Natural History of a Satellite DNA Family: From the Ancestral Genome Component to Species-Specific Sequences, Concerted and Non-Concerted Evolution

 ,
,  ,
,  ,
,
Abstract
1. Introduction
2. Results
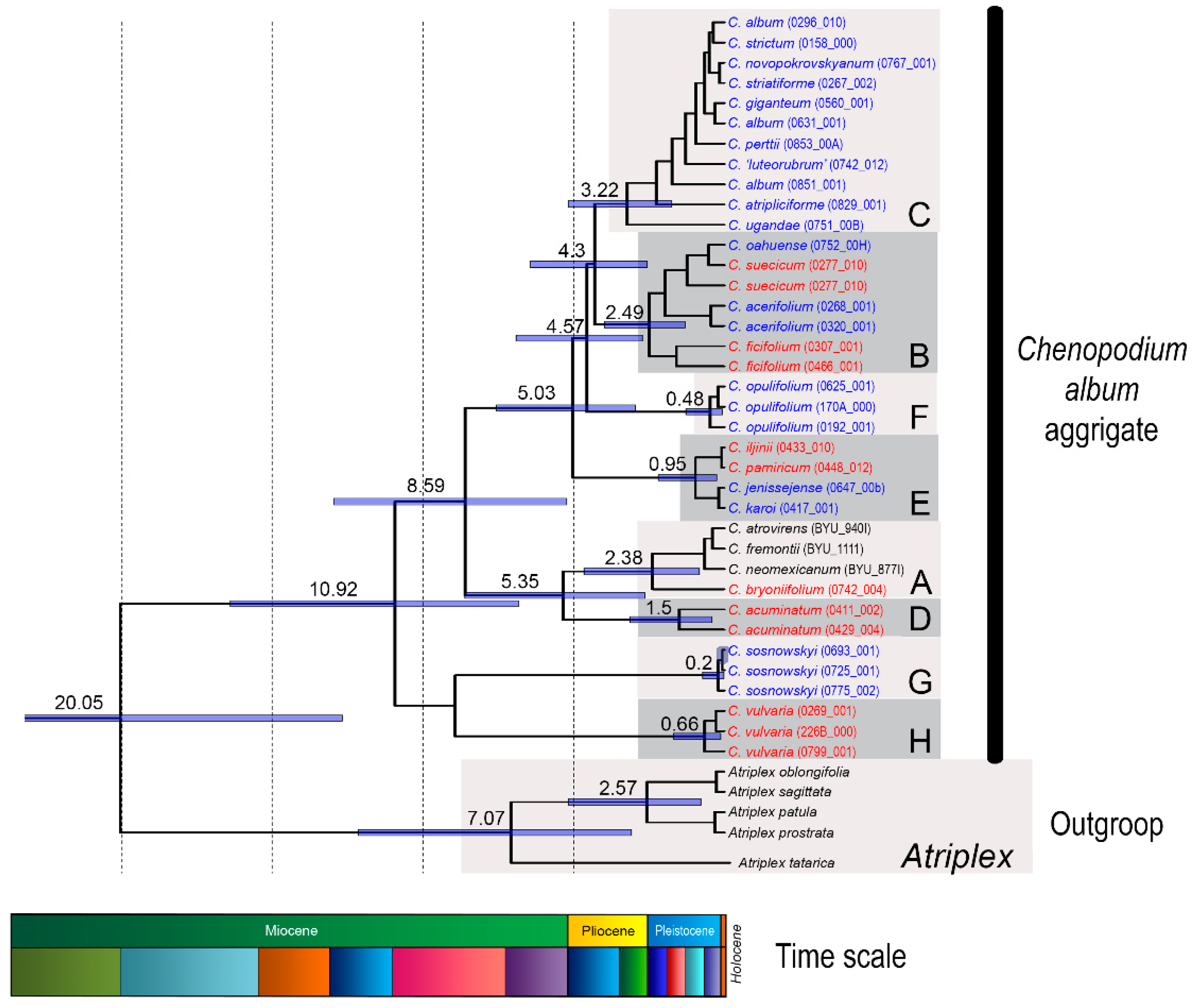
2.1. Clustering Results and Identification of satDNA Clusters
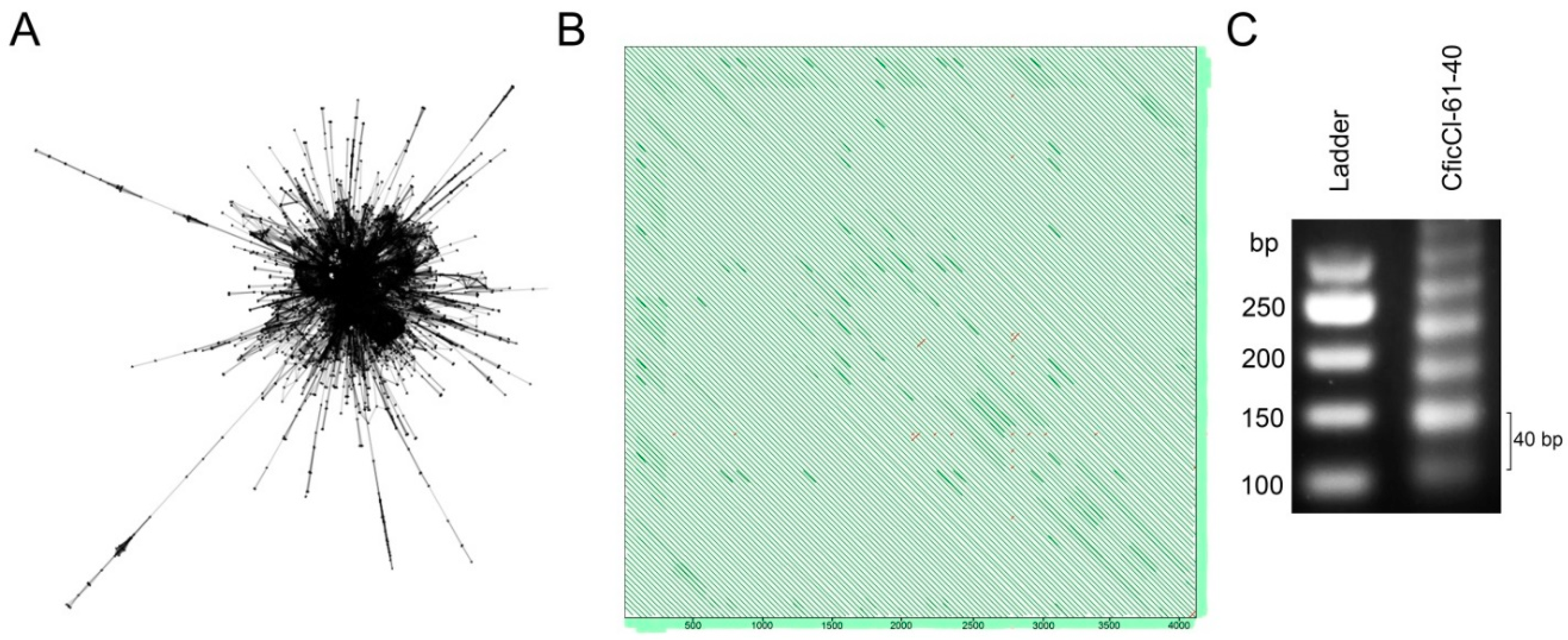
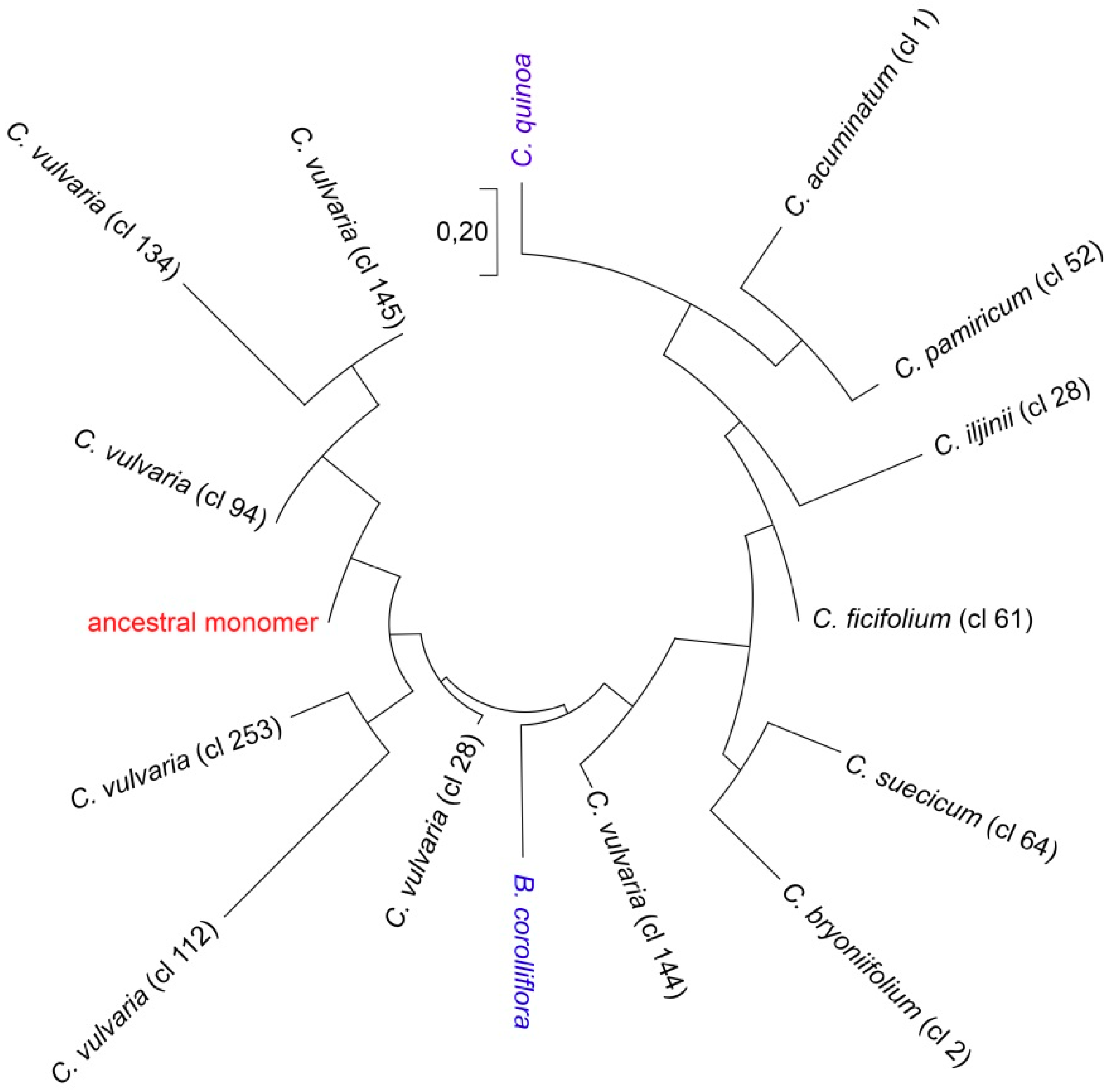
2.2. Sequence Analysis in the CficCl-61-40 satDNA Family
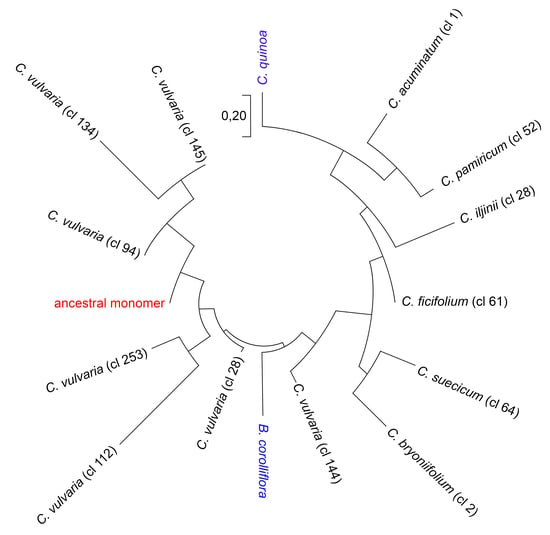
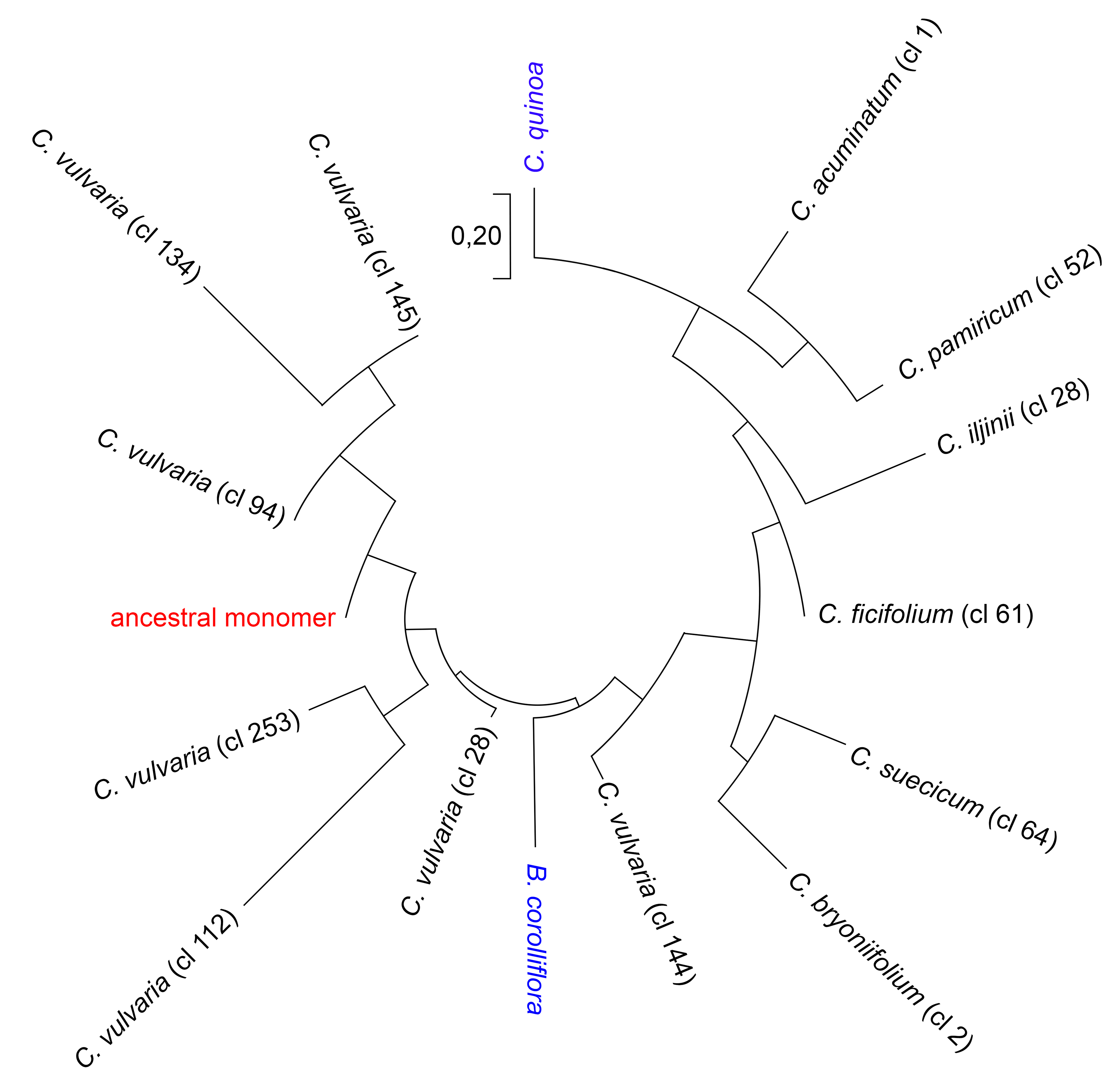
2.3. Reconstruction of the Ancestral Monomer
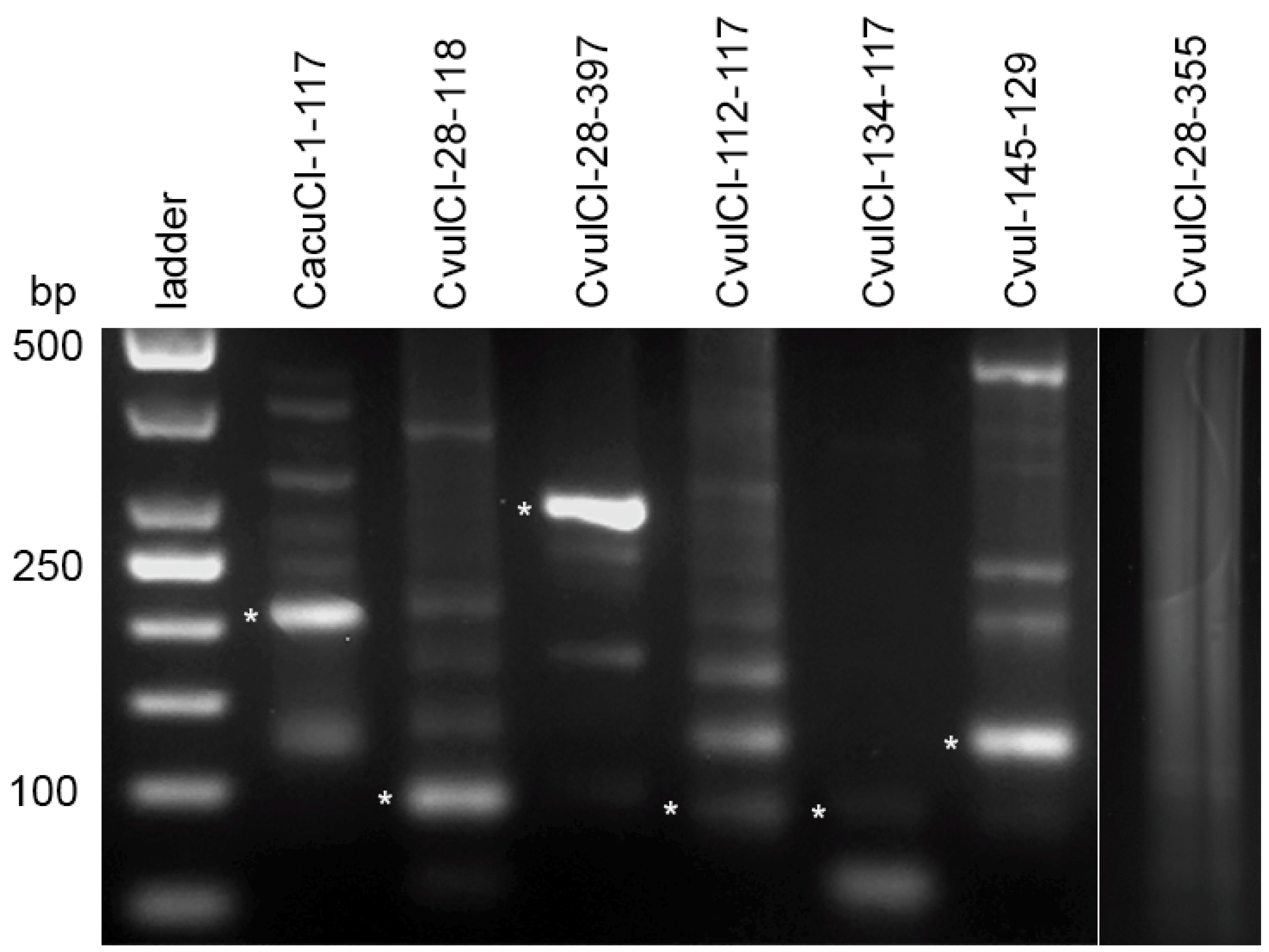
2.4. High Order Repeat (HOR) Detection in the CficCl-61-40 satDNA Family and Determination of Its Physical Counterpart
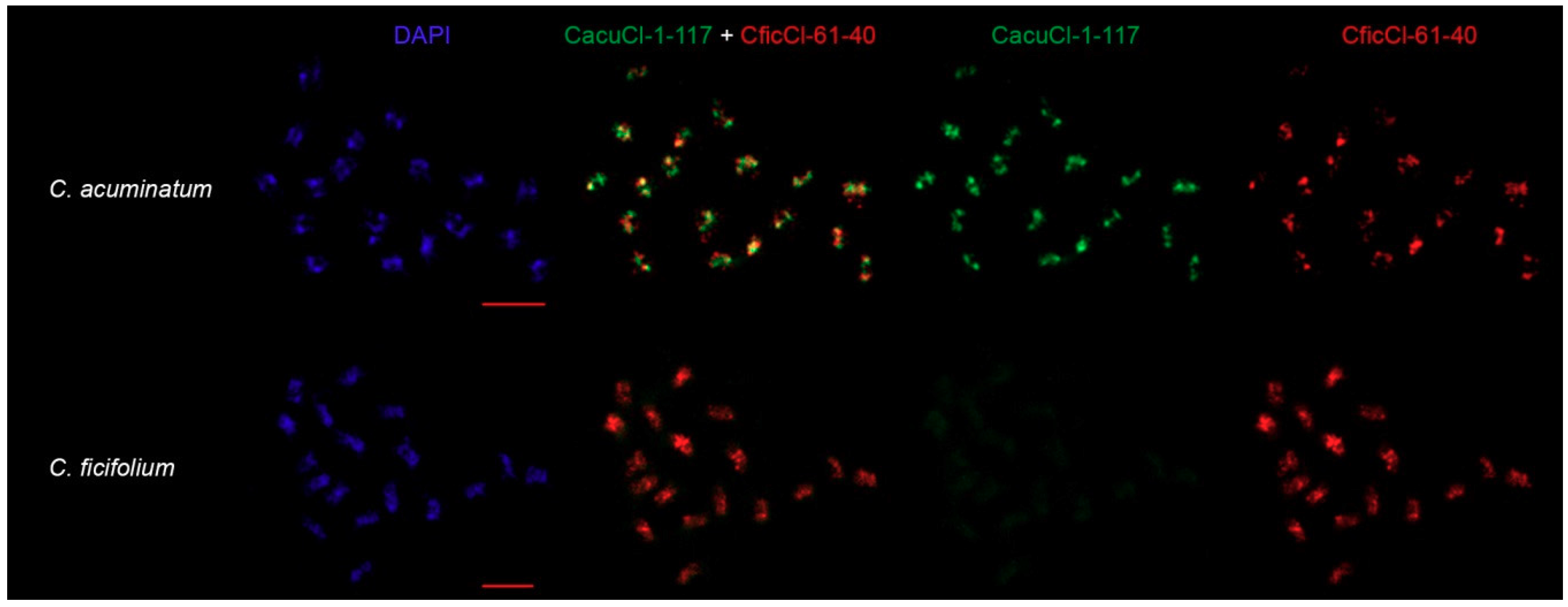
2.5. Comparison of CficCl-61-40 and Proposed HOR Unit CacuCl-1-117 Chromosomal Distribution
3. Discussion
4. Material and Methods
4.1. Plant Material, DNA Extraction, Library Preparation and Illumina Sequencing
4.2. Clustering of Repeatome Elements
4.3. Satellite DNA Clusters Screening for Tandem Repeats
4.4. Sequence Analysis
4.5. Detection Physical Counterparts of Basic Monomer and Proposed HOR Units
4.6. FISH Procedure
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bennetzen, J.L. The structure and evolution of angiosperm nuclear genomes. Curr. Opin. Plant Biol. 1998, 1, 103–108. [Google Scholar] [CrossRef]
- Maumus, F.; Quesneville, H. Ancestral repeats have shaped epigenome and genome composition for millions of years in Arabidopsis thaliana. Nat. Commun. 2014, 5, 4104. [Google Scholar] [CrossRef] [PubMed]
- Elder, J.F.; Turner, B.J. Concerted evolution of repetitive DNA sequences in eukaryotes. Q. Rev. Biol. 1995, 70, 297–320. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Ramos, M.A. Satellite DNA: An Evolving Topic. Genes 2017, 8, 230. [Google Scholar] [CrossRef] [PubMed]
- Biscotti, M.A.; Olmo, E.; Heslop-Harrison, J.S. Repetitive DNA in eukaryotic genomes. Chromosome Res. 2015, 23, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.H.-C.; Lower, S.E.; Caldas, I.V.; Sless, T.J.; Barbash, D.A.; Clark, A.G. Variable rates of simple satellite gains across the Drosophila phylogeny. Mol. Biol. Evol. 2018, 35, 925–941. [Google Scholar] [CrossRef]
- Satović, E.; Vojvoda Zeljko, T.; Luchetti, A.; Mantovani, B.; Plohl, M. Adjacent sequences disclose potential for intra-genomic dispersal of satellite DNA repeats and suggest a complex network with transposable elements. BMC Genom. 2016, 17, 997. [Google Scholar] [CrossRef]
- Charlesworth, B.; Sniegowski, P.; Stephan, W. The evolutionary dynamics of repetitive DNA in eukaryotes. Nature 1994, 371, 215–220. [Google Scholar] [CrossRef]
- Raskina, O.; Barber, J.C.; Nevo, E.; Belyayev, A. Repetitive DNA and chromosomal rearrangements: Speciation-related events in plant genomes. Cytogenet. Gen. Res. 2008, 120, 351–357. [Google Scholar] [CrossRef]
- Emadzade, K.; Jang, T.S.; Macas, J.; Kovařík, A.; Novák, P.; Parker, J.; Weiss-Schneeweiss, H. Differential amplification of satellite PaB6 in chromosomally hypervariable Prospero autumnale complex (Hyacinthaceae). Ann. Bot. 2014, 114, 1597–1608. [Google Scholar] [CrossRef]
- Dodsworth, S.; Chase, M.W.; Kelly, L.J.; Leitch, I.J.; Macas, J.; Novák, P.; Piednoël, M.; Weiss-Schneeweiss, H.; Leitch, A.R. Genomic repeat abundances contain phylogenetic signal. Syst. Biol. 2015, 64, 112–126. [Google Scholar] [CrossRef]
- Martienssen, R.A. Maintenance of heterochromatin by RNA interference of tandem repeats. Nat. Genet. 2003, 35, 213–214. [Google Scholar] [CrossRef] [PubMed]
- Kloc, A.; Martienssen, R. RNAi, heterochromatin and the cell cycle. Trends Genet. 2008, 24, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, S.; Goyal, V. Repetitive sequences in plant nuclear DNA: Types, distribution, evolution and function. Genom. Proteom. Bioinform. 2014, 12, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Ramos, M.A. SatDNA in plants: More than just rubbish. Cytogenet. Genome Res. 2015, 146, 153–170. [Google Scholar] [CrossRef] [PubMed]
- Meštrović, N.; Mravinac, B.; Pavlek, M.; Vojvoda-Zeljko, T.; Šatović, E.; Plohl, M. Structural and functional liaisons between transposable elements and satellite DNAs. Chromosome Res. 2015, 23, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Plohl, M.; Meštrović, N.; Mravinac, B. Satellite DNA evolution. Genome Dyn. 2012, 7, 126–152. [Google Scholar]
- Salser, W.; Bowen, S.; Browne, D.; el-Adli, F.; Fedoroff, N.; Fry, K.; Heindell, H.; Paddock, G.; Poon, R.; Wallace, B.; et al. Investigation of the organization of mammalian chromosomes at the DNA sequence level. Fed. Proc. 1976, 35, 23–35. [Google Scholar]
- Dover, G. Molecular drive. Trends Genet. 2002, 18, 587–589. [Google Scholar] [CrossRef]
- Plohl, M.; Luchetti, A.; Mestrovic, N.; Mantovani, B. Satellite DNAs between selfishness and functionality: Structure, genomics and evolution of tandem repeats in centromeric (hetero) chromatin. Gene 2008, 409, 72–82. [Google Scholar] [CrossRef]
- Samoluk, S.S.; Robledo, G.; Bertioli, D.; Seijo, J.G. Evolutionary dynamics of an at-rich satellite DNA and its contribution to karyotype differentiation in wild diploid Arachis species. Mol. Genet. Genom. 2017, 292, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Ugarkovic, D.; Plohl, M. Variation in satellite DNA profiles-causes and effects. EMBO J. 2002, 2, 5955–5959. [Google Scholar] [CrossRef]
- Novák, P.; Neumann, P.; Macas, J. Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinform. 2010, 11, 378. [Google Scholar] [CrossRef] [PubMed]
- Chu, G.-L.; Mosyakin, S.L.; Clemants, S.E. Chenopodiaceae. In Flora of China. Volume 5: Ulmaceae through Basellaceae; Wu, Z., Raven, P.H., Hong, D., Eds.; Missouri Botanical Garden Press: St. Louis, MI, USA, 2003; pp. 351–414. [Google Scholar]
- Habibi, F.; Vít, P.; Rahiminejad, M.; Mandák, B. Towards a better understanding of the C. album aggregate in the Middle East: A karyological, cytometric and morphometric investigation. J. Syst. Evol. 2018, 56, 231–242. [Google Scholar] [CrossRef]
- Mandák, B.; Krak, K.; Vít, P.; Pavlíková, Z.; Lomonosova, M.N.; Habibi, F.; Lei, W.; Jellen, E.N.; Douda, J. How genome size variation is linked with evolution within Chenopodium sensu lato. Perspect. Plant Ecol. Evol. System. 2016, 23, 18–32. [Google Scholar] [CrossRef]
- Mandák, B.; Krak, K.; Vít, P.; Lomonosova, M.N.; Belyayev, A.; Habibi, F.; Wang, L.; Douda, J.; Storchova, H. Hybridization and polyploidization within the Chenopodium album aggregate analyzed by means of cytological and molecular markers. Mol. Phylogenet. Evol. 2018, 129, 189–201. [Google Scholar] [CrossRef]
- Gao, D.; Schmidt, T.; Jung, C. Molecular characterization and chromosomal distribution of species-specific repetitive DNA sequences from Beta corolliflora, a wild relative of sugar beet. Genome 2000, 43, 1073–1080. [Google Scholar] [CrossRef]
- Kolano, B.; Gardunia, B.W.; Michalska, M.; Bonifacio, A.; Fairbanks, D.; Maughan, P.J.; Coleman, C.E.; Stevens, M.R.; Jellen, E.N.; Maluszynska, J. Chromosomal localization of two novel repetitive sequences isolated from the Chenopodium quinoa Willd. Genome 2011, 54, 710–717. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Gogarten, J.P.; Kibak, H.; Dittrich, P.; Taiz, L.; Bowman, E.J.; Bowman, B.J.; Manolson, M.F.; Poole, R.J.; Date, T.; Oshima, T.; et al. Evolution of the Vacuolar H+-Atpase: Implications for the Origin of Eukaryotes. Proc. Natl. Acad. Sci. USA 1989, 86, 6661–6665. [Google Scholar] [CrossRef]
- Iwabe, N.; Kuma, K.; Hasegawa, M.; Osawa, S.; Miyata, T. Evolutionary Relationship of Archaebacteria, Eubacteria, and Eukaryotes Inferred from Phylogenetic Trees of Duplicated Genes. Proc. Natl. Acad. Sci. USA 1989, 86, 9355–9359. [Google Scholar] [CrossRef] [PubMed]
- Kadereit, G.; Hohmann, S.; Kadereit, J.W. A synopsis of Chenopodiaceae subfam. Betoideae and notes on the taxonomy of Beta. Willdenowia 2006, 36, 9–19. [Google Scholar] [CrossRef]
- Koukalova, B.; Moraes, A.P.; Renny-Byfield, S.; Matyasek, R.; Leitch, A.; Kovarik, A. Fall and rise of satellite repeats in allopolyploids of Nicotiana over c. 5 million years. New Phytol. 2009, 186, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Plohl, M.; Petrović, V.; Luchetti, A.; Ricci, A.; Satović, E.; Passamonti, M.; Mantovani, B. Long-term conservation vs. high sequence divergence: The case of an extraordinarily old satellite DNA in bivalve mollusks. Heredity 2009, 104, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Willard, H.F.; Waye, J.S. Hierarchical order in chromosome-specific human alpha satellite DNA. Trends Genet. 1987, 3, 192–198. [Google Scholar] [CrossRef]
- Gallagher, D.S.; Modi, W.S.; Ivanov, S. Concerted Evolution and Higher-Order Repeat Structure of the 1.709 (Satellite IV) Family in Bovids. J. Mol. Evol. 2004, 58, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Adega, F.; Chaves, R.; Guedes-Pinto, H.; Heslop-Harrison, J.S. Physical organization of the 1.709 satellite IV DNA family in Bovini and Tragelaphini tribes of the Bovidae: Sequence and chromosomal evolution. Cytogenet. Genome Res. 2006, 114, 140–146. [Google Scholar] [CrossRef]
- Macas, J.; Navrátilová, A.; Koblížková, A. Sequence homogenization and chromosomal localization of VicTR-B satellites differ between closely related Vicia species. Chromosoma 2006, 115, 437–447. [Google Scholar] [CrossRef]
- Jarvis, D.E.; Ho, Y.S.; Lightfoot, D.J.; Schmöckel, S.M.; Li, B.; Borm, T.J.; Ohyanagi, H.; Mineta, K.; Michell, C.T.; Saber, N.; et al. The genome of Chenopodium quinoa. Nature 2017, 542, 307–312. [Google Scholar] [CrossRef]
- Belyayev, A.; Paštová, L.; Fehrer, J.; Josefiová, J.; Chrtek, J.; Mráz, P. Mapping of Hieracium (Asteraceae) chromosomes with genus-specific satDNA elements derived from next-generation sequencing data. Plant Syst. Evol. 2018, 304, 387–396. [Google Scholar] [CrossRef]
- Henikoff, S.; Ahmad, K.; Malik, H.S. The centromere paradox: Stable inheritance with rapidly evolving DNA. Science 2001, 293, 1098–1102. [Google Scholar] [CrossRef] [PubMed]
- Belyayev, A.; Josefiová, J.; Jandová, M.; Krak, K.; Mandák, B. Transposable elements dynamics in the evolution of Chenopodium album aggregate. in preparation.
- Kejnovský, E.; Michalovova, M.; Steflova, P.; Kejnovska, I.; Manzano, S.; Hobza, R.; Kubat, Z.; Kovarik, J.; Jamilena, M.; Vyskot, B. Expansion of microsatellites on evolutionary young Y chromosome. PLoS ONE 2013, 8, e45519. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-M.; Lee, B.S.; Mammadov, A.C.; Koo, B.C.; Mott, I.W.; Wang, R.R.-C. CAPS markers specific to Eb, Ee, and R genomes in the tribe Triticeae. Genome 2007, 50, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Luchetti, A.; Marini, M.; Mantovani, B. Non-concerted evolution of the RET76 satellite DNA family in Reticulitermes taxa (Insecta, Isoptera). Genetica 2006, 128, 123–132. [Google Scholar]
- Groom, Q.J. Piecing together the biogeographic history of Chenopodium vulvaria L. using botanical literature and collections. Peer J. 2015, 3, e723. [Google Scholar] [CrossRef] [PubMed]
- Mayr, E. Populations Species and Evolution: An Abridgment of Animal Species and Evolution; Belknap Press: Cambridge, UK, 1970. [Google Scholar]
- Grant, V. Plant Speciation, 2nd ed.; Columbia University Press: New York, NY, USA, 1981. [Google Scholar]
- Husband, B.C. Chromosomal variation in plant evolution. Am. J. Bot. 2004, 91, 621–625. [Google Scholar] [CrossRef]
- Belyayev, A. Bursts of transposable elements as an evolutionary driving force. J. Evol. Biol. 2014, 27, 2573–2584. [Google Scholar] [CrossRef]
- Vít, P.; Krak, K.; Trávníček, P.; Douda, J.; Lomonosova, M.N.; Mandák, B. Genome size stability across Eurasian Chenopodium species (Amaranthaceae). Bot. J. Linn. Soc. 2016, 182, 637–649. [Google Scholar] [CrossRef]
- Novák, P.; Neumann, P.; Pech, J.; Steinhaisl, J.; Macas, J. RepeatExplorer: A Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 2013, 29, 792–793. [Google Scholar] [CrossRef]
- Noe, L.; Kucherov, G. YASS: Enhancing the sensitivity of DNA similarity search. Nucleic Acids Res. 2005, 33, W540–W543. [Google Scholar] [CrossRef]
- Vinga, S.; Almeida, J. Alignment-free sequence comparison-a review. Bioinformatics 2003, 19, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2003, 5, 113. [Google Scholar]
- Kalendar, R.; Tselykh, T.; Khassenov, B.; Ramanculov, E.M. Introduction on using the FastPCR software and the related Java web tools for PCR, in silico PCR, and oligonucleotide assembly and analysis. Met. Mol. Biol. 2017, 1620, 33–64. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Pijnacker, L.P.; Ferwerda, M.A. Giemsa C-banding of potato chromosomes. Can. J. Genet. Cytol. 1984, 26, 415–419. [Google Scholar] [CrossRef]
- Belyayev, A.; Raskina, O.; Nevo, E. Chromosomal distribution of reverse transcriptase containing retroelements in two Triticeae species. Chromosome Res. 2001, 9, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Vogelstein, B. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal. Biochem. 1983, 132, 6–13. [Google Scholar] [CrossRef]
- Reeves, A. MicroMeasure: A new computer program for the collection and analysis of cytogenetic data. Genome 2001, 44, 439–443. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Clade | ID Number | Locality |
|---|---|---|---|
| C. acuminatum | D | 429/3 | China, Burquin |
| C. bryoniifolium | A | 742/4 | Russian Federation, Nakhodka |
| C. ficifolium | B | 330/2 | Czech Republic, Slatina |
| C. iljinii | E | 433/9 | China, Hoboksar |
| C. pamiricum | E | 830/3C | Tajikistan, Gorno-Badakhshan |
| C. suecicum | B | 328/10 | Czech republic, Švermov |
| C. vulvaria | H | 771/1 | Iran, Shahr |
| Species | Chr. Numb. 2n | Chr. Size μm | Genome Size 2C Values Mb [26] | RE Clusters # | RE Singlets # | CficCl-61-40 % in Genome |
|---|---|---|---|---|---|---|
| C. acuminatum | 18 | 0.8–1.5 | 960 | 393251 | 34269 | 3.80 |
| C. bryoniifolium | 18 | 0.7–0.9 | 1200 | 307778 | 38905 | 2.25 |
| C. ficifolium | 18 | 1.5–4.5 | 1785 | 369861 | 20661 | 0.31 |
| C. iljinii | 18 | 1.7–3.3 | 1144 | 327760 | 82679 | 0.42 |
| C. pamiricum | 18 | 1.2–2.5 | 1154 | 249599 | 42427 | 0.25 |
| C. suecicum | 18 | 2.5–5.0 | 1775 | 369583 | 72167 | 0.27 |
| C. vulvaria | 18 | 1.5–2.0 | 924 | 542674 | 93278 | 0.79 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belyayev, A.; Josefiová, J.; Jandová, M.; Kalendar, R.; Krak, K.; Mandák, B. Natural History of a Satellite DNA Family: From the Ancestral Genome Component to Species-Specific Sequences, Concerted and Non-Concerted Evolution. Int. J. Mol. Sci. 2019, 20, 1201. https://doi.org/10.3390/ijms20051201
Belyayev A, Josefiová J, Jandová M, Kalendar R, Krak K, Mandák B. Natural History of a Satellite DNA Family: From the Ancestral Genome Component to Species-Specific Sequences, Concerted and Non-Concerted Evolution. International Journal of Molecular Sciences. 2019; 20(5):1201. https://doi.org/10.3390/ijms20051201
Chicago/Turabian StyleBelyayev, Alexander, Jiřina Josefiová, Michaela Jandová, Ruslan Kalendar, Karol Krak, and Bohumil Mandák. 2019. "Natural History of a Satellite DNA Family: From the Ancestral Genome Component to Species-Specific Sequences, Concerted and Non-Concerted Evolution" International Journal of Molecular Sciences 20, no. 5: 1201. https://doi.org/10.3390/ijms20051201
APA StyleBelyayev, A., Josefiová, J., Jandová, M., Kalendar, R., Krak, K., & Mandák, B. (2019). Natural History of a Satellite DNA Family: From the Ancestral Genome Component to Species-Specific Sequences, Concerted and Non-Concerted Evolution. International Journal of Molecular Sciences, 20(5), 1201. https://doi.org/10.3390/ijms20051201