Comparative Genome-wide Analysis and Expression Profiling of Histone Acetyltransferase (HAT) Gene Family in Response to Hormonal Applications, Metal and Abiotic Stresses in Cotton

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Identification of HATs in Cotton
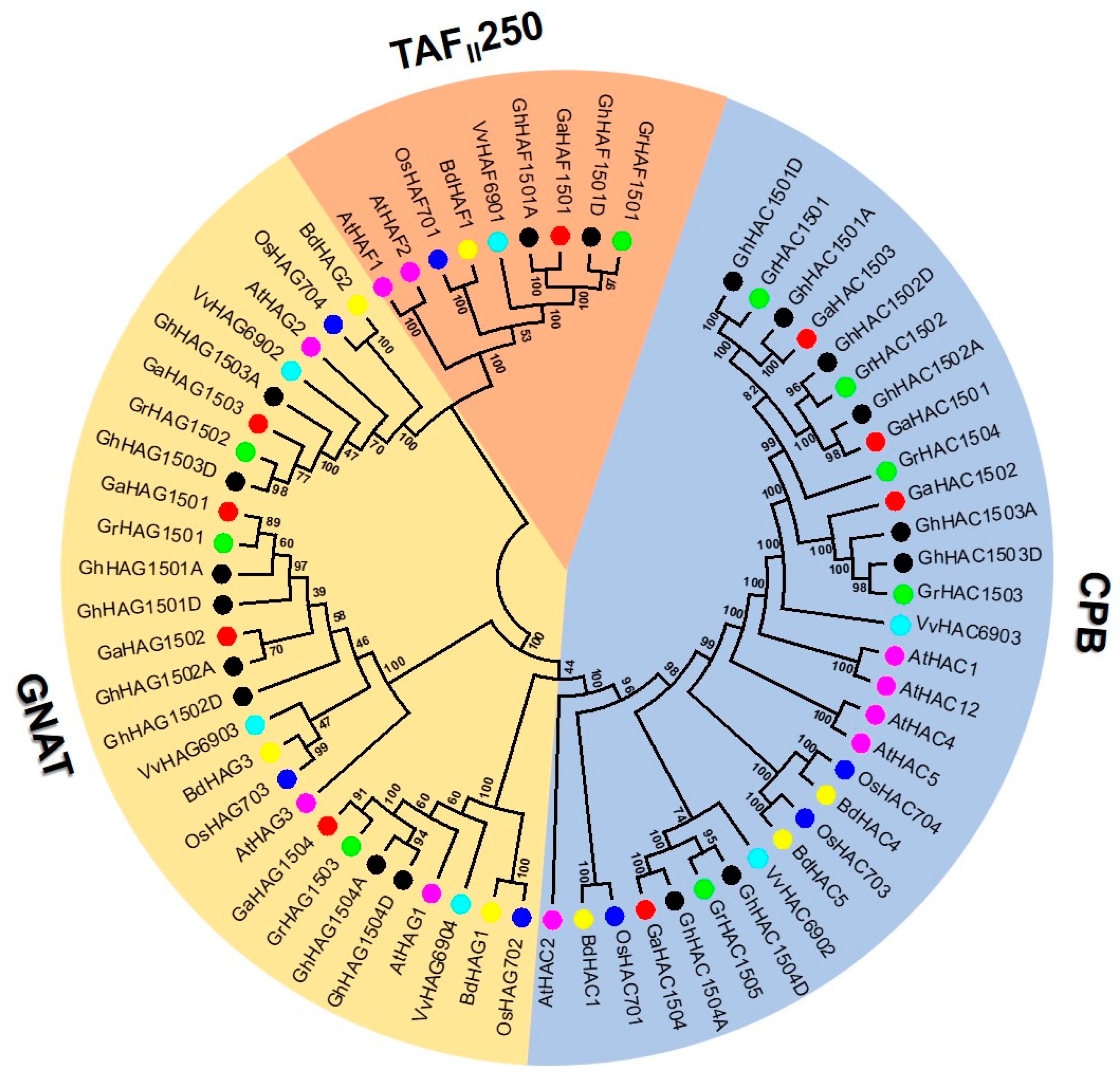
2.2. Phylogenetic Analysis of the HAT Gene Family
2.3. Phylogenetic Tree Construction, Conserved Motif and Gene Structure Analysis of Cotton HATs Genes
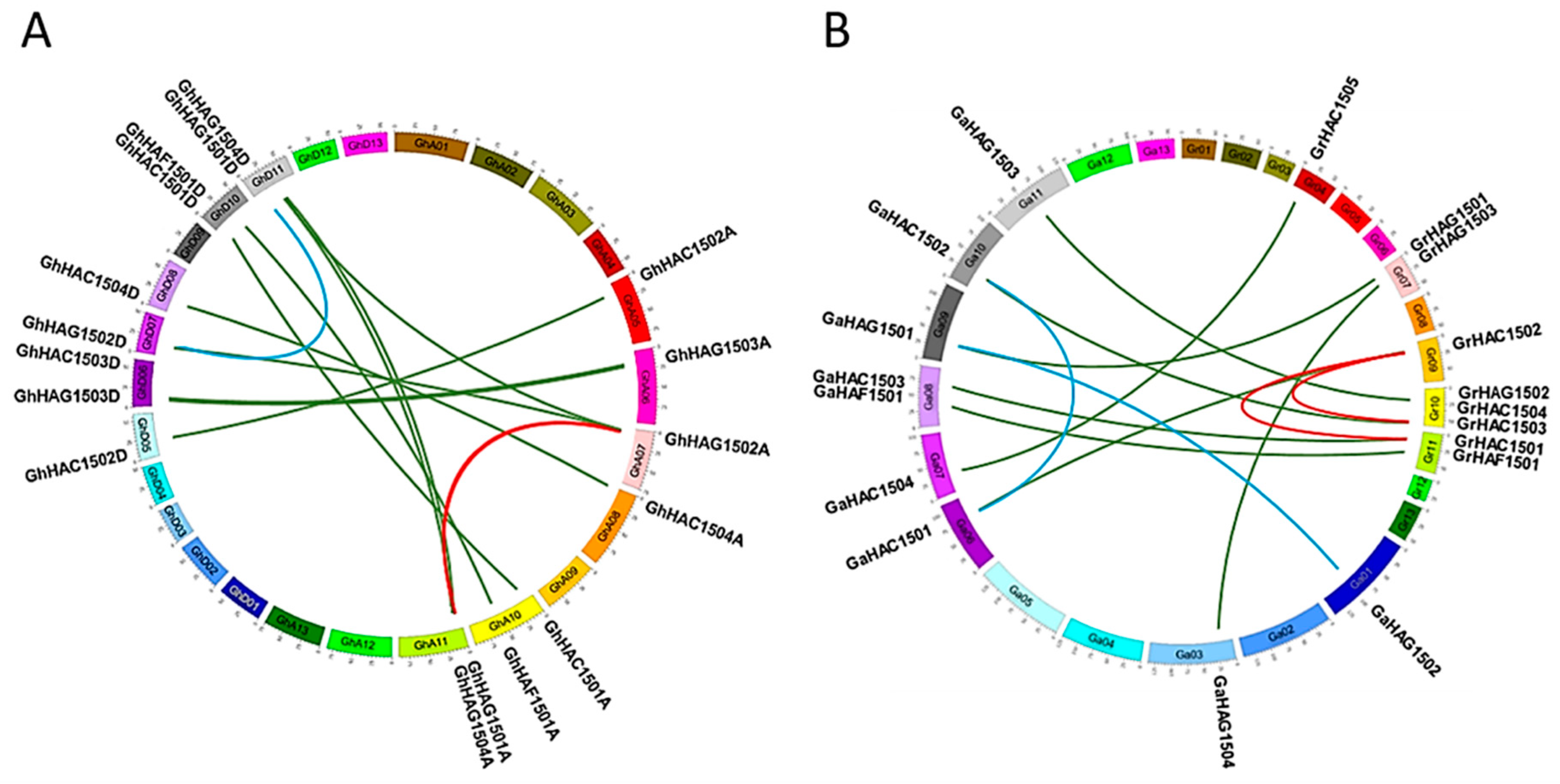
2.4. Chromosomal Distribution and Duplication Analysis of HATs
2.5. Putative Cis-Elements in the Promoter Regions of GhHATs
2.6. Gene Expression Analysis
2.6.1. Expression Analysis of GhHATs in Different Tissues, Developmental Stages and Multiple Abiotic Stresses by RNA-sequencing
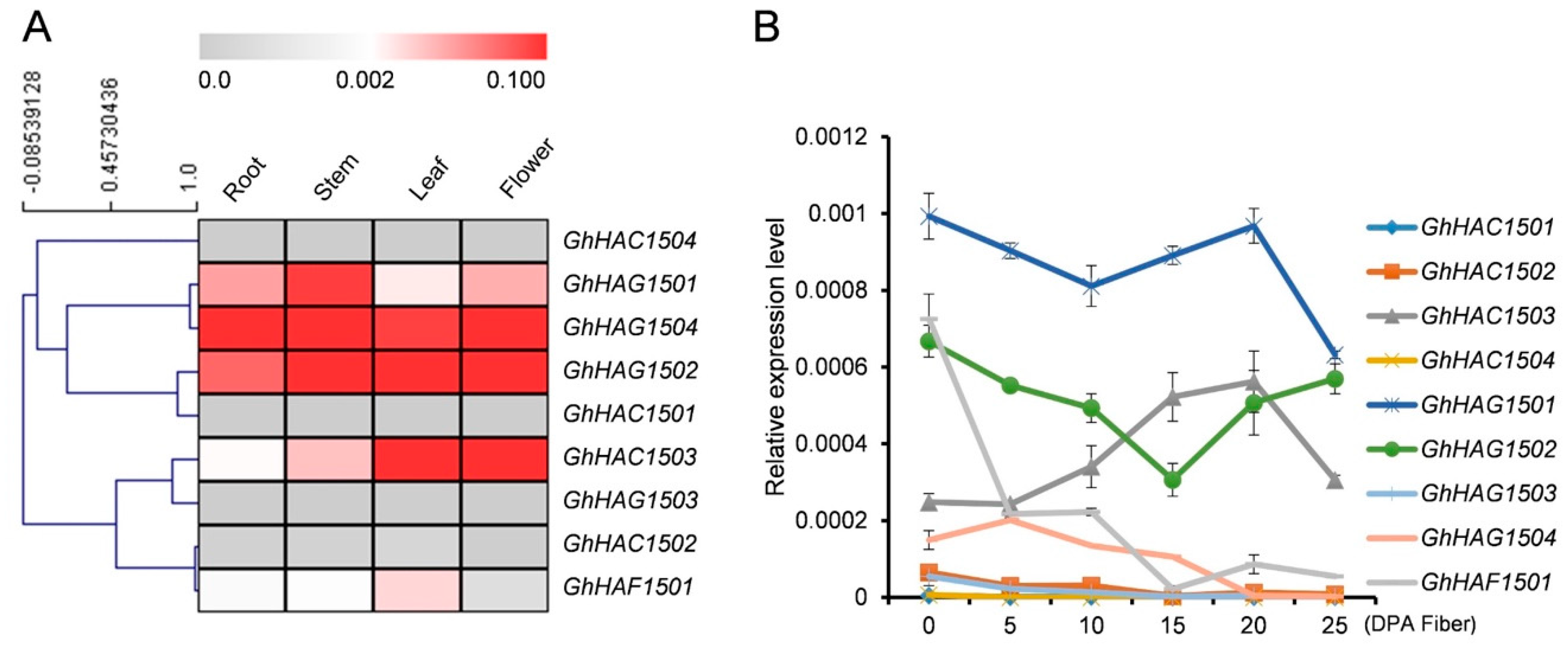
2.6.2. Expression Pattern of GhHATs Genes by qPCR
Tissue Specific Expression Patterns of HATs
Expression of HATs at Different Fiber Development Stages
Expression of HATs in Response to Heavy Metals and Abiotic Stresses
Expression of HATs in Response to MMS
Expression of HATs in Response to Phytohormones
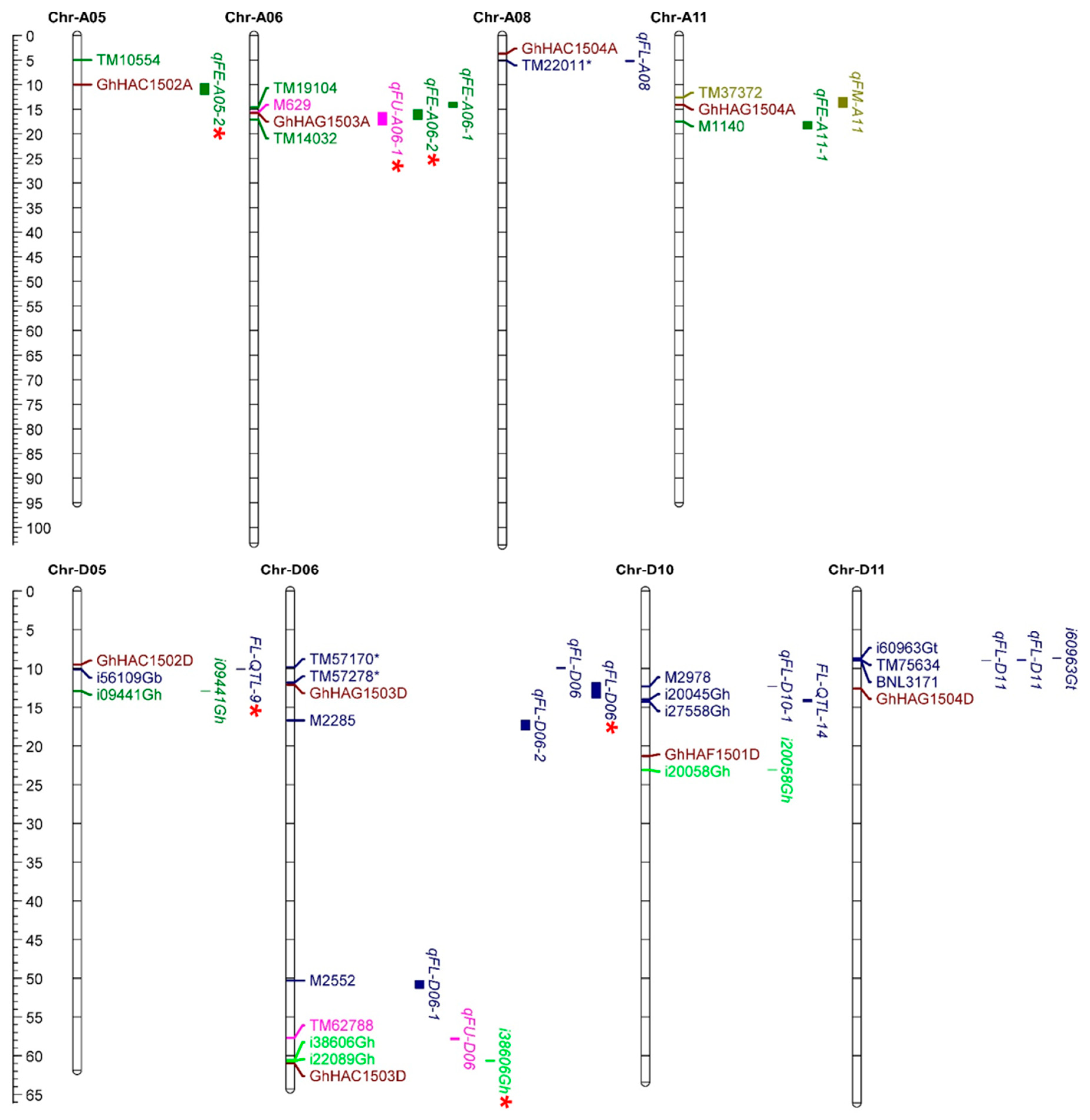
2.7. Co-Localization of HATs with QTLs of Fiber Development
3. Discussion
4. Materials and Methods
4.1. Identification of HATs Gene Family
4.2. Analysis of Chromosomal Location and Gene Duplication
4.3. Sequence Alignment and Phylogenetic Analyses
4.4. Gene Structure, Protein Motif, and Promoter Cis-Element Analysis
4.5. Transcriptome Data Analysis and Gene Expression Heatmap
4.6. Plant Materials, Stress Treatments, and qRT-PCR
4.7. Co-localization of HATs with Fiber Related QTLs
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Berr, A.; Shafiq, S.; Shen, W.-H. Histone modifications in transcriptional activation during plant development. Biochim. Et Biophys. Acta (BBA)-Gene Regul. Mech. 2011, 1809, 567–576. [Google Scholar] [CrossRef]
- Ho, L.; Crabtree, G.R. Chromatin remodelling during development. Nature 2010, 463, 474. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.J.; Wang, Z. Readout of epigenetic modifications. Annu. Rev. Biochem. 2013, 82, 81–118. [Google Scholar] [CrossRef] [PubMed]
- Earley, K.W.; Shook, M.S.; Brower-Toland, B.; Hicks, L.; Pikaard, C.S. In vitro specificities of Arabidopsis co-activator histone acetyltransferases: Implications for histone hyperacetylation in gene activation. Plant J. 2007, 52, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Sridhar, V.V.; Zhu, J.; Kapoor, A.; Zhu, J.-K. Distinctive core histone post-translational modification patterns in Arabidopsis thaliana. PLoS ONE 2007, 2, e1210. [Google Scholar] [CrossRef] [PubMed]
- Sterner, D.E.; Berger, S.L. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 2000, 64, 435–459. [Google Scholar] [CrossRef]
- Pandey, R.; MuÈller, A.; Napoli, C.A.; Selinger, D.A.; Pikaard, C.S.; Richards, E.J.; Bender, J.; Mount, D.W.; Jorgensen, R.A. Analysis of histone acetyltransferase and histone deacetylase families of Arabidopsis thaliana suggests functional diversification of chromatin modification among multicellular eukaryotes. Nucleic Acids Res. 2002, 30, 5036–5055. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Luo, M.; Zhang, W.; Zhao, J.; Zhang, J.; Wu, K.; Tian, L.; Duan, J. Histone acetyltransferases in rice (Oryza sativa L.): Phylogenetic analysis, subcellular localization and expression. BMC Plant Biol. 2012, 12, 145. [Google Scholar] [CrossRef]
- Aquea, F.; Timmermann, T.; Arce-Johnson, P. Analysis of histone acetyltransferase and deacetylase families of Vitis vinifera. Plant Physiol. Biochem. 2010, 48, 194–199. [Google Scholar] [CrossRef]
- Xu, J.; Xu, H.; Liu, Y.; Wang, X.; Xu, Q.; Deng, X. Genome-wide identification of sweet orange (Citrus sinensis) histone modification gene families and their expression analysis during the fruit development and fruit-blue mold infection process. Front. Plant Sci. 2015, 6, 607. [Google Scholar] [CrossRef]
- Deng, W.; Liu, C.; Pei, Y.; Deng, X.; Niu, L.; Cao, X. Involvement of the Histone Acetyltransferase AtHAC1 in the Regulation of Flowering Time via Repression of FLOWERING LOCUS C in Arabidopsis. Plant Physiol. 2007, 143, 1660–1668. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, C.; Bergounioux, C.; Domenichini, S.; Delarue, M.; Zhou, D.-X. Arabidopsis histone acetyltransferase AtGCN5 regulates the floral meristem activity through the WUSCHEL/AGAMOUS pathway. J. Biol. Chem. 2003, 278, 28246–28251. [Google Scholar] [CrossRef] [PubMed]
- Han, S.K.; Song, J.D.; Noh, Y.S.; Noh, B. Role of plant CBP/p300-like genes in the regulation of flowering time. Plant J. 2007, 49, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Kornet, N.; Scheres, B. Members of the GCN5 histone acetyltransferase complex regulate PLETHORA-mediated root stem cell niche maintenance and transit amplifying cell proliferation in Arabidopsis. Plant Cell 2009, 21, 1070–1079. [Google Scholar] [CrossRef]
- Latrasse, D.; Benhamed, M.; Henry, Y.; Domenichini, S.; Kim, W.; Zhou, D.-X.; Delarue, M. The MYST histone acetyltransferases are essential for gametophyte development in Arabidopsis. BMC Plant Biol. 2008, 8, 121. [Google Scholar] [CrossRef]
- Sokol, A.; Kwiatkowska, A.; Jerzmanowski, A.; Prymakowska-Bosak, M. Up-regulation of stress-inducible genes in tobacco and Arabidopsis cells in response to abiotic stresses and ABA treatment correlates with dynamic changes in histone H3 and H4 modifications. Planta 2007, 227, 245–254. [Google Scholar] [CrossRef]
- Stockinger, E.J.; Mao, Y.; Regier, M.K.; Triezenberg, S.J.; Thomashow, M.F. Transcriptional adaptor and histone acetyltransferase proteins in Arabidopsis and their interactions with CBF1, a transcriptional activator involved in cold-regulated gene expression. Nucleic Acids Res. 2001, 29, 1524–1533. [Google Scholar] [CrossRef]
- Bharti, K.; von Koskull-Döring, P.; Bharti, S.; Kumar, P.; Tintschl-Körbitzer, A.; Treuter, E.; Nover, L. Tomato heat stress transcription factor HsfB1 represents a novel type of general transcription coactivator with a histone-like motif interacting with the plant CREB binding protein ortholog HAC1. Plant Cell 2004, 16, 1521–1535. [Google Scholar] [CrossRef]
- Benhamed, M.; Bertrand, C.; Servet, C.; Zhou, D.-X. Arabidopsis GCN5, HD1, and TAF1/HAF2 interact to regulate histone acetylation required for light-responsive gene expression. Plant Cell 2006, 18, 2893–2903. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, H.; Jablonowski, D.; Zhou, X.; Ren, X.; Hong, X.; Schaffrath, R.; Zhu, J.-K.; Gong, Z. Mutations in ABO1/ELO2, a subunit of holo-Elongator, increase abscisic acid sensitivity and drought tolerance in Arabidopsis thaliana. Mol. Cell. Biol. 2006, 26, 6902–6912. [Google Scholar] [CrossRef]
- Nelissen, H.; De Groeve, S.; Fleury, D.; Neyt, P.; Bruno, L.; Bitonti, M.B.; Vandenbussche, F.; Van Der Straeten, D.; Yamaguchi, T.; Tsukaya, H. Plant Elongator regulates auxin-related genes during RNA polymerase II transcription elongation. Proc. Natl. Acad. Sci. USA 2010, 107, 1678–1683. [Google Scholar] [CrossRef] [PubMed]
- Cao, X. Whole genome sequencing of cotton—A new chapter in cotton genomics. Sci. China Life Sci. 2015, 58, 515–516. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J.; Scheffler, B.E.; Dennis, E.; Triplett, B.A.; Zhang, T.; Guo, W.; Chen, X.; Stelly, D.M.; Rabinowicz, P.D.; Town, C.D. Toward sequencing cotton (Gossypium) genomes. Plant Physiol. 2007, 145, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.-X.; Wright, R.J.; El-Zik, K.M.; Paterson, A.H. Polyploid formation created unique avenues for response to selection in Gossypium (cotton). Proc. Natl. Acad. Sci. USA 1998, 95, 4419–4424. [Google Scholar] [CrossRef]
- Wendel, J.F. New World tetraploid cottons contain Old World cytoplasm. Proc. Natl. Acad. Sci. USA 1989, 86, 4132–4136. [Google Scholar] [CrossRef]
- Wang, R.; Ji, S.; Zhang, P.; Meng, Y.; Wang, Y.; Chen, B.; Zhou, Z. Drought Effects on Cotton Yield and Fiber Quality on Different Fruiting Branches. Crop Sci. 2016, 56, 1265–1276. [Google Scholar] [CrossRef]
- Imran, M.; Shakeel, A.; Farooq, J.; Saeed, A.; Farooq, A.; Riaz, M. Genetic studies of fiber quality parameter and earliness related traits in upland cotton (Gossypium hirsutum L.). Adv. Agric. Bot. 2011, 3, 151–159. [Google Scholar]
- Imran, M.; Shakeel, A.; Azhar, F.; Farooq, J.; Saleem, M.; Saeed, A.; Nazeer, W.; Riaz, M.; Naeem, M.; Javaid, A. Combining ability analysis for within-boll yield components in upland cotton (Gossypium hirsutum L.). Genet. Mol. Res. 2012, 11, 2790–2800. [Google Scholar] [CrossRef]
- Tian, L.; Fong, M.P.; Wang, J.J.; Wei, N.E.; Jiang, H.; Doerge, R.; Chen, Z.J. Reversible histone acetylation and deacetylation mediate genome-wide, promoter-dependent and locus-specific changes in gene expression during plant development. Genetics 2005, 169, 337–345. [Google Scholar] [CrossRef]
- Chen, Z.J.; Tian, L. Roles of dynamic and reversible histone acetylation in plant development and polyploidy. Biochim. Et Biophys. Acta (BBA)-Gene Struct. Expr. 2007, 1769, 295–307. [Google Scholar] [CrossRef]
- Pavangadkar, K.; Thomashow, M.F.; Triezenberg, S.J. Histone dynamics and roles of histone acetyltransferases during cold-induced gene regulation in Arabidopsis. Plant Mol. Biol. 2010, 74, 183–200. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yan, S.; Zhao, L.; Tan, J.; Zhang, Q.; Gao, F.; Wang, P.; Hou, H.; Li, L. Histone acetylation associated up-regulation of the cell wall related genes is involved in salt stress induced maize root swelling. BMC Plant Biol. 2014, 14, 105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Fan, G.; Lu, C.; Xiao, G.; Zou, C.; Kohel, R.J.; Ma, Z.; Shang, H.; Ma, X.; Wu, J. Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 2015, 33, 524. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, Z.; Li, F.; Ye, W.; Wang, J.; Song, G.; Yue, Z.; Cong, L.; Shang, H.; Zhu, S. The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 2012, 44, 1098. [Google Scholar] [CrossRef]
- Gamsjaeger, R.; Liew, C.K.; Loughlin, F.E.; Crossley, M.; Mackay, J.P. Sticky fingers: Zinc-fingers as protein-recognition motifs. Trends Biochem. Sci. 2007, 32, 63–70. [Google Scholar] [CrossRef]
- Bienz, M. The PHD finger, a nuclear protein-interaction domain. Trends Biochem. Sci. 2006, 31, 35–40. [Google Scholar] [CrossRef]
- Lallous, N.; Legrand, P.; McEwen, A.G.; Ramón-Maiques, S.; Samama, J.-P.; Birck, C. The PHD finger of human UHRF1 reveals a new subgroup of unmethylated histone H3 tail readers. PLoS ONE 2011, 6, e27599. [Google Scholar] [CrossRef]
- Servet, C.; e Silva, N.C.; Zhou, D.-X. Histone acetyltransferase AtGCN5/HAG1 is a versatile regulator of developmental and inducible gene expression in Arabidopsis. Mol. Plant 2010, 3, 670–677. [Google Scholar] [CrossRef]
- Marmorstein, R.; Berger, S.L. Structure and function of bromodomains in chromatin-regulating complexes. Gene 2001, 272, 1–9. [Google Scholar] [CrossRef]
- Cannon, S.B.; Mitra, A.; Baumgarten, A.; Young, N.D.; May, G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.-H.; Li, D.-M.; Yin, M.-H.; Li, X.-B.; Zhang, M.; Wang, Y.-J.; Dong, J.; Zhao, J.; Luo, M.; Luo, X.-Y. Gibberellin 20-oxidase promotes initiation and elongation of cotton fibers by regulating gibberellin synthesis. J. Plant Physiol. 2010, 167, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhu, Y.; Hu, W.; Zhang, X.; Cai, C.; Guo, W. Comparative transcriptomics reveals jasmonic acid-associated metabolism related to cotton fiber initiation. PLoS ONE 2015, 10, e0129854. [Google Scholar] [CrossRef] [PubMed]
- Samuel Yang, S.; Cheung, F.; Lee, J.J.; Ha, M.; Wei, N.E.; Sze, S.H.; Stelly, D.M.; Thaxton, P.; Triplett, B.; Town, C.D. Accumulation of genome-specific transcripts, transcription factors and phytohormonal regulators during early stages of fiber cell development in allotetraploid cotton. Plant J. 2006, 47, 761–775. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.-H.; Zhu, S.-W.; Mao, X.-Z.; Feng, J.-X.; Qin, Y.-M.; Zhang, L.; Cheng, J.; Wei, L.-P.; Wang, Z.-Y.; Zhu, Y.-X. Transcriptome profiling, molecular biological, and physiological studies reveal a major role for ethylene in cotton fiber cell elongation. Plant Cell 2006, 18, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zheng, X.; Song, S.; Zeng, Q.; Hou, L.; Li, D.; Zhao, J.; Wei, Y.; Li, X.; Luo, M. Spatiotemporal manipulation of auxin biosynthesis in cotton ovule epidermal cells enhances fiber yield and quality. Nat. Biotechnol. 2011, 29, 453. [Google Scholar] [CrossRef] [PubMed]
- Manzano, C.; Ramirez-Parra, E.; Casimiro, I.; Otero, S.; Desvoyes, B.; De Rybel, B.; Beeckman, T.; Casero, P.; Gutierrez, C.; del Pozo, J.C. Auxin and epigenetic regulation of SKP2B, an F-box that represses lateral root formation. Plant Physiol. 2012, 160, 749–762. [Google Scholar] [CrossRef]
- Anzola, J.M.; Sieberer, T.; Ortbauer, M.; Butt, H.; Korbei, B.; Weinhofer, I.; Mullner, A.E.; Luschnig, C. Putative Arabidopsis transcriptional adaptor protein (PROPORZ1) is required to modulate histone acetylation in response to auxin. Proc. Natl. Acad. Sci. USA 2010, 107, 10308–10313. [Google Scholar] [CrossRef]
- Chailleux, C.; Tyteca, S.; Papin, C.; Boudsocq, F.; Puget, N.; Courilleau, C.; Grigoriev, M.; Canitrot, Y.; Trouche, D. Physical interaction between the histone acetyl transferase Tip60 and the DNA double-strand breaks sensor MRN complex. Biochem. J. 2010, 426, 365–371. [Google Scholar] [CrossRef]
- Dhar, S.; Gursoy-Yuzugullu, O.; Parasuram, R.; Price, B.D. The tale of a tail: Histone H4 acetylation and the repair of DNA breaks. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef]
- Murr, R.; Loizou, J.I.; Yang, Y.G.; Cuenin, C.; Li, H.; Wang, Z.Q.; Herceg, Z. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, L.; Liang, J.; Shi, L.; Yang, J.; Yi, X.; Zhang, D.; Han, X.; Yu, N.; Shang, Y. Histone acetyltransferase 1 promotes homologous recombination in DNA repair by facilitating histone turnover. J. Biol. Chem. 2013, 288, 18271–18282. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, Y. Cross-talk between the H3K36me3 and H4K16ac histone epigenetic marks in DNA double-strand break repair. J. Biol. Chem. 2017, 292, 11951–11959. [Google Scholar] [CrossRef] [PubMed]
- Campi, M.; D’Andrea, L.; Emiliani, J.; Casati, P. Participation of chromatin-remodeling proteins in the repair of ultraviolet-B-damaged DNA. Plant Physiol. 2012, 158, 981–995. [Google Scholar] [CrossRef] [PubMed]
- Fina, J.P.; Casati, P. HAG3, a Histone Acetyltransferase, Affects UV-B Responses by Negatively Regulating the Expression of DNA Repair Enzymes and Sunscreen Content in Arabidopsis thaliana. Plant Cell Physiol. 2015, 56, 1388–1400. [Google Scholar] [CrossRef]
- Kim, J.M.; To, T.K.; Ishida, J.; Morosawa, T.; Kawashima, M.; Matsui, A.; Toyoda, T.; Kimura, H.; Shinozaki, K.; Seki, M. Alterations of lysine modifications on the histone H3 N-tail under drought stress conditions in Arabidopsis thaliana. Plant Cell Physiol. 2008, 49, 1580–1588. [Google Scholar] [CrossRef]
- Asensi-Fabado, M.A.; Amtmann, A.; Perrella, G. Plant responses to abiotic stress: The chromatin context of transcriptional regulation. Biochim. Et Biophys. Acta. Gene Regul. Mech. 2017, 1860, 106–122. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, L.; Zhao, L.; Li, J.; He, S.; Zhou, K.; Yang, F.; Huang, M.; Jiang, L.; Li, L. Trichostatin A selectively suppresses the cold-induced transcription of the ZmDREB1 gene in maize. PLoS ONE 2011, 6, e22132. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Imran, M.; Tang, K.; Liu, J.-Y. Comparative Genome-Wide Analysis of the Malate Dehydrogenase Gene Families in Cotton. PLoS ONE 2016, 11, e0166341. [Google Scholar] [CrossRef][Green Version]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Liu, J.-Y. Genome-wide identification and expression analysis of the malate dehydrogenase gene family in Gossypium arboreum. Pak. J. Bot. 2016, 48, 1081–1090. [Google Scholar]
- Blanc, G.; Wolfe, K.H. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 2004, 16, 1667–1678. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Guo, A.; Zhu, Q.; Chen, X.; Luo, J. GSDS: A gene structure display server. Yi Chuan = Hered. 2007, 29, 1023–1026. [Google Scholar] [CrossRef]
- Bailey, T.L.; Williams, N.; Misleh, C.; Li, W.W. MEME: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006, 34, W369–W373. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Shafiq, S.; Chen, C.; Yang, J.; Cheng, L.; Ma, F.; Widemann, E.; Sun, Q. DNA Topoisomerase 1 prevents R-loop accumulation to modulate auxin-regulated root development in rice. Mol. Plant 2017, 10, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Braisted, J.; Klapa, M.; Currier, T.; Thiagarajan, M. TM4: A free, open-source system for microarray data management and analysis. Biotechniques 2003, 34, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Wang, X.; Liu, Z.; Gu, Q.; Zhang, Y.; Li, Z.; Ke, H.; Yang, J.; Wu, J.; Wu, L.; et al. Genome-wide association study discovered genetic variation and candidate genes of fibre quality traits in Gossypium hirsutum L. Plant Biotechnol. J. 2017, 15, 982–996. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imran, M.; Shafiq, S.; Farooq, M.A.; Naeem, M.K.; Widemann, E.; Bakhsh, A.; Jensen, K.B.; Wang, R.R.-C. Comparative Genome-wide Analysis and Expression Profiling of Histone Acetyltransferase (HAT) Gene Family in Response to Hormonal Applications, Metal and Abiotic Stresses in Cotton. Int. J. Mol. Sci. 2019, 20, 5311. https://doi.org/10.3390/ijms20215311
Imran M, Shafiq S, Farooq MA, Naeem MK, Widemann E, Bakhsh A, Jensen KB, Wang RR-C. Comparative Genome-wide Analysis and Expression Profiling of Histone Acetyltransferase (HAT) Gene Family in Response to Hormonal Applications, Metal and Abiotic Stresses in Cotton. International Journal of Molecular Sciences. 2019; 20(21):5311. https://doi.org/10.3390/ijms20215311
Chicago/Turabian StyleImran, Muhammad, Sarfraz Shafiq, Muhammad Ansar Farooq, Muhammad Kashif Naeem, Emilie Widemann, Ali Bakhsh, Kevin B. Jensen, and Richard R.-C. Wang. 2019. "Comparative Genome-wide Analysis and Expression Profiling of Histone Acetyltransferase (HAT) Gene Family in Response to Hormonal Applications, Metal and Abiotic Stresses in Cotton" International Journal of Molecular Sciences 20, no. 21: 5311. https://doi.org/10.3390/ijms20215311
APA StyleImran, M., Shafiq, S., Farooq, M. A., Naeem, M. K., Widemann, E., Bakhsh, A., Jensen, K. B., & Wang, R. R.-C. (2019). Comparative Genome-wide Analysis and Expression Profiling of Histone Acetyltransferase (HAT) Gene Family in Response to Hormonal Applications, Metal and Abiotic Stresses in Cotton. International Journal of Molecular Sciences, 20(21), 5311. https://doi.org/10.3390/ijms20215311