3390/ijms20081964