Cardiotonic Steroids—A Possible Link Between High-Salt Diet and Organ Damage


Abstract
1. Salt and Blood Pressure
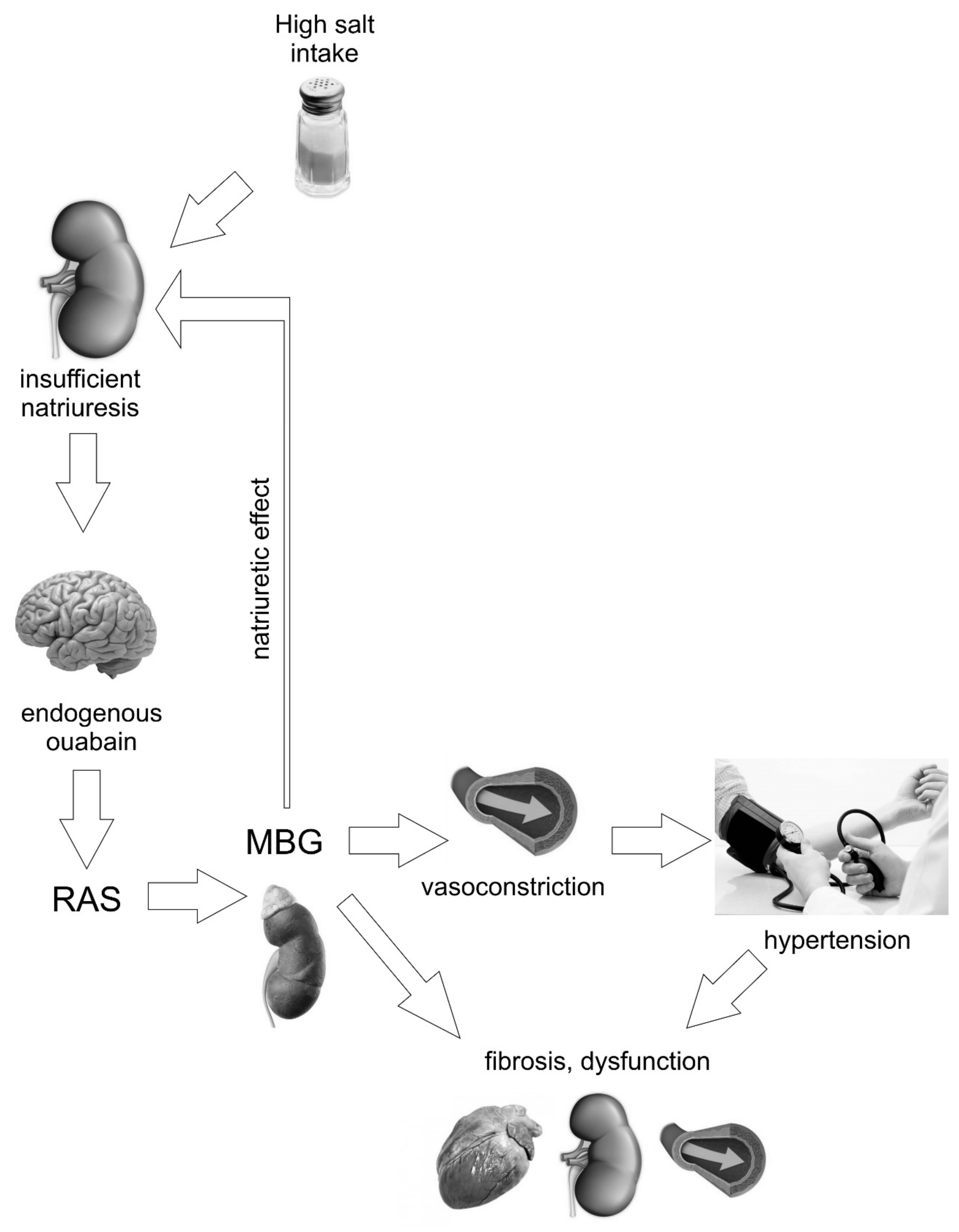
2. “Humoral Factor” Increases Blood Pressure in Response to Salt Intake
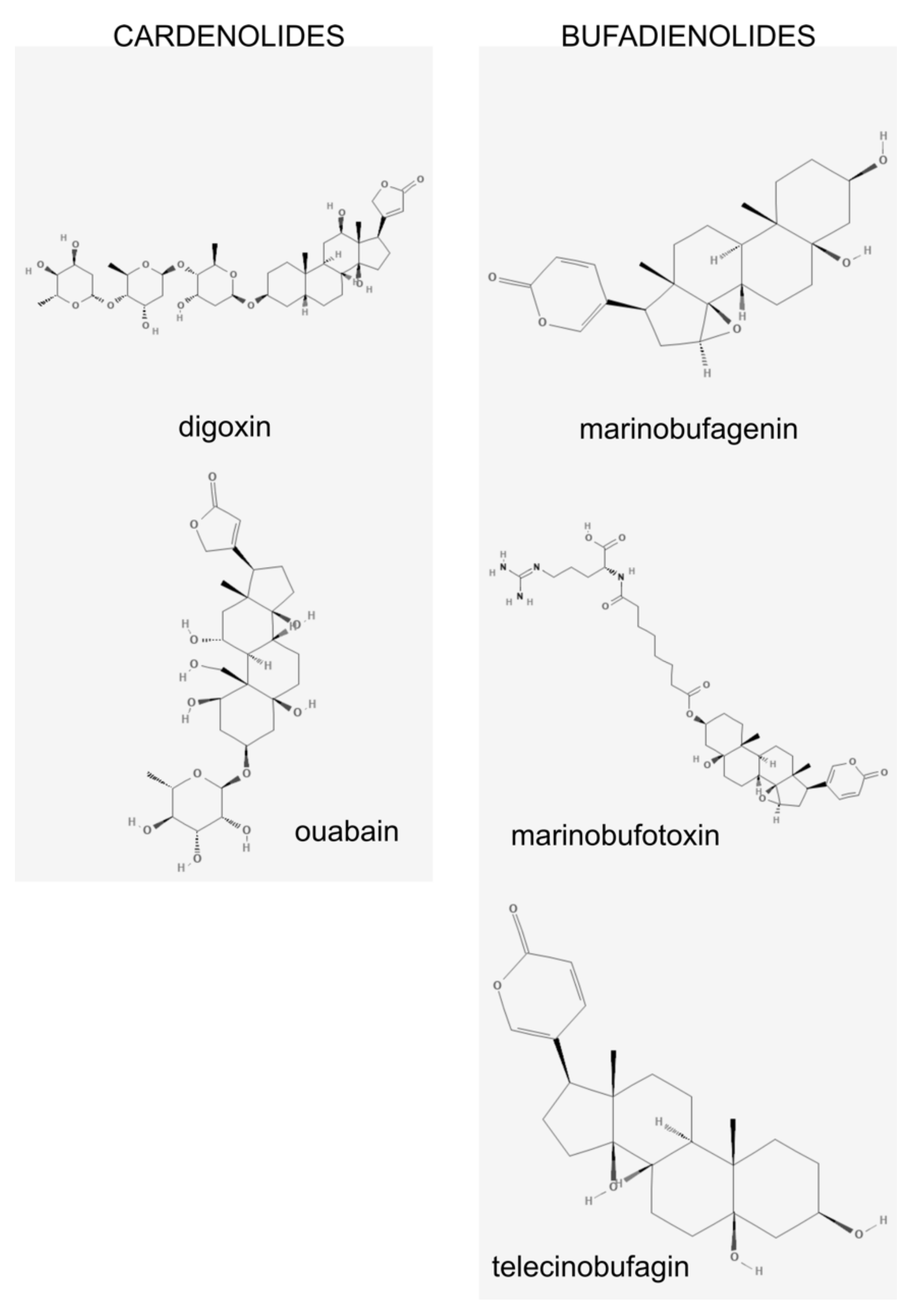
3. Na/K-ATPase: A Pump and a Receptor
4. Marinobufagenin is a Ligand for Na/K ATPase
5. Marinobufagenin and Fibrosis
6. Marinobufagenin and Cardiovascular Complications
7. Endogenous Ouabain and Other CTS
8. Summary
9. Future Perspectives
Funding
Conflicts of Interest
References
- GBD 2015 Risk Factors Collaborators. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990-2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1659–1724. [Google Scholar] [CrossRef]
- Intersalt: An international study of electrolyte excretion and blood pressure. Results for 24 hour urinary sodium and potassium excretion. Intersalt Cooperative Research Group. BMJ 1988, 297, 319–328. [CrossRef]
- Appel, L.J.; Moore, T.J.; Obarzanek, E.; Vollmer, W.M.; Svetkey, L.P.; Sacks, F.M.; Bray, G.A.; Vogt, T.M.; Cutler, J.A.; Windhauser, M.M.; et al. A clinical trial of the effects of dietary patterns on blood pressure. DASH Collaborative Research Group. N. Engl. J. Med. 1997, 336, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, M.H.; Miller, J.Z.; Luft, F.C.; Grim, C.E.; Fineberg, N.S. Definitions and characteristics of sodium sensitivity and blood pressure resistance. Hypertension 1986, 8, II127–II134. [Google Scholar] [CrossRef] [PubMed]
- De Wardener, H.E.; MacGregor, G.A. Sodium and blood pressure. Curr. Opin. Cardiol. 2002, 17, 360–367. [Google Scholar] [CrossRef]
- Bagrov, A.Y.; Lakatta, E.G. The dietary sodium-blood pressure plot “stiffens”. Hypertension 2004, 44, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Han, X.; Sun, N.; Chen, Y.; Jiang, S.; Li, M. Relationships between urinary electrolytes excretion and central hemodynamics, and arterial stiffness in hypertensive patients. Hypertens. Res. 2017, 40, 746–751. [Google Scholar] [CrossRef]
- Kaess, B.M.; Rong, J.; Larson, M.G.; Hamburg, N.M.; Vita, J.A.; Levy, D.; Benjamin, E.J.; Vasan, R.S.; Mitchell, G.F. Aortic stiffness, blood pressure progression, and incident hypertension. JAMA 2012, 308, 875–881. [Google Scholar] [CrossRef]
- Sutton-Tyrrell, K.; Najjar, S.S.; Boudreau, R.M.; Venkitachalam, L.; Kupelian, V.; Simonsick, E.M.; Havlik, R.; Lakatta, E.G.; Spurgeon, H.; Kritchevsky, S.; et al. Elevated aortic pulse wave velocity, a marker of arterial stiffness, predicts cardiovascular events in well-functioning older adults. Circulation 2005, 111, 3384–3390. [Google Scholar] [CrossRef]
- Jablonski, K.L.; Fedorova, O.V.; Racine, M.L.; Geolfos, C.J.; Gates, P.E.; Chonchol, M.; Fleenor, B.S.; Lakatta, E.G.; Bagrov, A.Y.; Seals, D.R. Dietary sodium restriction and association with urinary marinobufagenin, blood pressure, and aortic stiffness. Clin. J. Am. Soc. Nephrol. 2013, 8, 1952–1959. [Google Scholar] [CrossRef]
- Laragh, J.H.; Sealey, J.E.; Niarchos, A.P.; Pickering, T.G. The vasoconstriction-volume spectrum in normotension and in the pathogenesis of hypertension. Fed. Proc. 1982, 41, 2415–2423. [Google Scholar] [PubMed]
- Blaustein, M.P.; Hamlyn, J.M. Signaling mechanisms that link salt retention to hypertension: Endogenous ouabain, the Na(+) pump, the Na(+)/Ca(2+) exchanger and TRPC proteins. Biochim. Biophys. Acta 2010, 1802, 1219–1229. [Google Scholar] [CrossRef] [PubMed]
- Titze, J. Sodium balance is not just a renal affair. Curr. Opin. Nephrol. Hypertens. 2014, 23, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Oberleithner, H.; Kusche-Vihrog, K.; Schillers, H. Endothelial cells as vascular salt sensors. Kidney Int. 2010, 77, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Orlov, S.N.; Mongin, A.A. Salt-sensing mechanisms in blood pressure regulation and hypertension. Am. J. Physiol. 2007, 293, H2039–H2053. [Google Scholar] [CrossRef] [PubMed]
- Dahl, L.K. Possible role of chronic excess salt consumption in the pathogenesis of essential hypertension. Am. J. Cardiol. 1961, 8, 571–575. [Google Scholar] [CrossRef]
- Dahl, L.K.; Knudsen, K.D.; Iwai, J. Humoral transmission of hypertension: Evidence from parabiosis. Circ. Res. 1969, 24, 21–33. [Google Scholar]
- de Wardener, H.E.; Clarkson, E.M. Concept of natriuretic hormone. Physiol. Rev. 1985, 65, 658–759. [Google Scholar] [CrossRef] [PubMed]
- Bagrov, A.Y.; Shapiro, J.I.; Fedorova, O.V. Endogenous cardiotonic steroids: Physiology, pharmacology, and novel therapeutic targets. Pharmacol. Rev. 2009, 61, 9–38. [Google Scholar] [CrossRef] [PubMed]
- Fisch, C. William Withering: An account of the foxglove and some of its medical uses 1785-1985. J. Am. Coll. Cardiol. 1985, 5, 1A–2A. [Google Scholar] [CrossRef]
- Schoner, W.; Scheiner-Bobis, G. Endogenous and exogenous cardiac glycosides: Their roles in hypertension, salt metabolism, and cell growth. Am. J. Physiol. Cell Physiol. 2007, 293, C509–C536. [Google Scholar] [CrossRef] [PubMed]
- Blanco, G.; Mercer, R.W. Isozymes of the Na-K-ATPase: Heterogeneity in structure, diversity in function. Am. J. Physiol. 1998, 275, F633–F650. [Google Scholar] [CrossRef] [PubMed]
- Muller-Ehmsen, J.; Juvvadi, P.; Thompson, C.B.; Tumyan, L.; Croyle, M.; Lingrel, J.B.; Schwinger, R.H.; McDonough, A.A.; Farley, R.A. Ouabain and substrate affinities of human Na(+)-K(+)-ATPase alpha(1)beta(1), alpha(2)beta(1), and alpha(3)beta(1) when expressed separately in yeast cells. Am. J. Physiol. Cell Physiol. 2001, 281, C1355–C1364. [Google Scholar] [CrossRef] [PubMed]
- Ferrandi, M.; Molinari, I.; Barassi, P.; Minotti, E.; Bianchi, G.; Ferrari, P. Organ hypertrophic signaling within caveolae membrane subdomains triggered by ouabain and antagonized by PST 2238. J. Biol. Chem. 2004, 279, 33306–33314. [Google Scholar] [CrossRef]
- Khalaf, F.K.; Dube, P.; Mohamed, A.; Tian, J.; Malhotra, D.; Haller, S.T.; Kennedy, D.J. Cardiotonic Steroids and the Sodium Trade Balance: New Insights into Trade-Off Mechanisms Mediated by the Na(+)/K(+)-ATPase. Int. J. Mol. Sci. 2018, 19, 2576. [Google Scholar] [CrossRef]
- Liu, J.; Xie, Z.J. The sodium pump and cardiotonic steroids-induced signal transduction protein kinases and calcium-signaling microdomain in regulation of transporter trafficking. Biochim. Biophys. Acta 2010, 1802, 1237–1245. [Google Scholar] [CrossRef]
- Haas, M.; Wang, H.; Tian, J.; Xie, Z. Src-mediated inter-receptor cross-talk between the Na+/K+-ATPase and the epidermal growth factor receptor relays the signal from ouabain to mitogen-activated protein kinases. J. Biol. Chem. 2002, 277, 18694–18702. [Google Scholar] [CrossRef]
- Mohammadi, K.; Kometiani, P.; Xie, Z.; Askari, A. Role of protein kinase C in the signal pathways that link Na+/K+-ATPase to ERK1/2. J. Biol. Chem. 2001, 276, 42050–42056. [Google Scholar] [CrossRef]
- Liu, L.; Zhao, X.; Pierre, S.V.; Askari, A. Association of PI3K-Akt signaling pathway with digitalis-induced hypertrophy of cardiac myocytes. Am. J. Physiol. Cell Physiol. 2007, 293, C1489–C1497. [Google Scholar] [CrossRef]
- Tian, J.; Li, X.; Liang, M.; Liu, L.; Xie, J.X.; Ye, Q.; Kometiani, P.; Tillekeratne, M.; Jin, R.; Xie, Z. Changes in sodium pump expression dictate the effects of ouabain on cell growth. J. Biol. Chem. 2009, 284, 14921–14929. [Google Scholar] [CrossRef]
- Bagrov, A.Y.; Shapiro, J.I. Endogenous digitalis: Pathophysiologic roles and therapeutic applications. Nat. Clin. Pract. 2008, 4, 378–392. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, O.V.; Agalakova, N.I.; Talan, M.I.; Lakatta, E.G.; Bagrov, A.Y. Brain ouabain stimulates peripheral marinobufagenin via angiotensin II signalling in NaCl-loaded Dahl-S rats. J. Hypertens. 2005, 23, 1515–1523. [Google Scholar] [CrossRef] [PubMed]
- Blaustein, M.P. Sodium ions, calcium ions, blood pressure regulation, and hypertension: A reassessment and a hypothesis. Am. J. Physiol. 1977, 232, C165–C173. [Google Scholar] [CrossRef] [PubMed]
- Periyasamy, S.M.; Liu, J.; Tanta, F.; Kabak, B.; Wakefield, B.; Malhotra, D.; Kennedy, D.J.; Nadoor, A.; Fedorova, O.V.; Gunning, W.; et al. Salt loading induces redistribution of the plasmalemmal Na/K-ATPase in proximal tubule cells. Kidney Int. 2005, 67, 1868–1877. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liang, M.; Liu, L.; Malhotra, D.; Xie, Z.; Shapiro, J.I. Ouabain-induced endocytosis of the plasmalemmal Na/K-ATPase in LLC-PK1 cells requires caveolin-1. Kidney Int. 2005, 67, 1844–1854. [Google Scholar] [CrossRef] [PubMed]
- Elkareh, J.; Periyasamy, S.M.; Shidyak, A.; Vetteth, S.; Schroeder, J.; Raju, V.; Hariri, I.M.; El-Okdi, N.; Gupta, S.; Fedorova, L.; et al. Marinobufagenin induces increases in procollagen expression in a process involving protein kinase C and Fli-1: Implications for uremic cardiomyopathy. Am. J. Physiol. Renal. Physiol. 2009, 296, F1219–F1226. [Google Scholar] [CrossRef] [PubMed]
- Elkareh, J.; Kennedy, D.J.; Yashaswi, B.; Vetteth, S.; Shidyak, A.; Kim, E.G.; Smaili, S.; Periyasamy, S.M.; Hariri, I.M.; Fedorova, L.; et al. Marinobufagenin stimulates fibroblast collagen production and causes fibrosis in experimental uremic cardiomyopathy. Hypertension 2007, 49, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.C.; Burrell, L.M.; Black, M.J.; Wu, L.L.; Dilley, R.J.; Cooper, M.E.; Johnston, C.I. Salt induces myocardial and renal fibrosis in normotensive and hypertensive rats. Circulation 1998, 98, 2621–2628. [Google Scholar] [CrossRef]
- Ying, W.Z.; Aaron, K.; Sanders, P.W. Mechanism of dietary salt-mediated increase in intravascular production of TGF-beta1. Am. J. Physiol. Renal. Physiol. 2008, 295, F406–F414. [Google Scholar] [CrossRef]
- Ying, W.Z.; Aaron, K.; Wang, P.X.; Sanders, P.W. Potassium inhibits dietary salt-induced transforming growth factor-beta production. Hypertension 2009, 54, 1159–1163. [Google Scholar] [CrossRef]
- Fedorova, O.V.; Anderson, D.E.; Lakatta, E.G.; Bagrov, A.Y. Interaction of NaCl and behavioral stress on endogenous sodium pump ligands in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 281, R352–R358. [Google Scholar] [CrossRef] [PubMed]
- Grigorova, Y.N.; Juhasz, O.; Zernetkina, V.; Fishbein, K.W.; Lakatta, E.G.; Fedorova, O.V.; Bagrov, A.Y. Aortic Fibrosis, Induced by High Salt Intake in the Absence of Hypertensive Response, is Reduced by a Monoclonal Antibody to Marinobufagenin. Am. J. Hypertens. 2016, 29, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Grigorova, Y.N.; Wei, W.; Petrashevskaya, N.; Zernetkina, V.; Juhasz, O.; Fenner, R.; Gilbert, C.; Lakatta, E.G.; Shapiro, J.I.; Bagrov, A.Y.; et al. Dietary Sodium Restriction Reduces Arterial Stiffness, Vascular TGF-beta-Dependent Fibrosis and Marinobufagenin in Young Normotensive Rats. Int. J. Mol. Sci. 2018, 19, 3168. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, O.V.; Kolodkin, N.I.; Agalakova, N.I.; Namikas, A.R.; Bzhelyansky, A.; St-Louis, J.; Lakatta, E.G.; Bagrov, A.Y. Antibody to marinobufagenin lowers blood pressure in pregnant rats on a high NaCl intake. J. Hypertens. 2005, 23, 835–842. [Google Scholar] [CrossRef]
- La, J.; Reed, E.B.; Koltsova, S.; Akimova, O.; Hamanaka, R.B.; Mutlu, G.M.; Orlov, S.N.; Dulin, N.O. Regulation of myofibroblast differentiation by cardiac glycosides. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L815–L823. [Google Scholar] [CrossRef] [PubMed]
- La, J.; Reed, E.; Chan, L.; Smolyaninova, L.V.; Akomova, O.A.; Mutlu, G.M.; Orlov, S.N.; Dulin, N.O. Downregulation of TGF-beta Receptor-2 Expression and Signaling through Inhibition of Na/K-ATPase. PLoS ONE 2016, 11, e0168363. [Google Scholar] [CrossRef] [PubMed]
- Piecha, G.; Kujawa-Szewieczek, A.; Kuczera, P.; Skiba, K.; Sikora-Grabka, E.; Wiecek, A. Plasma marinobufagenin immunoreactivity in patients with chronic kidney disease: A case control study. Am. J. Physiol. Renal. Physiol. 2018, 315, F637–F643. [Google Scholar] [CrossRef]
- Strauss, M.; Smith, W.; Wei, W.; Bagrov, A.Y.; Fedorova, O.V.; Schutte, A.E. Large artery stiffness is associated with marinobufagenin in young adults: The African-PREDICT study. J. Hypertens. 2018, 36, 2333–2339. [Google Scholar] [CrossRef]
- Strauss, M.; Smith, W.; Kruger, R.; van der Westhuizen, B.; Schutte, A.E. Large artery stiffness is associated with salt intake in young healthy black but not white adults: The African-PREDICT study. Eur. J. Nutr. 2018, 57, 2649–2656. [Google Scholar] [CrossRef]
- Strauss, M.; Smith, W.; Kruger, R.; Wei, W.; Fedorova, O.V.; Schutte, A.E. Marinobufagenin and left ventricular mass in young adults: The African-PREDICT study. Eur. J. Prev. Cardiol. 2018, 25, 1587–1595. [Google Scholar] [CrossRef]
- Koleganova, N.; Piecha, G.; Ritz, E.; Becker, L.E.; Muller, A.; Weckbach, M.; Nyengaard, J.R.; Schirmacher, P.; Gross-Weissmann, M.L. Both high and low maternal salt intake in pregnancy alter kidney development in the offspring. Am. J. Physiol. Renal. Physiol. 2011, 301, F344–F354. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.E.; Fedorova, O.V.; Morrell, C.H.; Longo, D.L.; Kashkin, V.A.; Metzler, J.D.; Bagrov, A.Y.; Lakatta, E.G. Endogenous sodium pump inhibitors and age-associated increases in salt sensitivity of blood pressure in normotensives. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R1248–R1254. [Google Scholar] [CrossRef] [PubMed]
- Manunta, P.; Messaggio, E.; Ballabeni, C.; Sciarrone, M.T.; Lanzani, C.; Ferrandi, M.; Hamlyn, J.M.; Cusi, D.; Galletti, F.; Bianchi, G. Plasma ouabain-like factor during acute and chronic changes in sodium balance in essential hypertension. Hypertension 2001, 38, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, O.V.; Lakatta, E.G.; Bagrov, A.Y. Endogenous Na,K pump ligands are differentially regulated during acute NaCl loading of Dahl rats. Circulation 2000, 102, 3009–3014. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, P.; Ferrandi, M.; Tripodi, G.; Torielli, L.; Padoani, G.; Minotti, E.; Melloni, P.; Bianchi, G. PST 2238: A new antihypertensive compound that modulates Na,K-ATPase in genetic hypertension. J. Pharmacol. Exp. Ther. 1999, 288, 1074–1083. [Google Scholar]
- Staessen, J.A.; Thijs, L.; Stolarz-Skrzypek, K.; Bacchieri, A.; Barton, J.; Espositi, E.D.; de Leeuw, P.W.; Dluzniewski, M.; Glorioso, N.; Januszewicz, A.; et al. Main results of the ouabain and adducin for Specific Intervention on Sodium in Hypertension Trial (OASIS-HT): A randomized placebo-controlled phase-2 dose-finding study of rostafuroxin. Trials 2011, 12, 13. [Google Scholar] [CrossRef] [PubMed]
- Lewis, L.K.; Yandle, T.G.; Hilton, P.J.; Jensen, B.P.; Begg, E.J.; Nicholls, M.G. Endogenous ouabain is not ouabain. Hypertension 2014, 64, 680–683. [Google Scholar] [CrossRef]
- Baecher, S.; Kroiss, M.; Fassnacht, M.; Vogeser, M. No endogenous ouabain is detectable in human plasma by ultra-sensitive UPLC-MS/MS. Clin. Chim. Acta 2014, 431, 87–92. [Google Scholar] [CrossRef]
- Takahashi, H.; Yoshika, M.; Komiyama, Y.; Nishimura, M. The central mechanism underlying hypertension: A review of the roles of sodium ions, epithelial sodium channels, the renin-angiotensin-aldosterone system, oxidative stress and endogenous digitalis in the brain. Hypertens. Res. 2011, 34, 1147–1160. [Google Scholar] [CrossRef]
- Yoshika, M.; Komiyama, Y.; Takahashi, H. Isolation of marinobufotoxin from the supernatant of cultured PC12 cells. Clin. Exp. Pharmacol. Physiol. 2011, 38, 334–337. [Google Scholar] [CrossRef]
- Komiyama, Y.; Nishimura, N.; Munakata, M.; Mori, T.; Okuda, K.; Nishino, N.; Hirose, S.; Kosaka, C.; Masuda, M.; Takahashi, H. Identification of endogenous ouabain in culture supernatant of PC12 cells. J. Hypertens. 2001, 19, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Goto, A.; Ishiguro, T.; Yamada, K.; Ishii, M.; Yoshioka, M.; Eguchi, C.; Shimora, M.; Sugimoto, T. Isolation of a urinary digitalis-like factor indistinguishable from digoxin. Biochem. Biophys. Res. Commun. 1990, 173, 1093–1101. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paczula, A.; Wiecek, A.; Piecha, G. Cardiotonic Steroids—A Possible Link Between High-Salt Diet and Organ Damage. Int. J. Mol. Sci. 2019, 20, 590. https://doi.org/10.3390/ijms20030590
Paczula A, Wiecek A, Piecha G. Cardiotonic Steroids—A Possible Link Between High-Salt Diet and Organ Damage. International Journal of Molecular Sciences. 2019; 20(3):590. https://doi.org/10.3390/ijms20030590
Chicago/Turabian StylePaczula, Aneta, Andrzej Wiecek, and Grzegorz Piecha. 2019. "Cardiotonic Steroids—A Possible Link Between High-Salt Diet and Organ Damage" International Journal of Molecular Sciences 20, no. 3: 590. https://doi.org/10.3390/ijms20030590
APA StylePaczula, A., Wiecek, A., & Piecha, G. (2019). Cardiotonic Steroids—A Possible Link Between High-Salt Diet and Organ Damage. International Journal of Molecular Sciences, 20(3), 590. https://doi.org/10.3390/ijms20030590