DockNmine, a Web Portal to Assemble and Analyse Virtual and Experimental Interaction Data

 ,
,
Abstract
:1. Introduction
2. Results
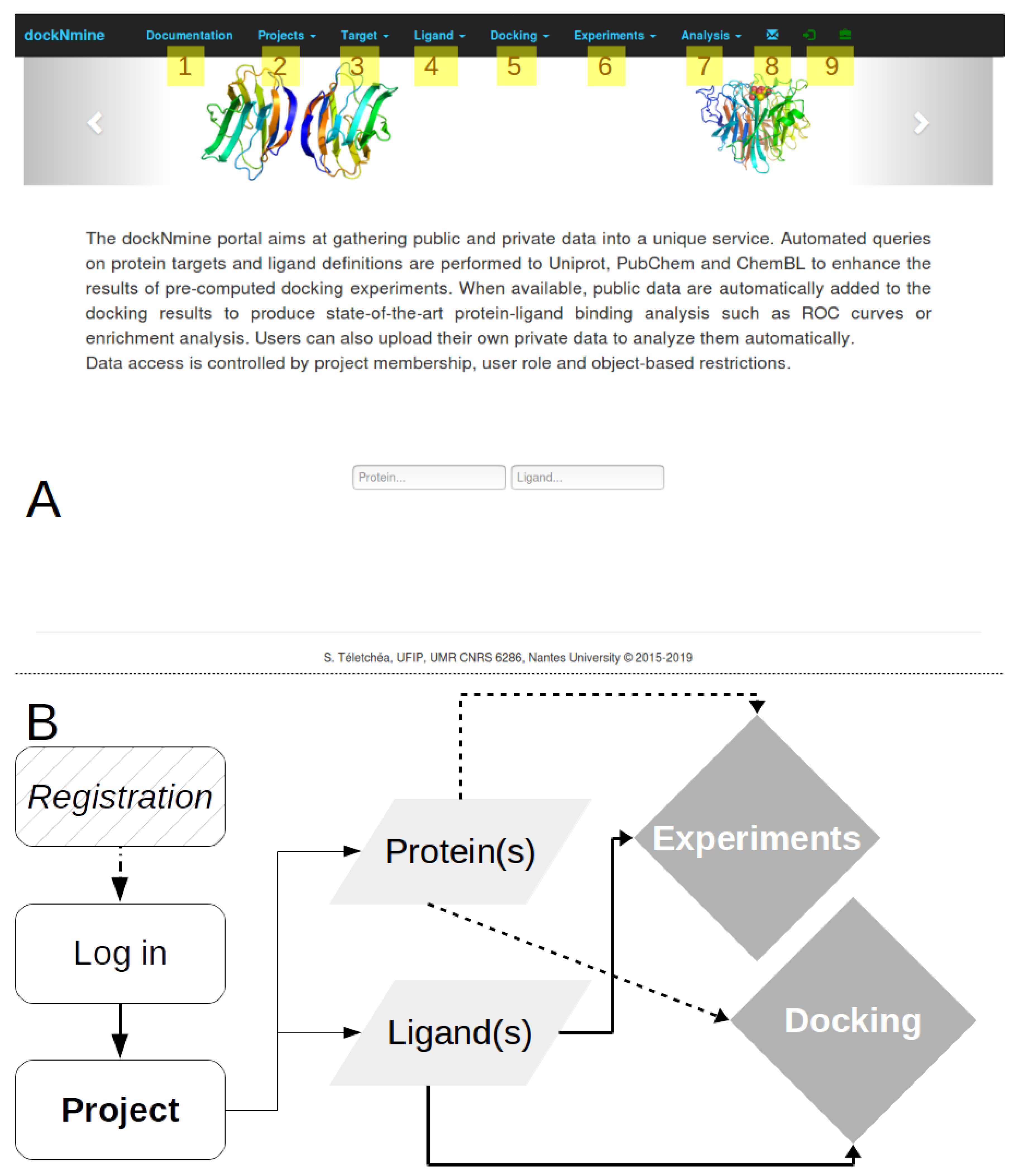
2.1. DockNmine Overview
2.2. Target Management
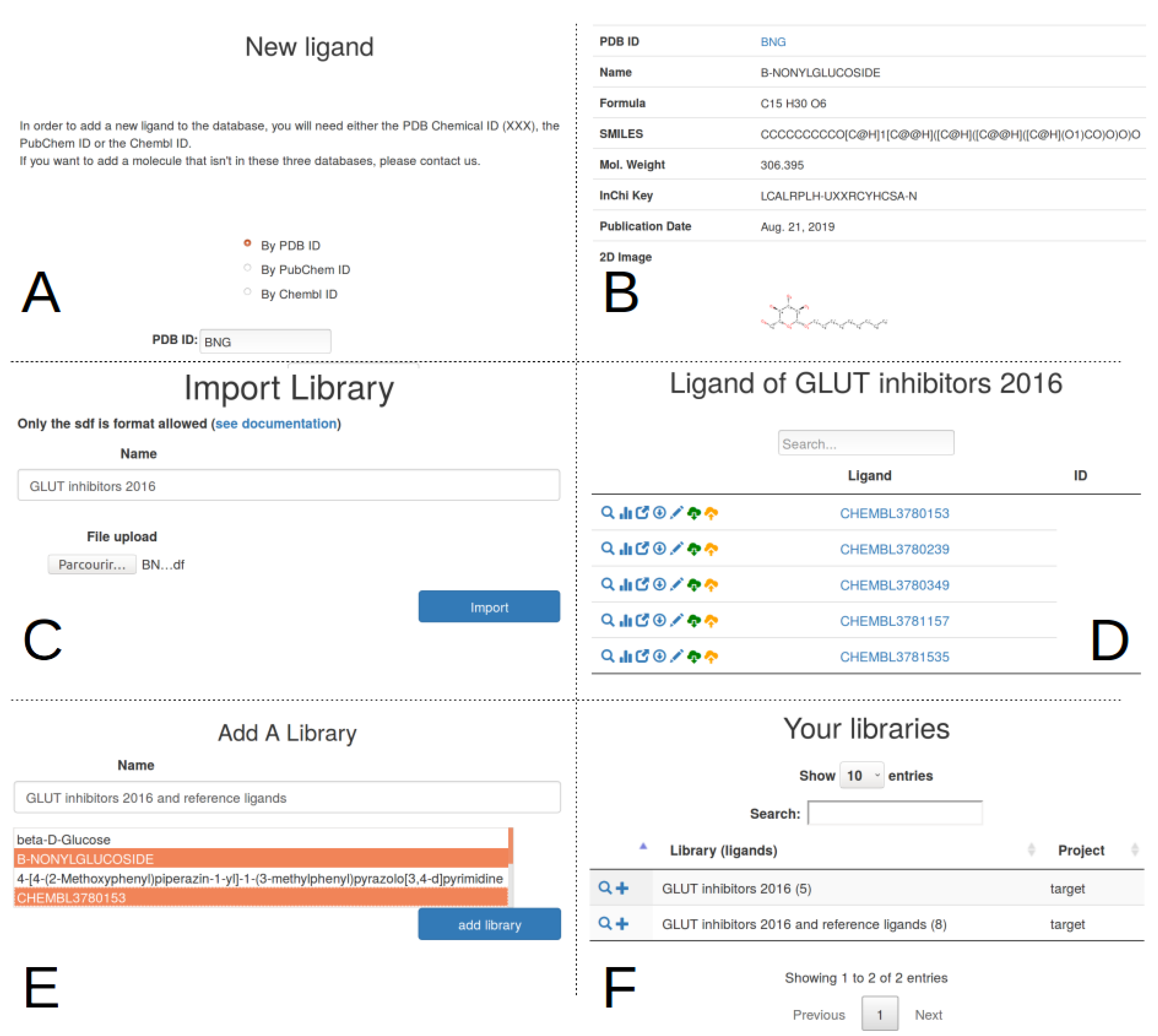
2.3. Ligand Import And Management
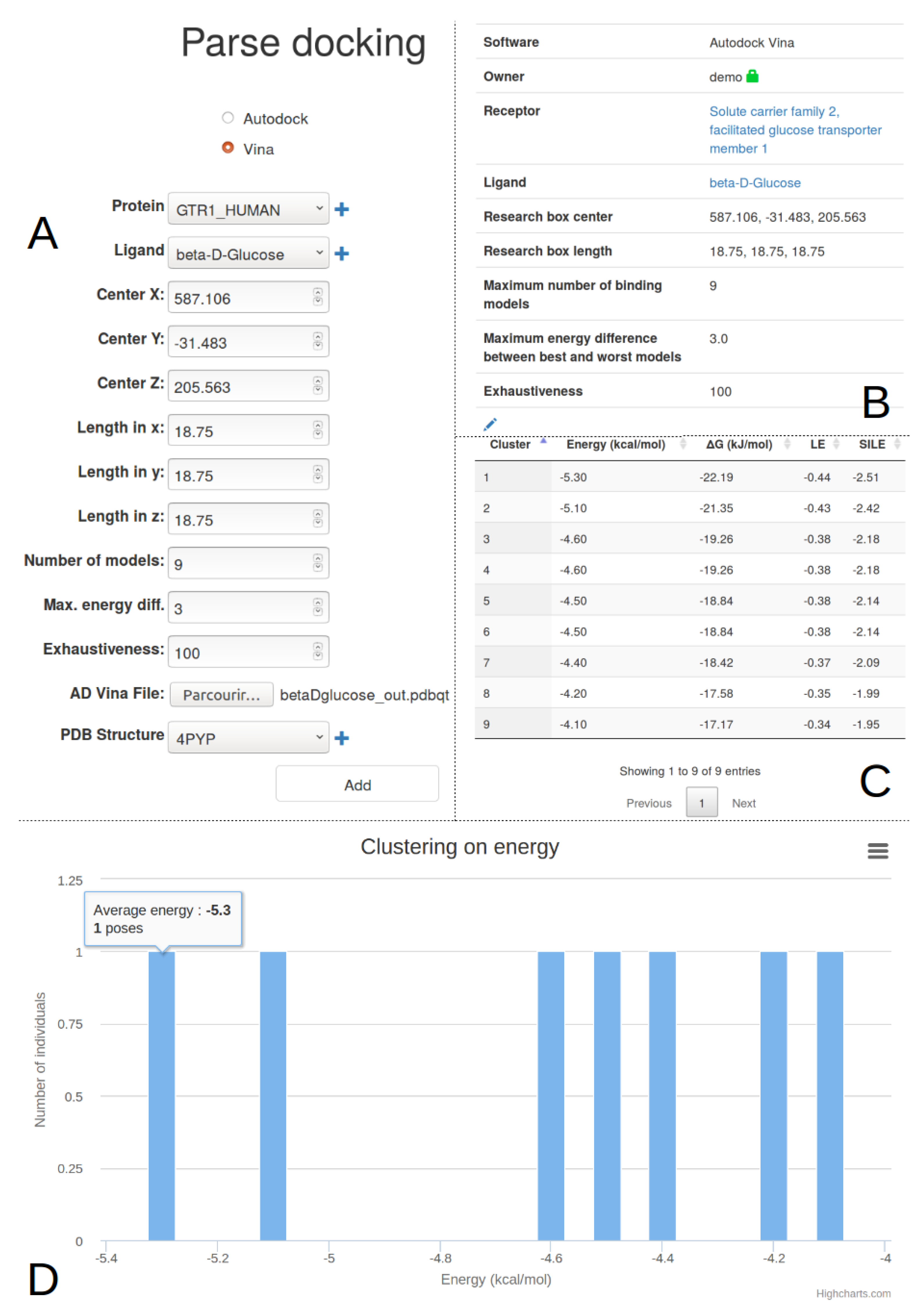
2.4. Docking Import And Management
2.5. Experimental Data
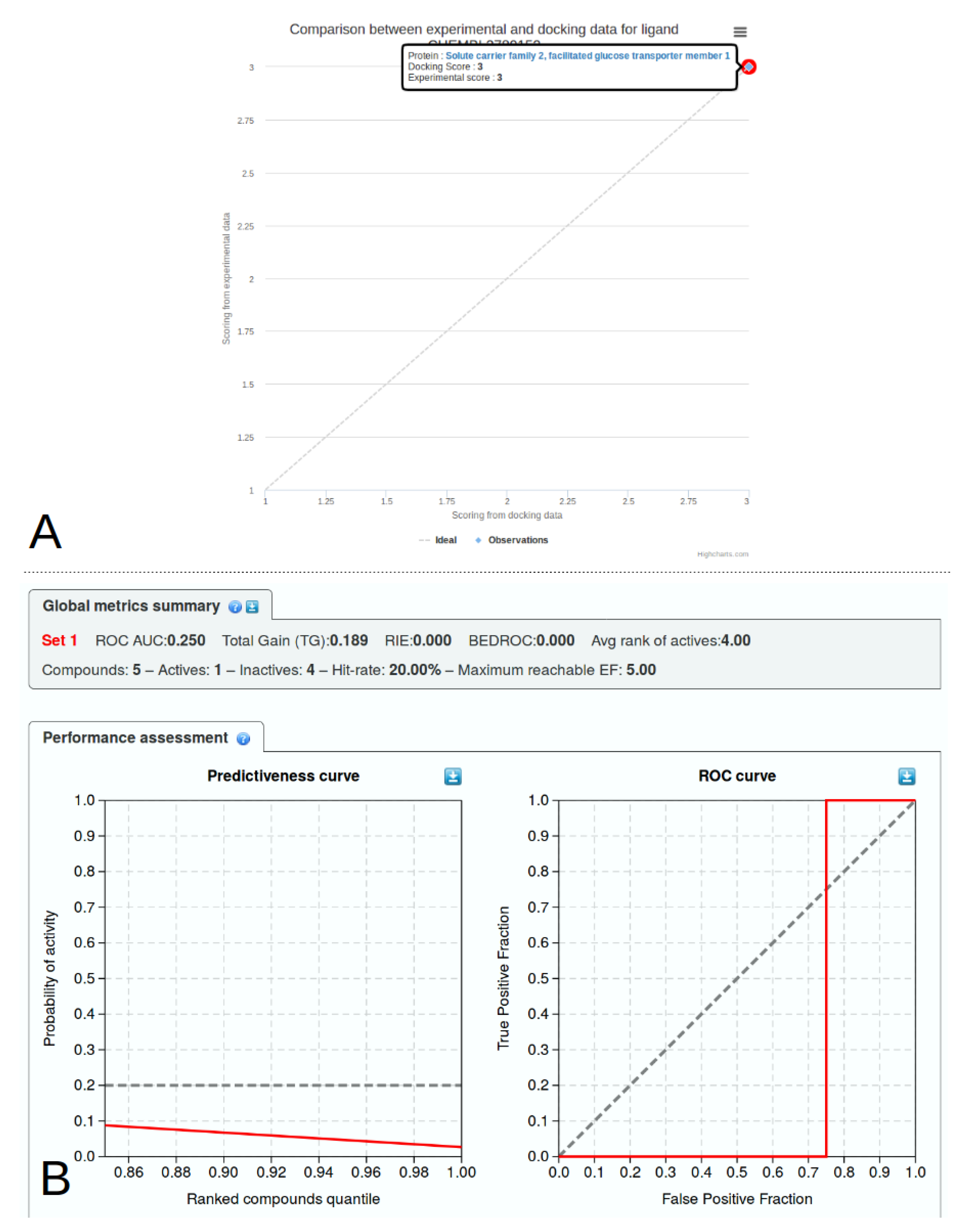
2.6. Library Analysis
2.7. Access Controls
2.8. Extending Docknmine
3. Discussion
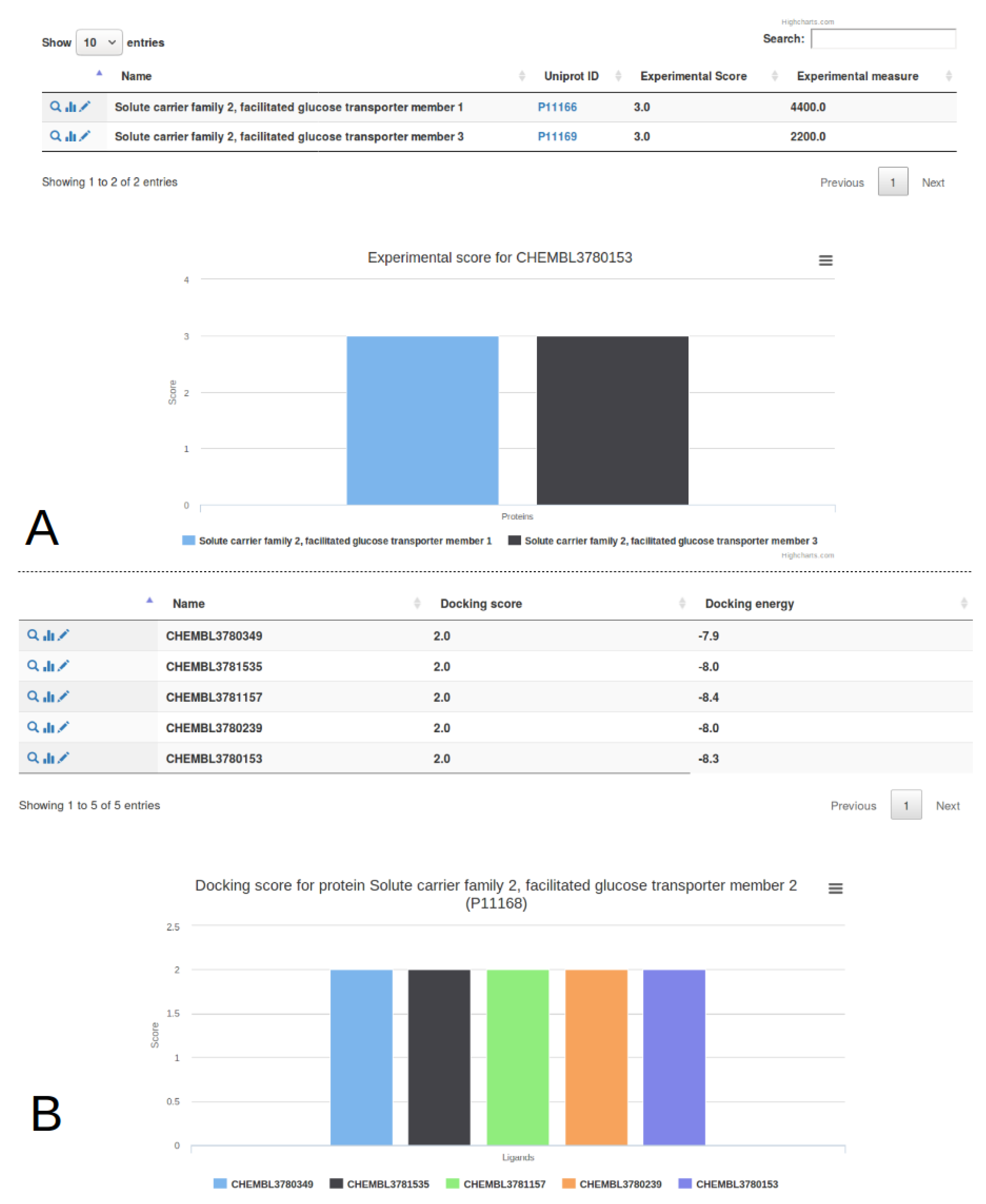
3.1. Single Protein Analysis
3.2. Multiple Proteins Analysis
3.3. Advanced Analysis
4. Materials and Methods
4.1. Server Design, Implementation And Security
4.2. Data Retrieval And Processing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AUC | Area Under the Curve |
| CADD | Computer-Aided Drug Design |
| CRUD | Create, Read, Update, Delete |
| ITC | isothermal titration calorimetry |
| LE | Ligand Efficiency |
| NMR | Nuclear Magnetic Resonance |
| PDB | Protein Data Bank |
| ROC | Receiver Operating Characteristics |
| SILE | Size-Independent Ligand Efficiency |
| SPR | Surface Plasmon Resonance |
References
- Pacanowski, M.; Huang, S.M. Precision Medicine. Clin. Pharmacol. Ther. 2016, 99, 124–129. [Google Scholar] [CrossRef]
- Schütte, M.; Ogilvie, L.A.; Rieke, D.T.; Lange, B.M.H.; Yaspo, M.L.; Lehrach, H. Cancer Precision Medicine: Why More Is More and DNA Is Not Enough. Public Health Genom. 2017, 20, 70–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzagalli, M.; Raimondi, M.; Fontana, F.; Montagnani Marelli, M.; Moretti, R.M.; Limonta, P. Cellular and molecular biology of cancer stem cells in melanoma: Possible therapeutic implications. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Zaman, A.; Wu, W.; Bivona, T.G. Targeting Oncogenic BRAF: Past, Present, and Future. Cancers 2019, 11, 1197. [Google Scholar] [CrossRef] [PubMed]
- Villoutreix, B.O.; Eudes, R.; Miteva, M.A. Structure-Based Virtual Ligand Screening: Recent Success Stories. Comb. Chem. High Throughput Screen. 2009, 12, 1000–10016. [Google Scholar] [CrossRef] [PubMed]
- Forli, S. Charting a Path to Success in Virtual Screening. Molecules 2015, 20, 18732–18758. [Google Scholar] [CrossRef] [Green Version]
- Rognan, D. Proteome-scale docking: Myth and reality. Drug Discov. Today Technol. 2013, 10, e403–e409. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Guertin, D.A.; Sabatini, D.M. Defining the Role of mTOR in Cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Deng, D.; Xu, C.; Sun, P.; Wu, J.; Yan, C.; Hu, M.; Yan, N. Crystal structure of the human glucose transporter GLUT1. Nature 2014, 510, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Galochkina, T.; Chong, M.N.F.; Challali, L.; Abbar, S.; Etchebest, C. New insights into GluT1 mechanics during glucose transfer. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Téletchéa, S.; Santuz, H.; Léonard, S.; Etchebest, C. Repository of Enriched Structures of Proteins Involved in the Red Blood Cell Environment (RESPIRE). PLoS ONE 2019, 14, e0211043. [Google Scholar] [CrossRef] [PubMed]
- Siebeneicher, H.; Bauser, M.; Buchmann, B.; Heisler, I.; Müller, T.; Neuhaus, R.; Rehwinkel, H.; Telser, J.; Zorn, L. Identification of novel GLUT inhibitors. Bioorganic Med. Chem. Lett. 2016, 26, 1732–1737. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.; Nowotka, M.; Papadatos, G.; Dedman, N.; Gaulton, A.; Atkinson, F.; Bellis, L.; Overington, J.P. ChEMBL web services: Streamlining access to drug discovery data and utilities. Nucleic Acids Res. 2015, 43, W612–W620. [Google Scholar] [CrossRef]
- Nowotka, M.M.; Gaulton, A.; Mendez, D.; Bento, A.P.; Hersey, A.; Leach, A. Using ChEMBL web services for building applications and data processing workflows relevant to drug discovery. Expert Opin. Drug Discov. 2017, 12, 757–767. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Costanzo, L.D.; Christie, C.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. Protein Data Bank: The single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2019, 47, D520–D528. [Google Scholar] [CrossRef]
- Heller, S.R.; McNaught, A.; Pletnev, I.; Stein, S.; Tchekhovskoi, D. InChI, the IUPAC International Chemical Identifier. J. Cheminform. 2015, 7, 23. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- RDKit: Open-Source Cheminformatics. Available online: http://www.rdkit.org (accessed on 28 August 2019).
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosconati, S.; Forli, S.; Perryman, A.L.; Harris, R.; Goodsell, D.S.; Olson, A.J. Virtual screening with AutoDock: Theory and practice. Expert Opin. Drug Discov. 2010, 5, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Nissink, J.W.M. Simple Size-Independent Measure of Ligand Efficiency. J. Chem. Inf. Model. 2009, 49, 1617–1622. [Google Scholar] [CrossRef] [PubMed]
- Kenny, P.W. The nature of ligand efficiency. J. Cheminform. 2019, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, E.A.; Baltos, J.A.; Nguyen, A.T.N.; Christopoulos, A.; White, P.J.; May, L.T. New paradigms in adenosine receptor pharmacology: Allostery, oligomerization and biased agonism. Br. J. Pharmacol. 2018, 175, 4036–4046. [Google Scholar] [CrossRef] [PubMed]
- Triballeau, N.; Acher, F.; Brabet, I.; Pin, J.P.; Bertrand, H.O. Virtual Screening Workflow Development Guided by the “Receiver Operating Characteristic” Curve Approach. Application to High-Throughput Docking on Metabotropic Glutamate Receptor Subtype 4. J. Med. Chem. 2005, 48, 2534–2547. [Google Scholar] [CrossRef]
- Empereur-mot, C.; Guillemain, H.; Latouche, A.; Zagury, J.F.; Viallon, V.; Montes, M. Predictiveness curves in virtual screening. J. Cheminform. 2015, 7, 52. [Google Scholar] [CrossRef]
- Lam, S.D.; Das, S.; Sillitoe, I.; Orengo, C. An overview of comparative modelling and resources dedicated to large-scale modelling of genome sequences. Acta Crystallogr. Sect. D 2017, 73, 628–640. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Marschall, T.; Marz, M.; Abeel, T.; Dijkstra, L.; Dutilh, B.E.; Ghaffaari, A.; Kersey, P.; Kloosterman, W.P.; Mäkinen, V.; Novak, A.M.; et al. Computational pan-genomics: Status, promises and challenges. Brief. Bioinform. 2018, 19, 118–135. [Google Scholar] [CrossRef]
- Kuhlman, B.; Bradley, P. Advances in protein structure prediction and design. Nat. Rev. Mol. Cell Biol. 2019, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K.; Mysinger, M.M.; Huang, N.; Colizzi, F.; Wassam, P.; Cao, Y. Automated Docking Screens: A Feasibility Study. J. Med. Chem. 2009, 52, 5712–5720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, R.G.; Sharp, K.A. Protein Pockets: Inventory, Shape, and Comparison. J. Chem. Inf. Model. 2010, 50, 589–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullock, C.; Cornia, N.; Jacob, R.; Remm, A.; Peavey, T.; Weekes, K.; Mallory, C.; Oxford, J.T.; McDougal, O.M.; Andersen, T.L. DockoMatic 2.0: High Throughput Inverse Virtual Screening and Homology Modeling. J. Chem. Inf. Model. 2013, 53, 2161–2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with PyRx. In Chemical Biology: Methods and Protocols; Hempel, J.E., Williams, C.H., Hong, C.C., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2015; pp. 243–250. [Google Scholar] [CrossRef]
- Backman, T.W.H.; Cao, Y.; Girke, T. ChemMine tools: An online service for analyzing and clustering small molecules. Nucleic Acids Res. 2011, 39, W486–W491. [Google Scholar] [CrossRef] [PubMed]
- Capuzzi, S.J.; Kim, I.S.J.; Lam, W.I.; Thornton, T.E.; Muratov, E.N.; Pozefsky, D.; Tropsha, A. Chembench: A Publicly Accessible, Integrated Cheminformatics Portal. J. Chem. Inf. Model. 2017, 57, 105–108. [Google Scholar] [CrossRef] [Green Version]
- Deng, D.; Sun, P.; Yan, C.; Ke, M.; Jiang, X.; Xiong, L.; Ren, W.; Hirata, K.; Yamamoto, M.; Fan, S.; et al. Molecular basis of ligand recognition and transport by glucose transporters. Nature 2015, 526, 391–396. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Schmidl, S.; Iancu, C.V.; Choe, J.Y.; Oreb, M. Ligand Screening Systems for Human Glucose Transporters as Tools in Drug Discovery. Front. Chem. 2018, 6, 183. [Google Scholar] [CrossRef]
- Holman, G.D. Chemical biology probes of mammalian GLUT structure and function. Biochem. J. 2018, 475, 3511–3534. [Google Scholar] [CrossRef] [Green Version]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Wójcikowski, M.; Zielenkiewicz, P.; Siedlecki, P. Open Drug Discovery Toolkit (ODDT): A new open-source player in the drug discovery field. J. Cheminform. 2015, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Leung, K.S.; Nakane, T.; Wong, M.H. iview: An interactive WebGL visualizer for protein-ligand complex. BMC Bioinform. 2014, 15, 56. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound ID | IC50 (nM) | ChEMBL ID | PubChem ID | Vina Energy (kcal/mol) |
|---|---|---|---|---|
| 13 | 1 | 3780239 | 72547759 | −6.2 |
| 3 | 25 | 3781157 | 1977736 | −7.4 |
| 66 | 80 | 3781535 | 127030174 | −5.3 |
| 63 | 510 | 3780349 | 52149799 | −6.3 |
| 41 | 44,000 | 3780153 | 127030188 | −5.3 |
| Project | Target | Ligand | Experimental Method | Docking | Library | |
|---|---|---|---|---|---|---|
| SuperUser | CRUD | CRUD | CRUD | CRUD | CRUD | CRUD |
| Manager | CRU | CRU | CRU | CRU | CRU | CRU |
| Member | R | R | CR | CR | CR | CR |
| Anonymous | R | R | R | R | R | R |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gheyouche, E.; Launay, R.; Lethiec, J.; Labeeuw, A.; Roze, C.; Amossé, A.; Téletchéa, S. DockNmine, a Web Portal to Assemble and Analyse Virtual and Experimental Interaction Data. Int. J. Mol. Sci. 2019, 20, 5062. https://doi.org/10.3390/ijms20205062
Gheyouche E, Launay R, Lethiec J, Labeeuw A, Roze C, Amossé A, Téletchéa S. DockNmine, a Web Portal to Assemble and Analyse Virtual and Experimental Interaction Data. International Journal of Molecular Sciences. 2019; 20(20):5062. https://doi.org/10.3390/ijms20205062
Chicago/Turabian StyleGheyouche, Ennys, Romain Launay, Jean Lethiec, Antoine Labeeuw, Caroline Roze, Alan Amossé, and Stéphane Téletchéa. 2019. "DockNmine, a Web Portal to Assemble and Analyse Virtual and Experimental Interaction Data" International Journal of Molecular Sciences 20, no. 20: 5062. https://doi.org/10.3390/ijms20205062