3.1. Known sRNAs and Their Target Genes Are Involved in Regulating Flower Development in Yellow Lupine
Among the known and conserved miRNAs a number of miRNAs commonly associated with flower morphogenesis and development, belonging to,
inter alia, the MIR156/157, MIR159, MIR165/166, MIR167 and MIR172 families [
10] were spotted.
Studies have shown that miR156 is necessary for maintaining anther fertility in
Arabidopsis, by orchestrating the development of primary tapetum cells and primary sporogenous cells [
61]. In
A. thaliana,
SPL13B expression is strictly limited by miR156 to anther tapetum in young buds, while
SPL2 is weakly expressed in parietal and sporogenous cells and the surrounding cell layers in young flowers [
61], where it is targeted by miR156 to regulate pollen maturation [
62]. MiR159 was shown to target the conserved
GAMYB-like genes that are a part of the GA signaling pathway [
63,
64]. In
A. thaliana miR159 regulates the morphogenesis of the stamen, and male fertility [
65]. Two transcription factors involved in pistil and stamen development in various plant species,
ARF6 and
ARF8, contain the target site for miR167 [
66,
67,
68]. For
Arabidopsis, it has been proven that both these genes are involved in stamen filament elongation, anther dehiscence, stamen maturation and anthesis [
69]. In tomato, a reduction in the accumulation of the miR167-targeted
ARF6 and
ARF8 leads to the lack of trichomes on the style surface, failed pollen germination and, consequently, sterility [
11]. Recent research into multiple plant species has shown that miR172 targets genes belonging to the
APETALA2 (
AP2,
TOE1,
TOE2,
TOE3) family. MiR172 is part of the photoperiodic flower induction pathway and is associated with the functioning of the ABCDE model of floral development [
70]. Overexpression of
MIR172 causes formation of a phenotype characterized by the absence of perianth, transformation of sepals into pistils and early flowering [
70].
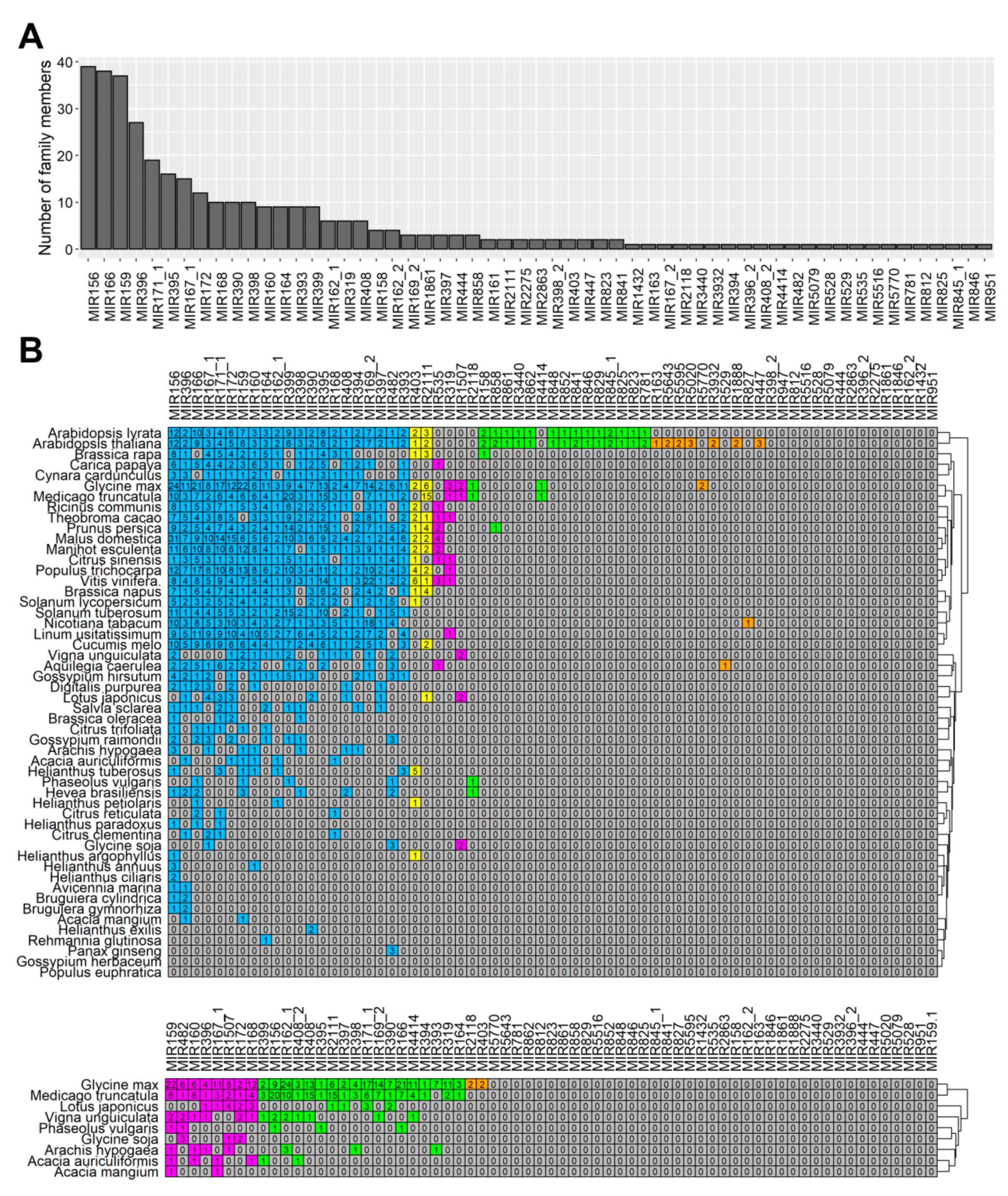
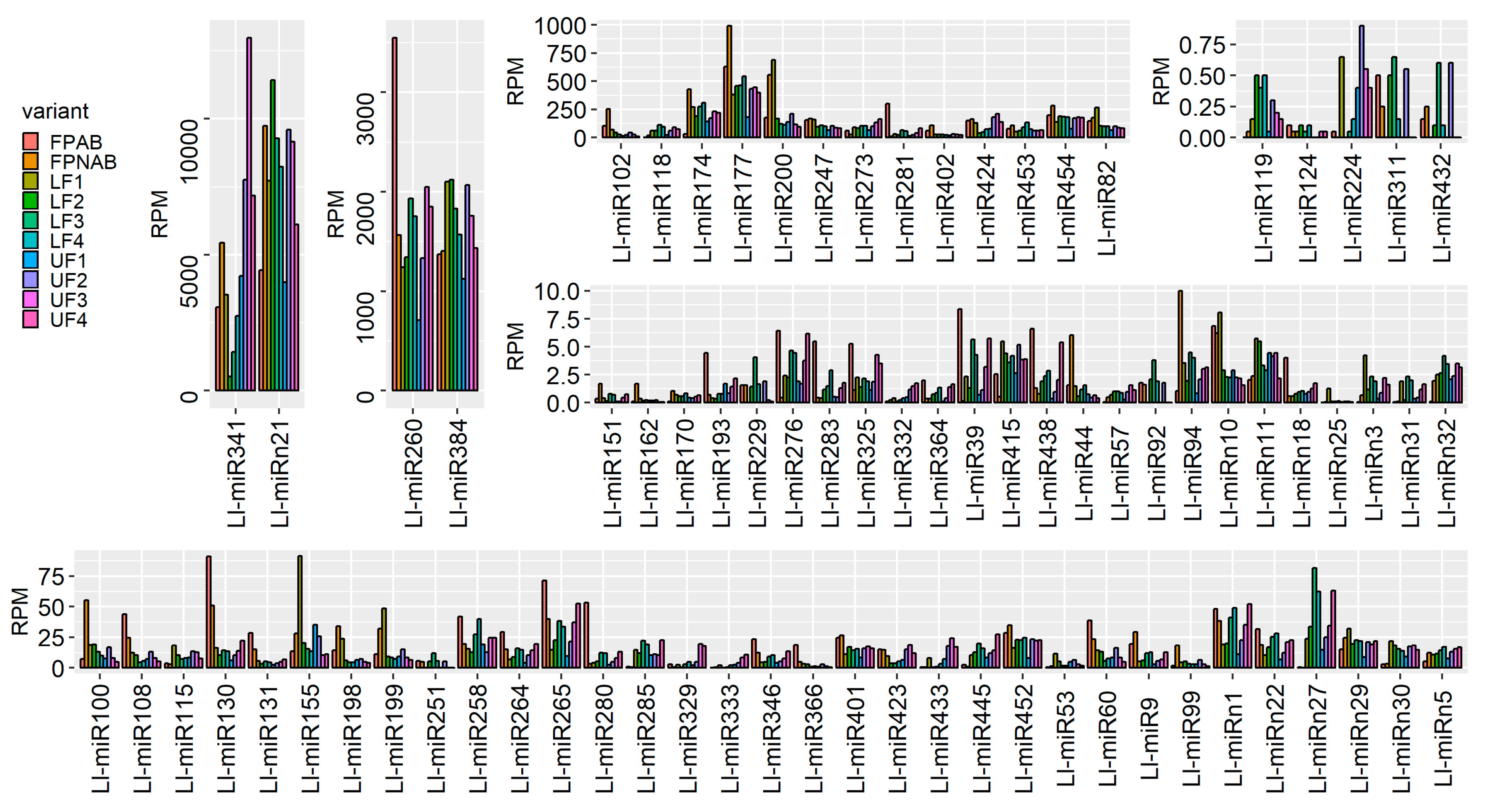
Our study showed the presence of at least one member of all these families in flowers (
Figure 3,
Table S5), which indicated that in lupine how crucial the families were for generative development in lupine, as well. MIR156 and MIR159 are the most numerous families in
L. luteus, which suggests they play fundamental roles in its flower development processes.
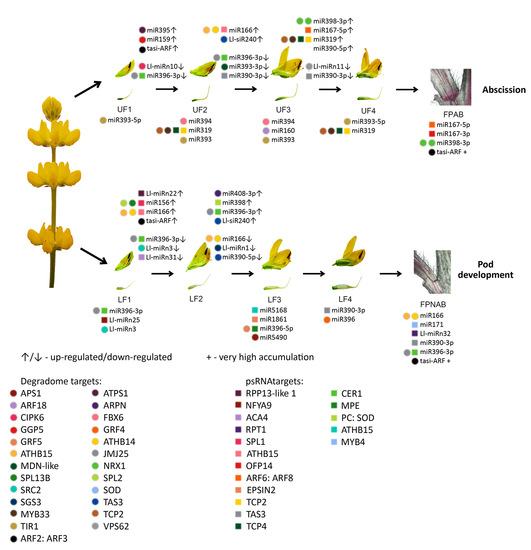
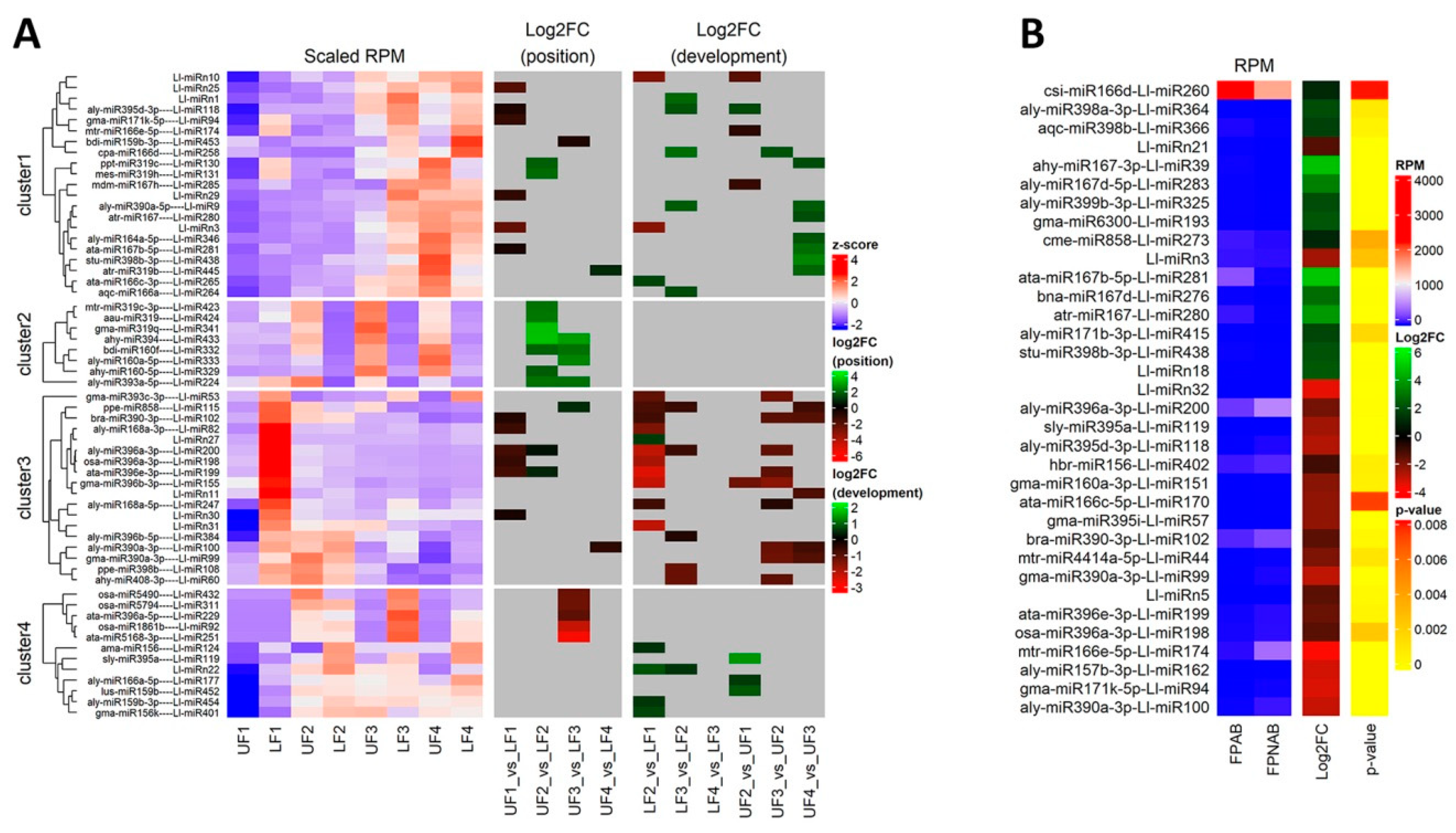
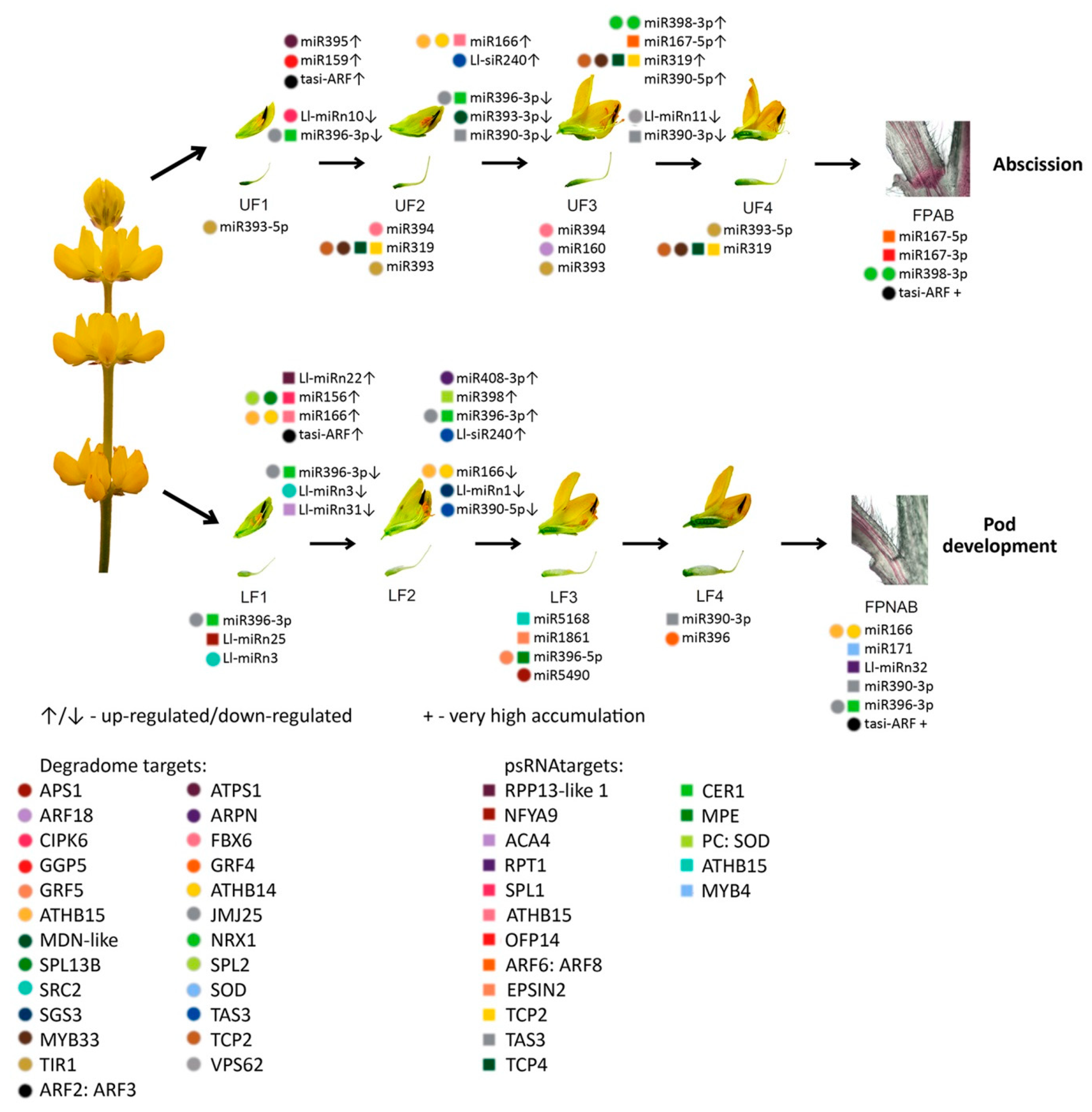
The differentially expressed miRNAs identified in yellow lupine flowers were clustered by the dynamics of their expression (
Figure 5). The first cluster comprised miRNAs, the accumulation of which increased as the flowers developed, and contained miRNAs belonging to the MIR166, MIR167, MIR319, MIR390, and MIR395 families. The first of these families include Ll-miR177, which guides the cleavage of
RADIALIS, a transcription factor from the MYB family that controls the asymmetric flower shape in
Antirrhinum majus [
71,
72], as well as Ll-miR258 and Ll-miR265, which probably target the Homeobox-leucine zipper protein ATHB-15. In
Arabidopsis, both miR165 and miR166 target the same
HD-ZIP III genes:
ATHB15,
ATHB8,
REVOLUTA (REV),
PHABULOSA (
PHB), and
PHAVOLUTA (
PHV) to regulate gynoecium and microspore development [
28,
73]. In lupine the MIR167 family members that accumulate in larger quantities during flower development are Ll-miR280, Ll-miR281, and Ll-miR285, which probably target
ARF6 and
ARF8. Ll-miR445 and Ll-miR130 are members of the MIR319 family, while their putative target genes are
TCP4 and
MYB33, respectively. In
Arabidopsis, the miR319a/TCP4 regulatory module is necessary for petal growth and development. Moreover, the overexpression of
MIR319 reduces male fertility, and this defect is hypothesized to be caused by the cross-regulation of
MYB33 and
MYB65 by miR319 and miR159. As the miR319 target site within the
MYB33 and
MYB65 transcripts exhibit a lower match with miRNA than the miR159 target site, the latter is more efficient at regulating these genes and miR319 is their secondary regulator [
74]. This regulatory network is even more complex. In
A. thaliana, cooperation of three miRNAs and their target genes, namely miR159/
MYB, miR167/
ARF6/ARF8, and miR319/
TCP4, is a prerequisite for proper sepal, petal and anther development, and maturation. miR159 and miR319 influence the expression of
MIR167 genes, which in turn affect each other. These miRNAs orchestrate plant development by regulating the activity of the phytohormones GA, JA, and auxin [
75]. Increased accumulation of miR167 and miR319 in the late stages of yellow lupine flower development could also be associated with regulating the growth and development of petals and anthers. Another miRNA showing a similar expression profile is Ll-miR9/miR390-5p. In lupine, it targets the
TAS3 transcript, which in turn is a source of tasiR-ARF, a negative regulator of
ARF2,
ARF3 and
ARF4 activity. This regulatory cascade plays a vivid role in development of many plant species [
76]. The expression level of miR390 derived from
MIR390b reflects auxin concentration in organs, while the repression of
ARF2,
ARF3, and
ARF4 by tasiR-ARF are important for lateral organ development [
18,
77], and flower formation [
78]. Ll-miR118 and Ll-miR119, which target ATP sulfurylase (
ATPS) according to our degradome data, belong to the MIR395 family. In
Arabidopsis, miR395 targets two gene families, ATP sulfurylases and sulfate transporter 2:1 (
SULTR2:1), which are elements of the sulfate metabolism pathway [
79]. ATPS regulates glutathione synthesis and is an essential enzyme in the sulfur-assimilatory pathway [
80]. In cotton, the miR395-APS1 module is engaged in drought and salt stress response [
81]. Sulfate is the main source of sulfur and is taken up by roots, transported throughout the plant and used for assimilation. Sulfate limitation forces a significant up-regulation of miR395 expression [
82]. Presumably, during yellow lupine flower development, the demand for sulfur increases and the plant activates mechanisms for its efficient uptake.
Within the cluster of miRNAs, the expression of which decreased as the flowers developed, there were homologues of miR390-3p, miR858, miR396-3p, miR168, miR408-3p and miR398 (
Figure 5). Ll-miR99, Ll-miR100, and Ll-miR102 are identical to miR390-3p (the so-called passenger strand, former star strand). However, their expression showed an opposite trend to that of miR390-5p. The differential expression and functioning of passenger miRNAs have already been described. The research carried out by Xie and Zhang in 2015 on cotton showed that the formation of some miRNA*s, such as miR172* and miR390*, was associated with the phases of the plant’s growth [
83]. Therefore, miRNA*s can be specifically expressed in various tissues to maintain the steady state of the organism. Our degradome analysis for yellow lupine showed that Ll-miR9/miR390-5p was able to guide the cleavage of the
TAS3 transcript. There is no certainty as to the status of its passenger strand, which suggests its locally limited activity or its involvement in regulation of other targets and further research is required to identify its accumulation and function in the organs concerned. Another miRNA from the cluster is Ll-miR155/miR396-3p (passenger strand), which guides cleavage of JMJ25 demetyhylase mRNA (confirmed in degradomes), involved in preserving the active chromatin state [
84].
ECERIFERUM1 (
CER1), the target gene in lupine for another two homologues of miR396-3p, Ll-miR199 and Ll-miR200, is a homologue encoding an enzyme involved in alkane biosynthesis, and in cucumber is engaged both in wax synthesis and ensuring pollen viability [
85]. This cluster also included a miRNA that negatively regulates elements involved in miRNA and ta-siRNA functioning, namely Ll-miR247/miR168 targeting
AGO1 mRNA [
86]. Another miRNA clustered here was the highly conserved Ll-miR60/miR408-3p, which guides the processing of the mRNA of the copper-binding Basic Blue protein homologue (plantacyanin, PC). In
Arabidopsis, PC plays a role in fertility, exhibiting the highest expression in the inflorescence, especially in the transmitting tract. [
87]. Transgenic
Arabidopsis plants over-expressing
MIR408 displayed altered morphology, including significantly enlarged organs, resulting in enhanced biomass and seed yield. Plant enlargement was shown to be primarily caused by cell expansion rather than cell proliferation, and in transgenic plants it was correlated with stronger accumulation of the myosin-encoding transcript and gibberellic acid [
88]. It seems that high expression levels of miRNAs grouped in the cluster are correlated with intensive growth and differentiation of young floral tissues.
Among the miRNAs identified in yellow lupine several that seemed to be crucial in particular stages of the plant’s development were spotted (
Figure 4,
Table 4,
Table S7). For example, the largest quantities of miR159 (Ll-miR452 and Ll-miR454) were accumulated in stages 2 and 3 of the plant’s development. According to degradome data they targeted
GGP-5 (
GAMMA-GLUTAMYL PEPTIDASE 5) of an undefined function in plants, and an evolutionarily conserved target for
GAMYB, respectively. As already mentioned, this could be associated with miRNA family cooperating with miR167 and miR319 in regulating
L. luteus anther maturation. The accumulation of Ll-miR251/miR5168-3p, Ll-miR92/miR1861b, Ll-miR229/miR369-5p, and Ll-miR311/miR5794 increased in stage 2 upper and lower flowers, while – interestingly – in the later stages these miRNAs were only present in lower flowers. According to degradome analysis, Ll-miR251/miR5168 guides cleavage of the mRNAs of the genes encoding the Homeobox-leucine zipper protein ATHB-14 and the chaperone protein dnaJ 13. The miR5168 sequence displays a great similarity to that of miR166, thanks to which they may perhaps share the same target gene
ATHB-14, the putative transcription factor engaged in the adaxial-abaxial polarity determination in the ovule primordium in
A. thaliana [
89]. As confirmed by yellow lupine degradome sequencing, Ll-miR229/miR396-5p targets
GROWTH-REGULATING FACTOR 5 (
GRF5) and
GRF4 transcripts. In
Arabidopsis,
GRF5 is expressed in anthers at early stages of flower development and in gynoecia throughout the whole flower development, and transcripts of
GRF4 accumulate later in sepals, tapetum, and endocarpic tissues of ovary valves [
90]. Transgenic rice with Os-miR396 overexpression and
GRF6 knock-down suffers from open husks and sterile seeds [
91].
GRF6 cooperates with
GRF10 to transactivate the
JMJC gene
706 (
OsJMJ706) and
CRINKLY4 RECEPTOR-LIKE KINASE (
OsCR4) responding to GA, which is a prerequisite for the flower to successfully develop into a normal seed [
91]. An increased share of miRNAs involved in cell division, namely miR396, miR319, and miR164, in NGS analyses was also observed in early grain development in wheat [
92].The presence of these miRNAs in yellow lupine flowers suggests that their regulation of cell proliferation also plays an important role in development of generative organs.
3.2. Involvement of New miRNAs in L. luteus Flower Development
Using ShortStack [
53] software we predicted 28 candidates for new miRNAs (
Table 3). Interestingly, many of these novel miRNAs showed similarity to precursor miRNAs from miRBase, which leads to the conclusion that they might be new members of the already known families, for example MIR167 (Ll-miRn12 and Ll-miRn27), MIR172 (Ll-miRn4), MIR393 (Ll-miRn19) or MIR169 (Ll-miRn3, Ll-miRn11, and Ll-miRn15) (
Table S6).The other 13 had no homologues among known miRNAs and were recognized as lupine-specific miRNAs. Some of the new miRNAs displayed differential expression during
L. luteus flower development. Ll-miRn3, which shows similarity to pre-miR169, displayed differential expression in UF1 vs LF1 and LF2 vs LF1 library comparisons, wherein it is the most accumulated in LF1, and in flower pedicels (up-regulated in FPNAB). According to degradome data, this miRNA targets
SCARECROW2 (
SCR2) homologue, a putative activator of the calcium-dependent activation of
RBOHF that enhances reactive oxygen species (ROS) production and may be involved in cold stress response [
93]. In rice
SCR2 expression is relatively high in flower buds and flowers, and after flowering rises in the leaves and roots [
94]. In yellow lupine, this gene may be involved in intense cell divisions during early flower development and is down-regulated in the pedicels with an active AZ to stop its growth. Another frequently encountered novel DEmiR was Ll-miRn22, which shows sequence similarity to pre-miR1507, is up-regulated in LF3 vs LF2 and LF2 vs LF1 library comparisons, and its expression escalates with flower development in the bottom whorl. The MiR1507 family is annotated as legume-specific [
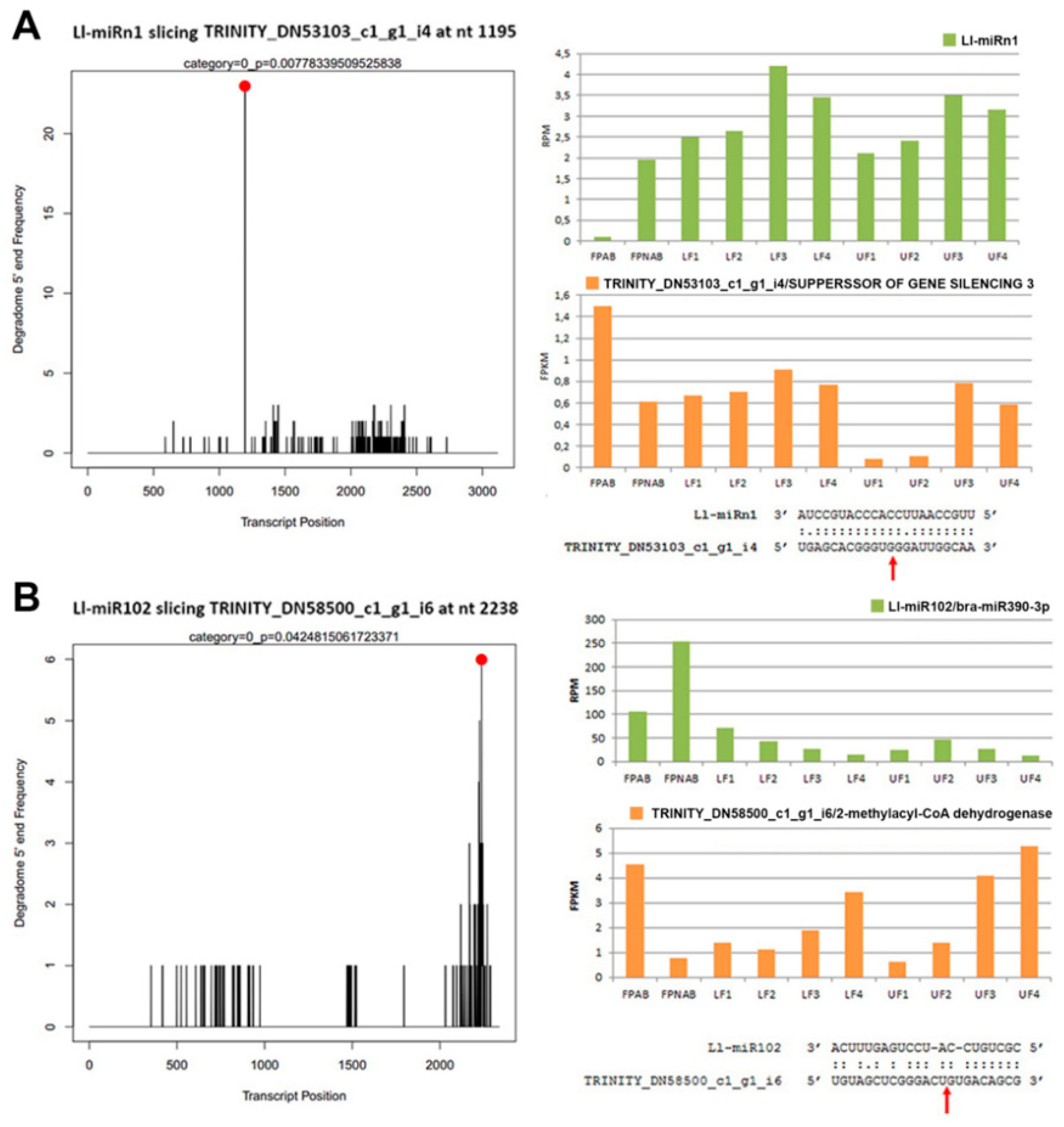
95]. Through analyses of our degradome data we have not found its target gene, and the psRNATarget hit was the putative disease resistance RPP13-like protein 1. Unfortunately, this protein has been poorly described, therefore it is difficult to determine its function in yellow lupine flowers. Noteworthily, the target genes of Ll-miRn1 and Ll-miRn30 identified through degradome sequencing are
SGS3 and
DCL2, respectively, and the miRNAs are up-regulated in LF3 vs LF2 comparisons and down-regulated in UF1 vs LF1 comparisons, respectively.
SGS3- and
DCL2-encoded proteins are involved in sRNA biogenesis [
96]. Importantly, novel miRNA identified in soybean Soy_25 displays high sequence similarity to Ll-miRn1 and also targets
SGS3, which indicates that this regulatory feedback loop for sRNA biogenesis is common for
Fabaceae [
97]. These results indicate that
L. luteus miRNAs play a regulatory role in siRNA biogenesis in early flower development.
3.3. miRNA Accumulation Varies in Lower and Upper Flowers in Different Stages of Development
One of our goals was to identify the sRNAs engaged in yellow lupine flower development, with a particular emphasis on the differences between flowers from lower and upper parts of the inflorescence, in order to gain an insight into how early the flower fate is determined.
In our study, we spotted differences in miRNA accumulation patterns as early as the first stage of flower development.
Flowers collected from the lower whorls displayed higher accumulations of sequences corresponding to miR5490, miR5794, miR1861, miR396-5p, miR395, miR166, and miR159-3p (
Table 5). miR1861 and miR396 were recognized as positive cell proliferation and development regulators [
98,
99,
100]. In rice, for example, miR1861 exhibited differential expression during grain filling [
101], and its expression was higher in superior grains in comparison to inferior ones [
102]. This is consistent with our hypothesis, that a higher occurrence of miR1861 and miR396 in lower flowers may be an indication of the plant investing more supplies in this part of the inflorescence.
From the second stage until the end of their development, upper flowers accumulated more miRNAs corresponding to miR319, miR394, miR160, and miR393 (
Figure 4,
Table 5). MiR393 regulates the accumulation of transcripts encoding auxin receptors belonging to the TAAR family. Changes in receptor abundance affect the sensitivity of the given tissue to auxin and this is how this molecule influences plant development [
102]. In
A. thaliana, miR160 directly controls three
ARF genes, namely:
ARF10, ARF16 and
ARF17 [
103]. In tomato, sly-miR160 is abundant in ovaries, and changes in its expression affect plant fertility [
12]. Down-regulation of sly-miR160 caused improper ovary patterning and thinning of the placenta already prior to anthesis [
12]. In view of these facts, higher expression of miR160 in lupine upper flowers in their development means that a slightly different organization of the gynoecia may be one of the crucial determinants of flower fate. Additionally, the elevated expression levels of miR160 and miR393 in upper flowers of lupine suggest a reduction in the abundance of the transcripts of their target genes encoding auxin receptors and auxin response factors. This, in turn, may have led to a reduction in auxin sensitivity. Decreasing the number of transcription factors belonging to the TCP family (targeted by miR319), probably caused different cell proliferation profiles in flowers collected from the upper whorls.
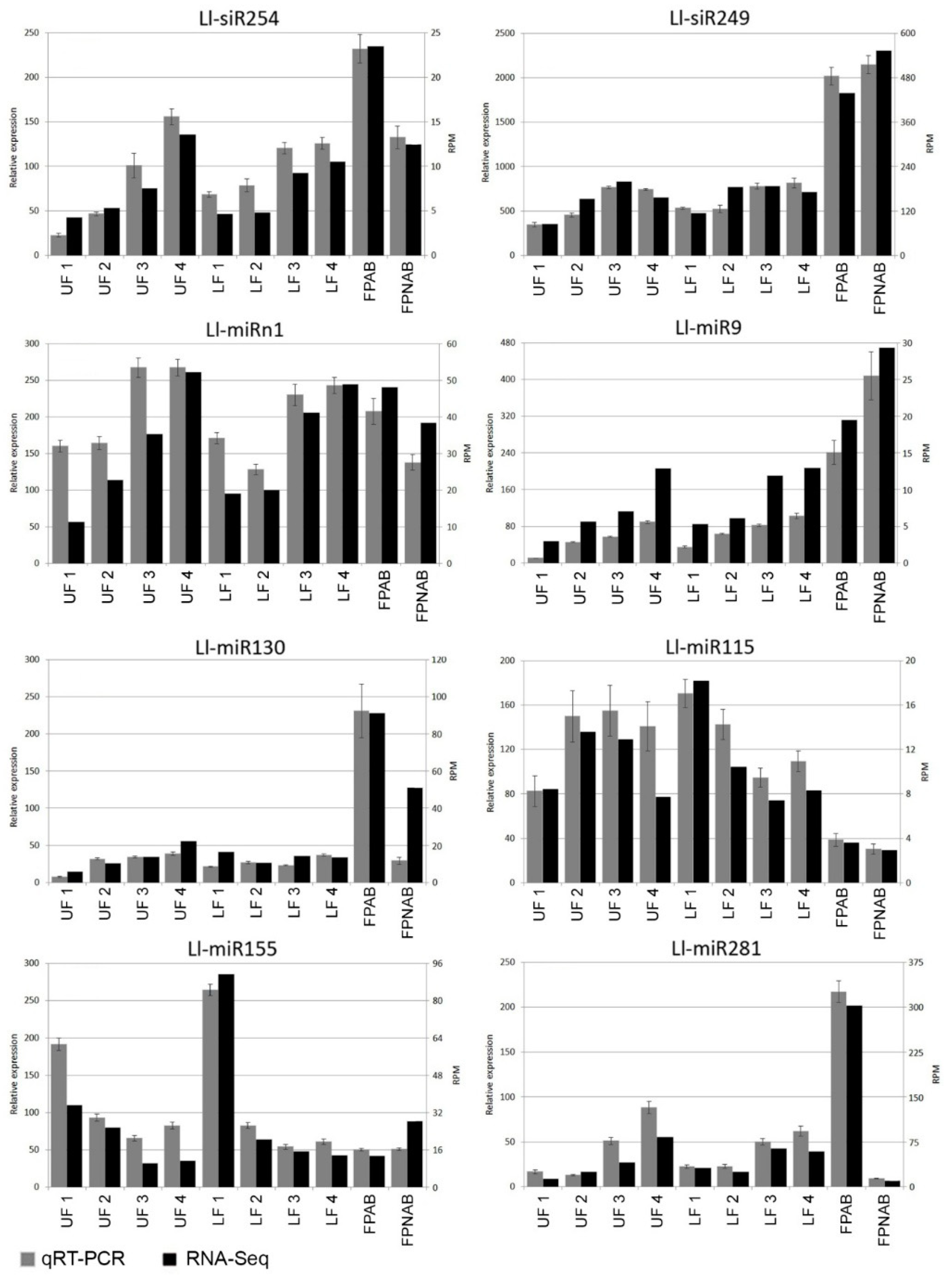
Additional expression studies of selected miRNA (Ll-miR281/miR167, Ll-miR224/miR393, Ll-miR333/miR160, Ll-miR329/miR160) carried out in the upper flowers of yellow lupin developing after removal of the lower ones (UFR) (
Figure S2), and consequently with a changed, when compared to the original, fate, provide additional confirmation of the results obtained from RNA-seq analysis (
Figure S3).
3.4. sRNAs Are Involved in Flower Abscission in L. luteus
Little is known about sRNA engagement in flower abscission. Research on the involvement of miRNAs in this process has been already carried out in cotton [
104], tomato [
12,
105], and sugarcane [
106]. For a genome-wide investigation of miRNAs involved in the formation of the abscission layer in cotton, two sRNA libraries were constructed using the abscission zones (AZ) of cotton pedicels treated with ethephon or water. Among the 460 identified miRNAs, only gra-MIR530b and seven novels showed differential expression in abscission tissues [
104], and these miRNAs have no homologues in our dataset.
Besides ovary patterning in tomato, sly–miR160 regulates other two auxin-mediated developmental processes: floral organ abscission and lateral organ lamina outgrowth [
12]. In that study, down-regulation of sly-miR160 and the resulting higher expression of its target genes, transcriptional repressors of auxin response
ARF10 and
ARF17, also resulted in the narrowing of leaves, sepals and petals and an impeded shedding of the perianth after successful pollination [
12]. This was consistent with the higher accumulation of Ll-miR329/miR160-5p, Ll-miR332/miR160-5p, and Ll-miR333/miR160-5p in upper flowers designated to fall off in yellow lupine. As these miRNAs showed no differential expression in flower pedicels, it probably does not play a role in the executory module of abscission itself but is rather a part of a mechanism that determines flower fate.
Another research on tomato using sRNA and degradome sequencing libraries explored the roles of sRNAs in AZ formation in the early and late stages of the process additionally accelerated or not by ethylene or control treatment [
107]. The study showed that in tomato pedicels, the accumulation levels of,
inter alia, miR156, miR166, miR167, miR169, miR171, and miR172 rose in late stages of abscission, while the abundance of miR160, miR396 and miR477 dropped [
107]. Although it is difficult to compare ethylene-treated tomato pedicel results to our data, it is worth noting that in the corresponding FPAB vs. FPNAB comparison in our study, the accumulation of some miRNAs was similar: miR396 level was lower, and the levels of miRNAs annotated as miR167 and miR166 were higher in FPAB (
Table 6).
It has been proven for sugarcane that among others both mature (5p) and passenger (3p) miRNAs from MIR167 family were up-regulated in ‘leaf abscission sugarcane plants’ comparing to ‘leaf packaging sugarcane plants’ (which corresponds to the FPAB vs. FPNAB comparison in our study) [
106]. In our study, both mature and passenger members of the MIR167 family were leaders among DEmiRs, too, (
Table 6) pointing to their crucial role in both vegetative and generative organ abscission. Significantly, this applies to evolutionarily distant taxa: both monocots and dicots.
In our paper, among the up-regulated miRNAs, the most numerous family besides already mentioned MIR167 was MIR398 with 3 members being among top-regulated ones. Among the down-regulated miRNAs, the members of MIR390, MIR396 and MIR395 families were most abundant. It was shown for other plant species, that these miRNAs are engaged in the regulation of auxin signal transduction pathway (miR167 and miR390 [
108]), regulation of cell division (miR396 [
100]) and stress response (miR395 [
81,
82]).
It is worth noting, that in comparisons of
Lupinus pedicel libraries there are novel miRNAs: three are down-regulated in FPAB and one is up-regulated. Furthermore, Ll-miRn3 is up-regulated in both, young flowers designated to be maintained on the plant (LF1) and pedicels with inactive AZ (
Table 6), which may indicate its role in preventing flower abscission. In the future, it is worth examining the role of its target gene, which encodes a protein that does not resemble any known protein.
With regard to siRNAs, the most up-regulated ones in FPNAB were: Ll-siR173, Ll-siR4 and Ll-siR13, and the most down-regulated one were Ll-siR208. Unfortunately, the lack of literature data on their targets makes it impossible for the specifics of their function in the studied process. However, it is worth mentioning, that in pedicels high levels of accumulation are displayed by siR249/tasiR-ARF and siR308/tasiR-ARF, which target transcripts encoding ARF2, ARF3 (confirmed in degradomes). These results strongly suggest the involvement of siRNAs in the functioning of lupine pedicels.
3.5. Possible miRNA-dependent Regulatory Pathways That Participate in Development and Abscission of Yellow Lupine Flowers
Recent studies have shown that sRNA activity is associated with the hormonal regulation of plant development through influencing the spatio-temporal localization of the hormone response pathway [
109].
The auxin signal transduction pathway mainly consists of three elements. Auxin is perceived by members of the TAAR family. There are AUX/IAA repressor proteins and ARF transcription factors downstream of these receptors [
110,
111,
112]. The expression of
TAAR receptors is regulated by miR393 and secondary ta-siRNA derived from their own transcripts [
20]. miR167 and miR160 affect the
ARF6,
ARF8 [
67]
ARF10,
ARF16 and
ARF17 [
113] transcript accumulation, respectively. It has been proven that the expression of
ARF2, together with
ARF3 and
ARF4, is regulated by the ta-siRNA/miR390 module [
114]. In the two-hit model, ta-siRNA-containing the
TAS transcript is recognized by two miR390 molecules, one of which guides its cleavage, and the other, in a complex with AGO7, serves as a primer for complementary strand synthesis, with its subsequent processing ultimately resulting in ARF-targeting siRNA biogenesis [
115].
In our study, among the differentially expressed sRNAa in flowers and flower pedicels, there were members of the MIR167, MIR160, MIR393 and MIR390 families, as well as phased siRNAs targeting
ARF2,
ARF3, and
ARF4. This fact suggests a vivid role of auxin-related sRNAs in flower development and abscission in
L. luteus and confirms our previously published results of transcriptome-wide analyses, where we observed differences in expression levels of genes encoding several elements of the auxin signal transduction pathway [
17]. The relatively high number of members of the MIR167 family showing differential expression in the studied variants indicates that miR167 is one of the key regulators of flower development and abscission in yellow lupine.
Lupinus LlARF2,
LlARF3, and
LlARF4 transcripts are possibly down-regulated in the processing that is guided by Ll-siR249 and Ll-siR308 (
Table 4), which are identical to tasiR-ARFs in many plant species according to the tasiRNAdb database [
116]. These tasiR-ARFs probably originate from
TAS3 transcript (TRINITY_DN55534_c4_g1) containing two binding sites for miR390 (
Figure S9a). Ll-miR9/miR390and surprisingly also Ll-siR240, guide the cleavage of another
TAS3 mRNA (TRINITY_DN54998_c6_g5_i2) (
Figure S10) which contains only one target site for miR390 (
Figure S9b). This is the first report on
TAS3 processing regulated by siRNA. The target site for Ll-siR240 is shifted by 10 nucleotides relative to the target site for Ll-miR9/miR390 (
Figure S10). The expression of Ll-siR249, Ll-siR308, and Ll-miR9 showed a similar profile, as it rose during flower development and was the highest in the pedicels (
Figure 7). Ll-siR240 accumulated proportionally to
TAS3 with only one target site for miR390, which means that it was least expressed in the pedicels, while in flowers its expression increased with time (
Table S18). The identified target transcripts belonging to the
ARF2,
ARF3, and
ARF4 gene families showed differential expression but with no clear trend (
Table S18). This may indicate that these siRNAs act locally, repressing only a pool of transcripts expressed in a given tissue, while in other flower parts activity of these genes is regulated in other ways. The presence of all the elements of the miR390/TAS3/tasiR-ARF module among the DE sRNAs in yellow lupine suggests that alterations in its functioning have a great impact on
L. luteus flower development. The additional element in the form of siRNA that processes
TAS3 mRNA seems to be a new species-specific adjuster of this regulation module.
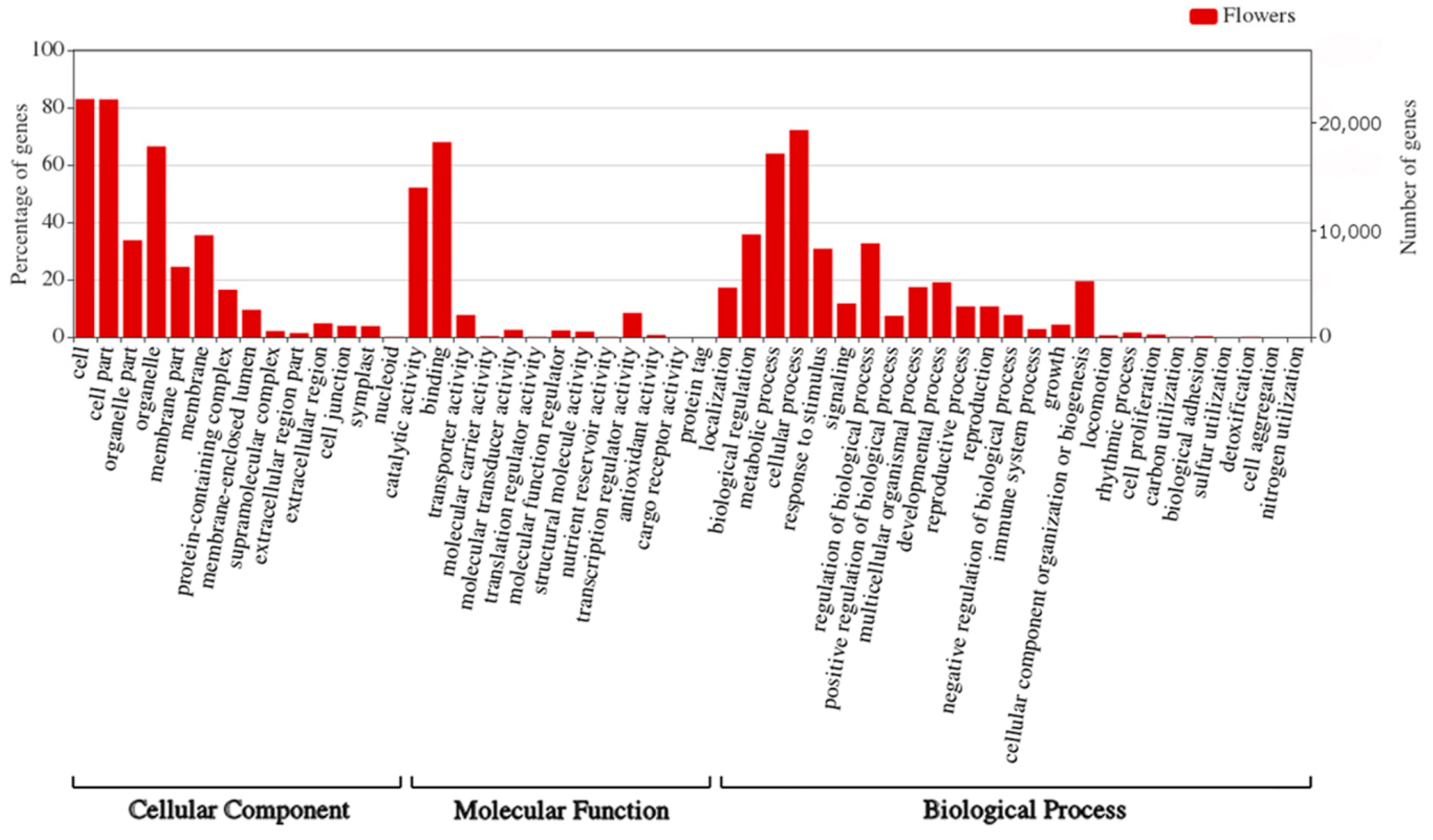
We have also performed GO enrichment analysis of the target genes for sRNAs identified in flowers of yellow lupine (
Figure 9,
Figure S4a,b,
Table S10). What is most interesting is that quite a considerable number of target genes fell within the ‘response to stimulus’ and ‘signaling’ categories, which means that miRNAs modulated the way the plant adapted to environmental stimuli (
Figure 9). An in-depth analysis of GO terms concerning plant hormones (
Figure S4a) showed that most of the miRNAs identified in yellow lupine modulated more than one hormone signaling pathway. For example, Ll-miR181 belonging to the MIR166 family modulated processes associated with four hormones, namely auxin, gibberellin, jasmonic acid, and salicylic acid, by targeting not only transcription factor AS1, a central cell division regulator [
117], but also Cullin-3A, an element of the ubiquitination complex [
118]. Another two members of this family, Ll-miR173 and Ll-miR177, targeted the same gene,
26S PROTEASOME NON-ATPASE REGULATORY SUBUNIT 8 HOMOLOG A (
RPN12A), involved in the ATP-dependent degradation of ubiquitinated proteins during auxin and cytokinin response [
119]. Our GO analysis for yellow lupine flowers additionally showed that miRNAs were responsible for guiding the processing of genes simultaneously involved in multiple processes associated with flower development (
Figure S4b). For example, in many plants
AP2 is involved in the specification of floral organ identity [
120], as well as ovule [
121] and seed development [
122,
123], and in our study, it was targeted by ten lupine miRNAs. On the other hand, seven of these miRNAs additionally targeted a homologue of negative flower development regulator,
LIKE HETEROCHROMATIN PROTEIN 1 (
LHP1) [
124]. This highly degenerated and ambiguous model of gene regulation by lupine miRNAs shows that in this plant the adjustment of key biological processes related to fertility is a complex network of interconnected factors.
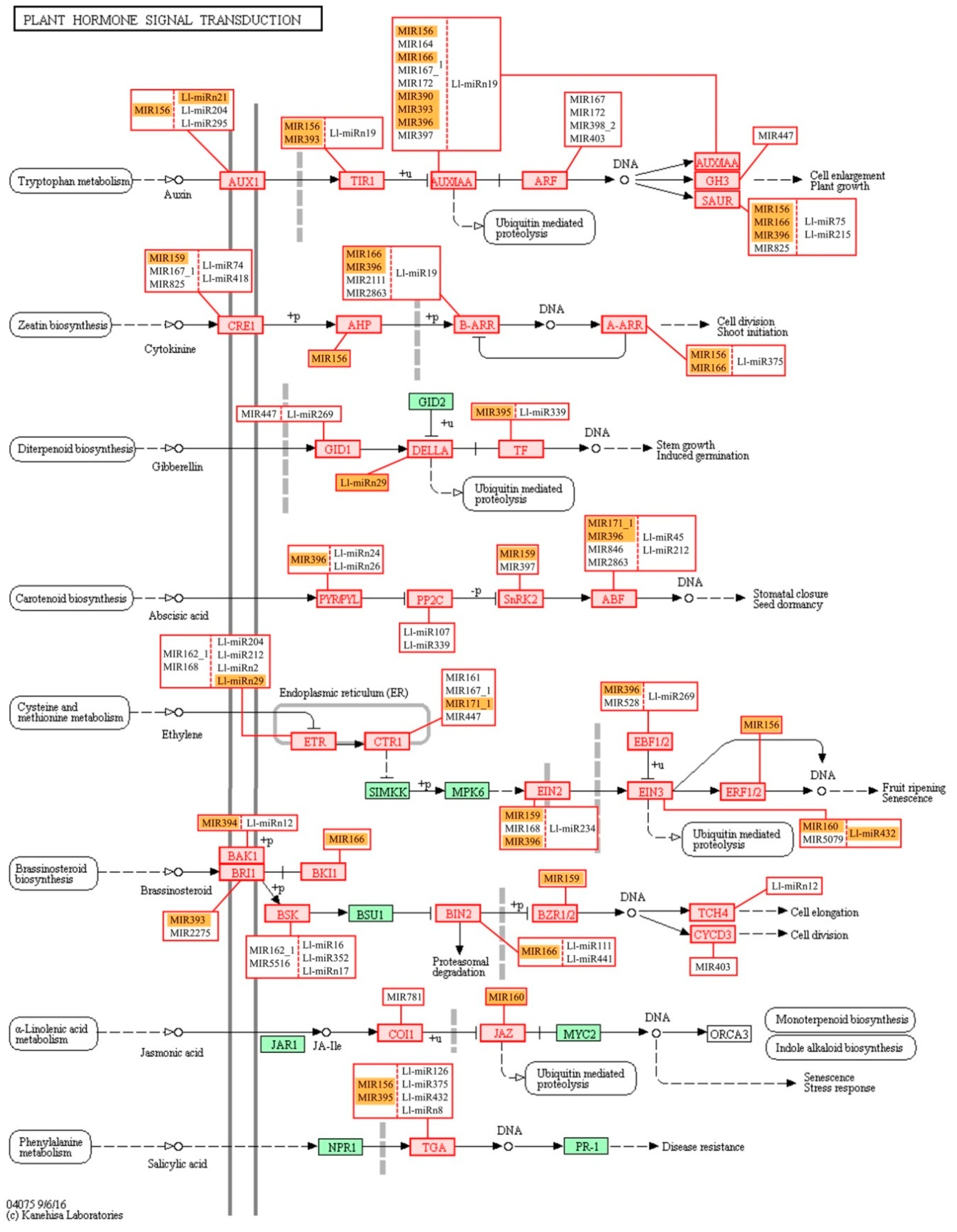
We have also conducted KEGG functional analysis of the putative targets identified for miRNAs in lupine which indicated their engagement in regulating a number of metabolic pathways—especially ‘carbohydrate metabolism’ and ‘nucleotide metabolism’ (
Figure S5). ‘Carbohydrate metabolism’ was also one of the most enriched KEGG pathways in our previous
L. luteus transcriptome analysis [
17], and its activation may be an indication of cell walls being rebuilt or changes in nutrient supply. The next most numerous group of miRNA targets was categorized into the ‘Genetic information processing’ KEGG pathways, namely, ‘spliceosome’, ‘RNA transport’, and ‘ubiquitin proteolysis’. This suggests that in yellow lupine flowers most miRNAs regulate processes related to post-transcriptional events and protein degradation. Three KEGG categories within the ‘Environmental information processing’ category is extremely important in terms of plant development, and they are ‘Signal transduction pathways’ comprising the MAPK cascade, ‘phosphatidylinositol’ and ‘plant hormone’ signaling pathways (
Figure 10,
Figure S6, S7, S8). The MAPK pathway is involved in regulating several processes, such as biotic and abiotic stress response (reviewed in [
125,
126]), and associated with the functioning of hormones such as ethylene [
127] and abscisic acid, engaged in organ abscission and other processes (reviewed in [
128,
129]). The MAPK cascade is also an element of the positive feedback loop amplifying the abscission signal [
130]. Auxin seems to be major target of sRNAs in yellow lupine. However, KEGG enrichment analyses of the identified target genes for lupine miRNAs indicated that the signal transduction pathways of gibberellin, cytokinin, the already mentioned ethylene, and ABA were potentially modulated by miRNAs in
L. luteus, as well, but in less extent (
Figure 10).
Interestingly, like in the case of GO analysis, KEGG analysis for the MIR166 family showed that it was involved in the auxin, cytokinin, and brassinosteroid signal transduction pathways (
Figure 10). These data show again how the fine-tuning of expression of phytohormone-related genes by sRNAs is important for growth and development regulation.

 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}