Degradation of High Energy Materials Using Biological Reduction: A Rational Way to Reach Bioremediation



Abstract
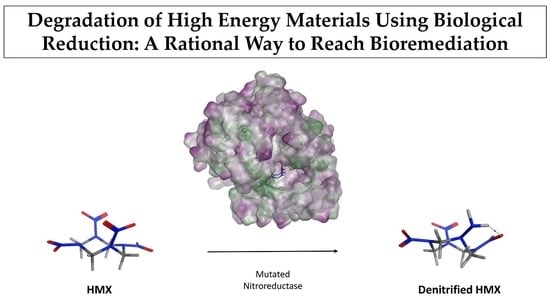
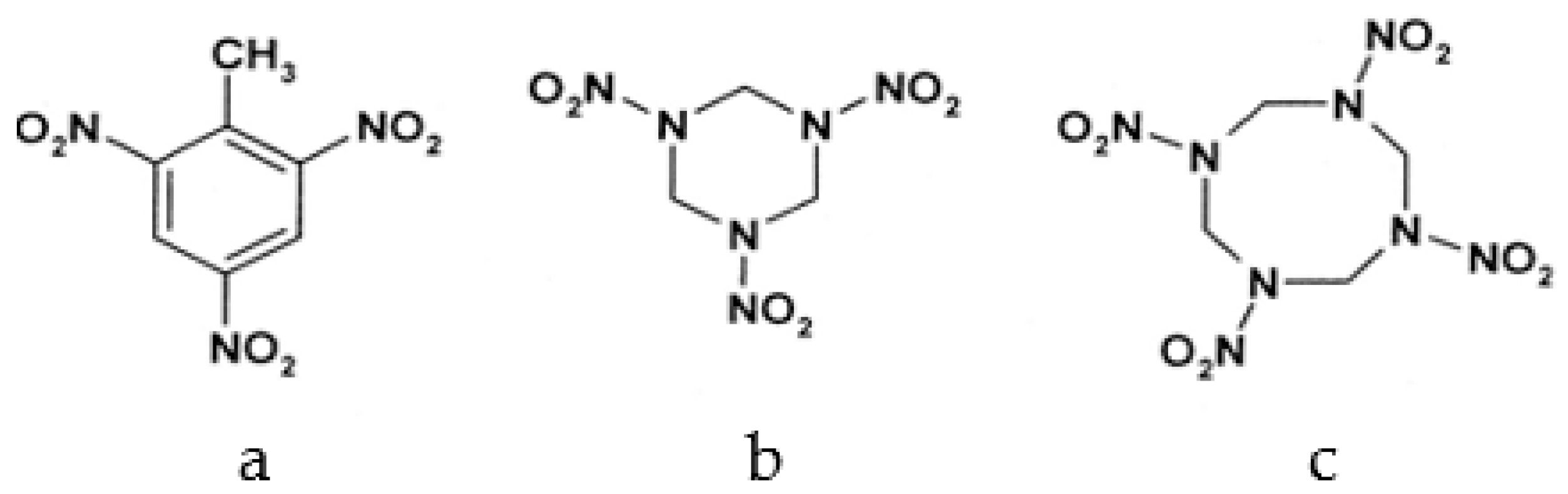
1. Introduction
2. Results
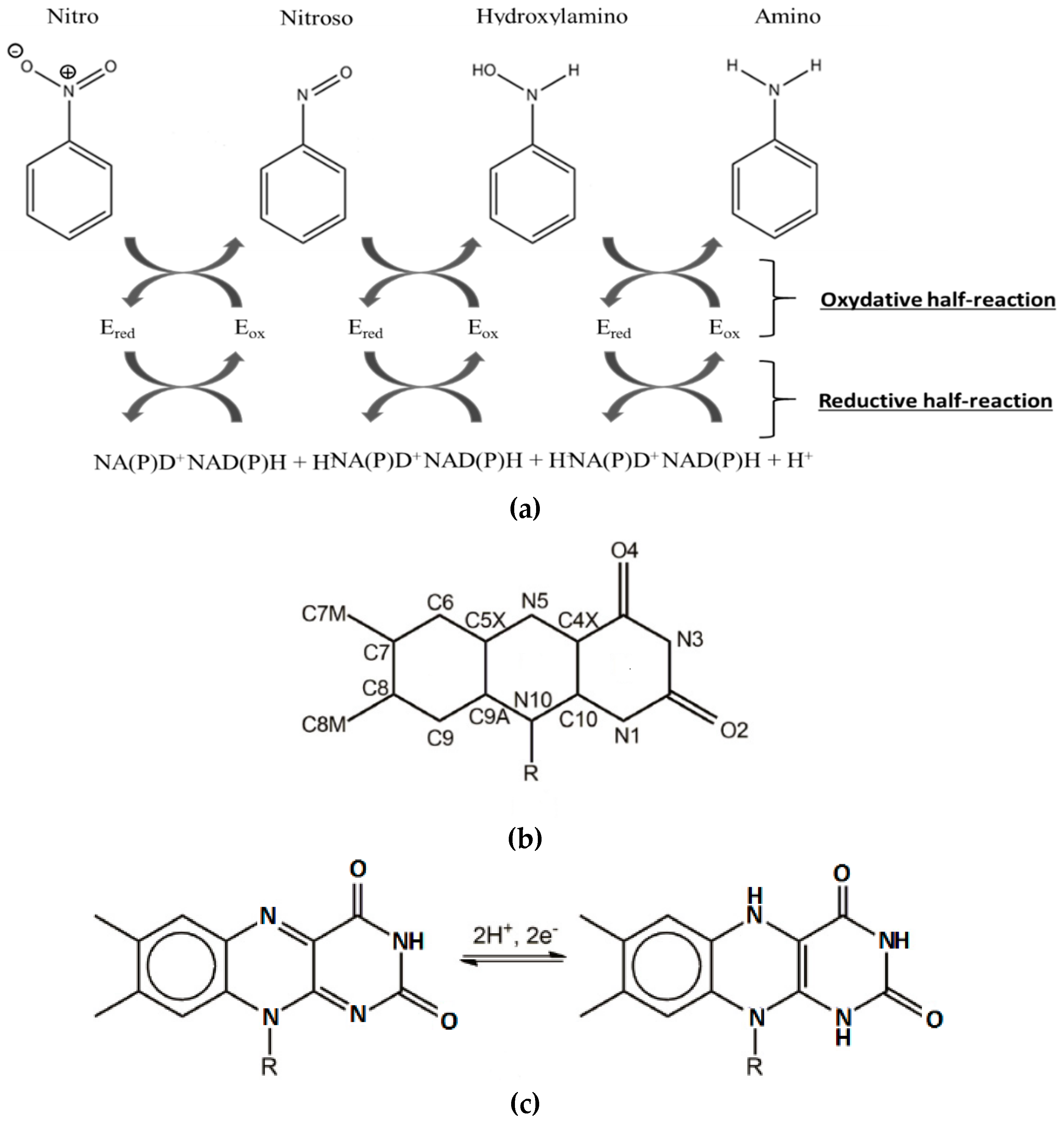
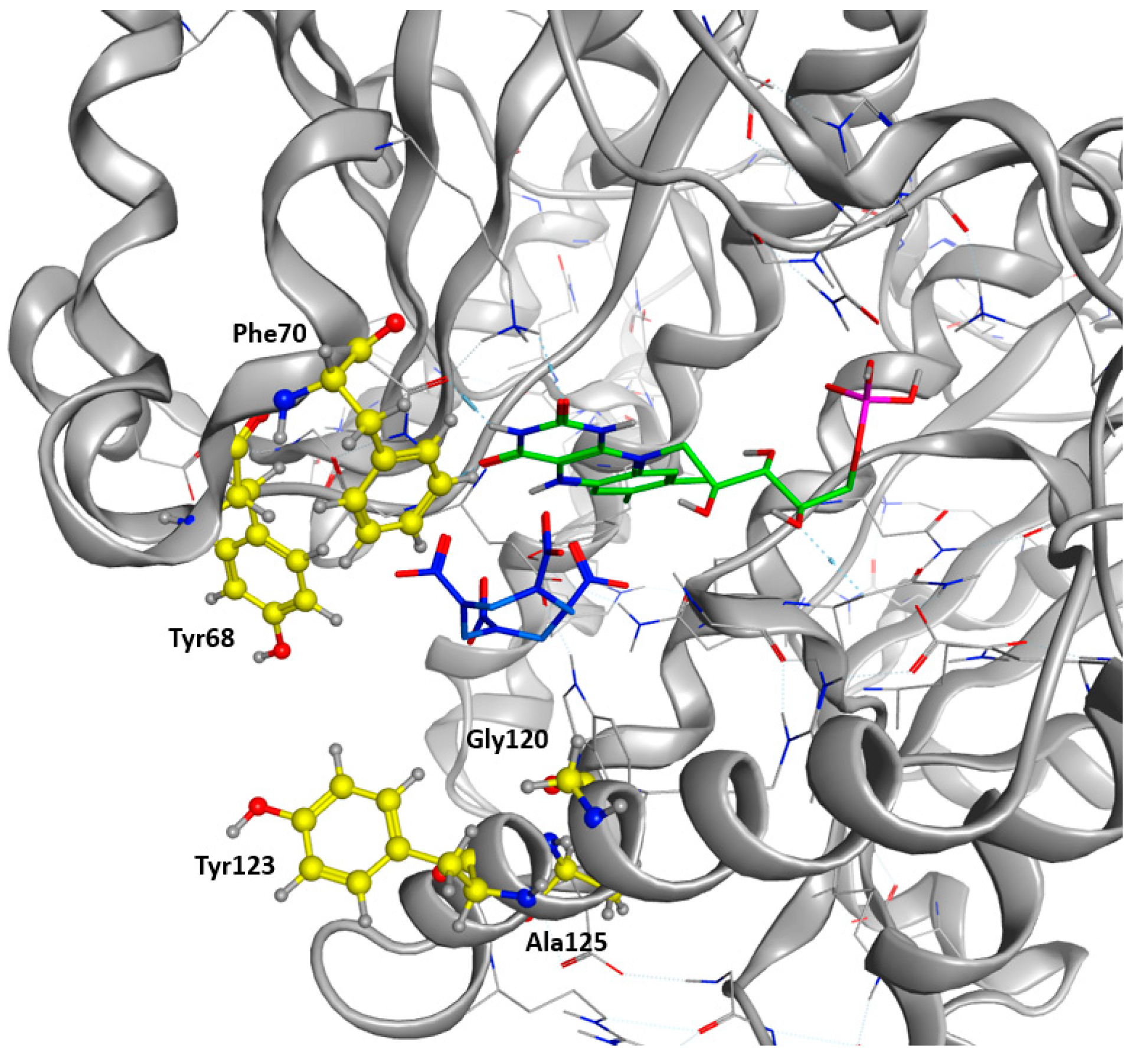
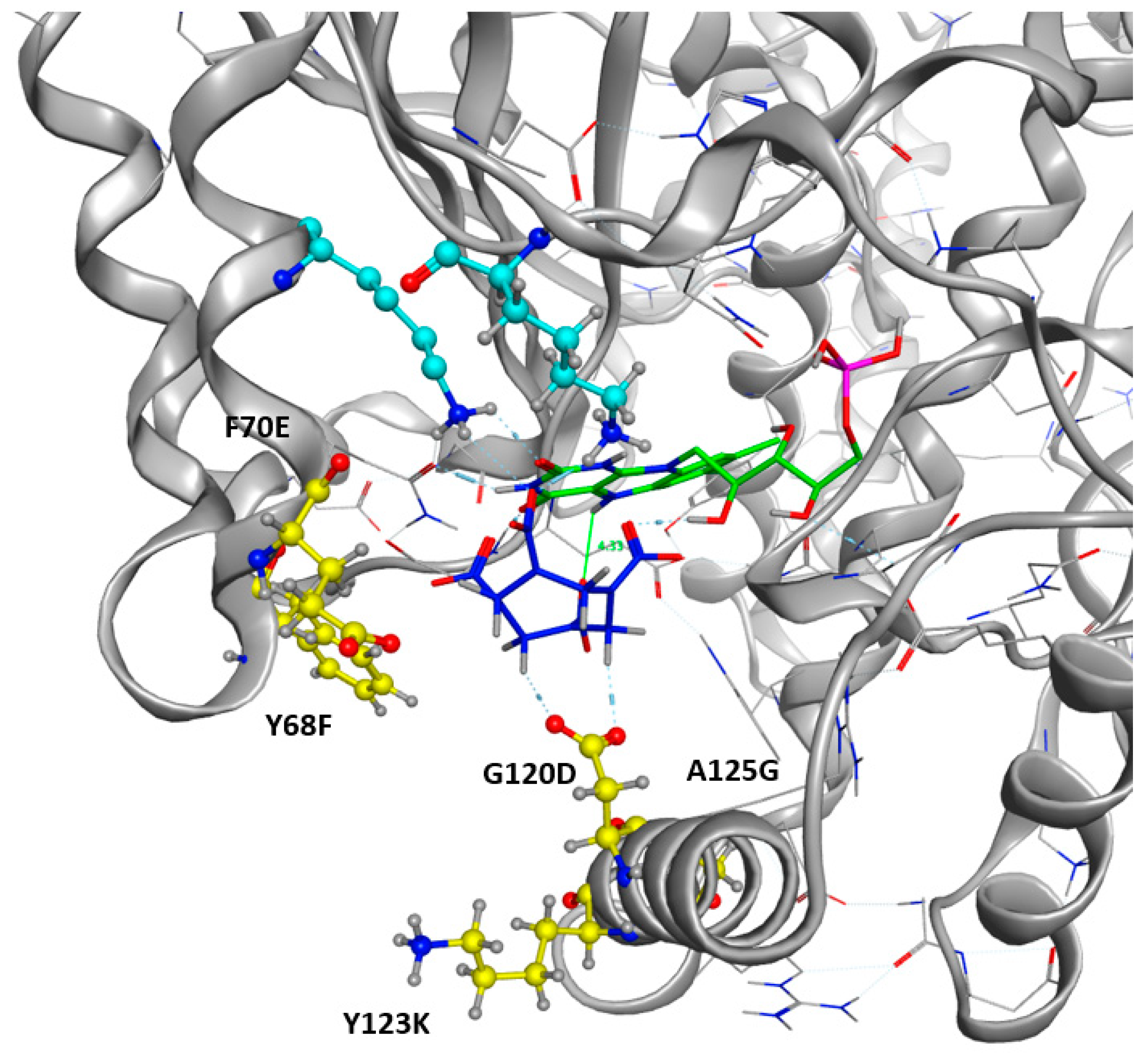
2.1. Design of the Active Site
2.2. Molecular Dynamic Simulations
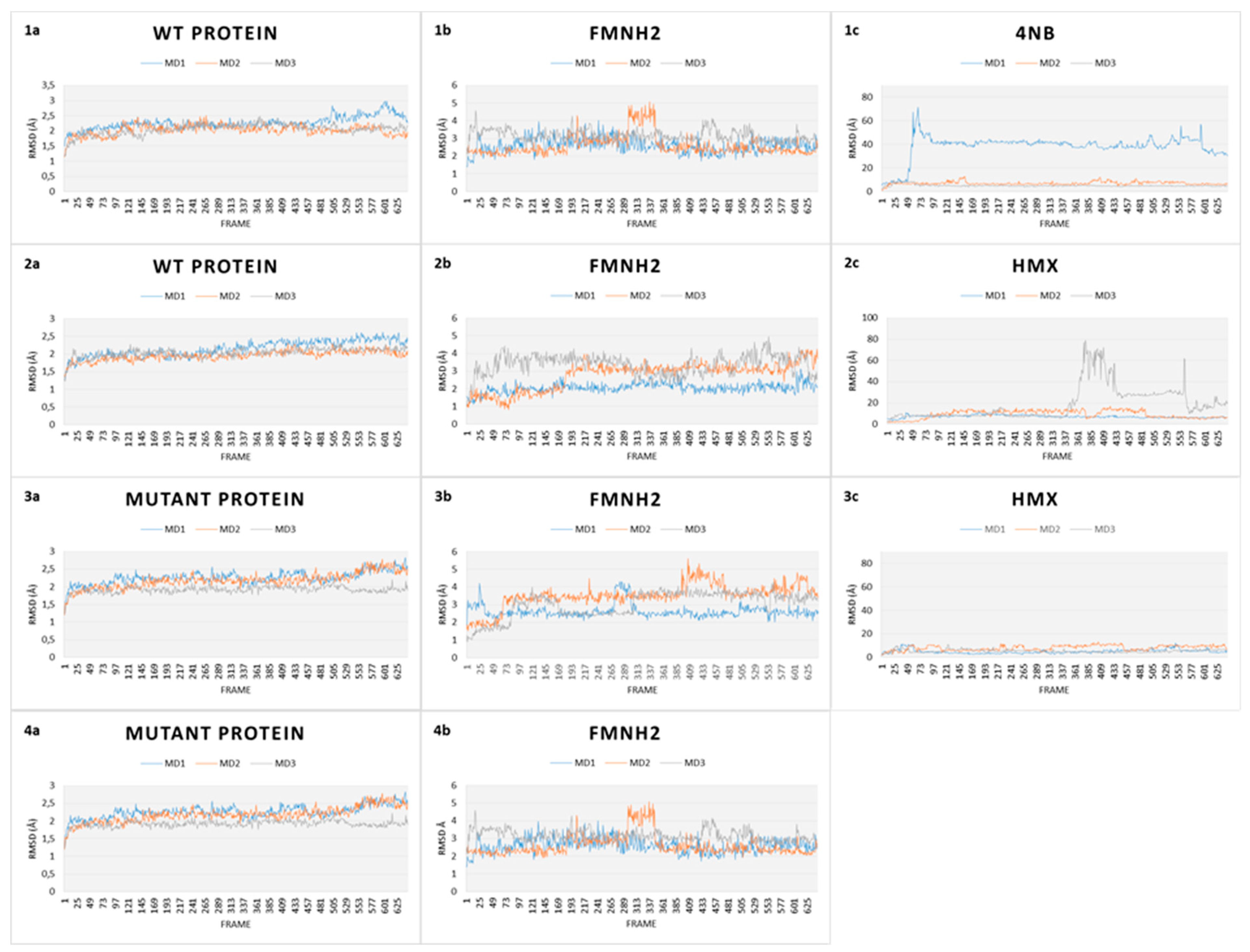
2.2.1. Global MD Analysis
2.2.2. Stability Studies
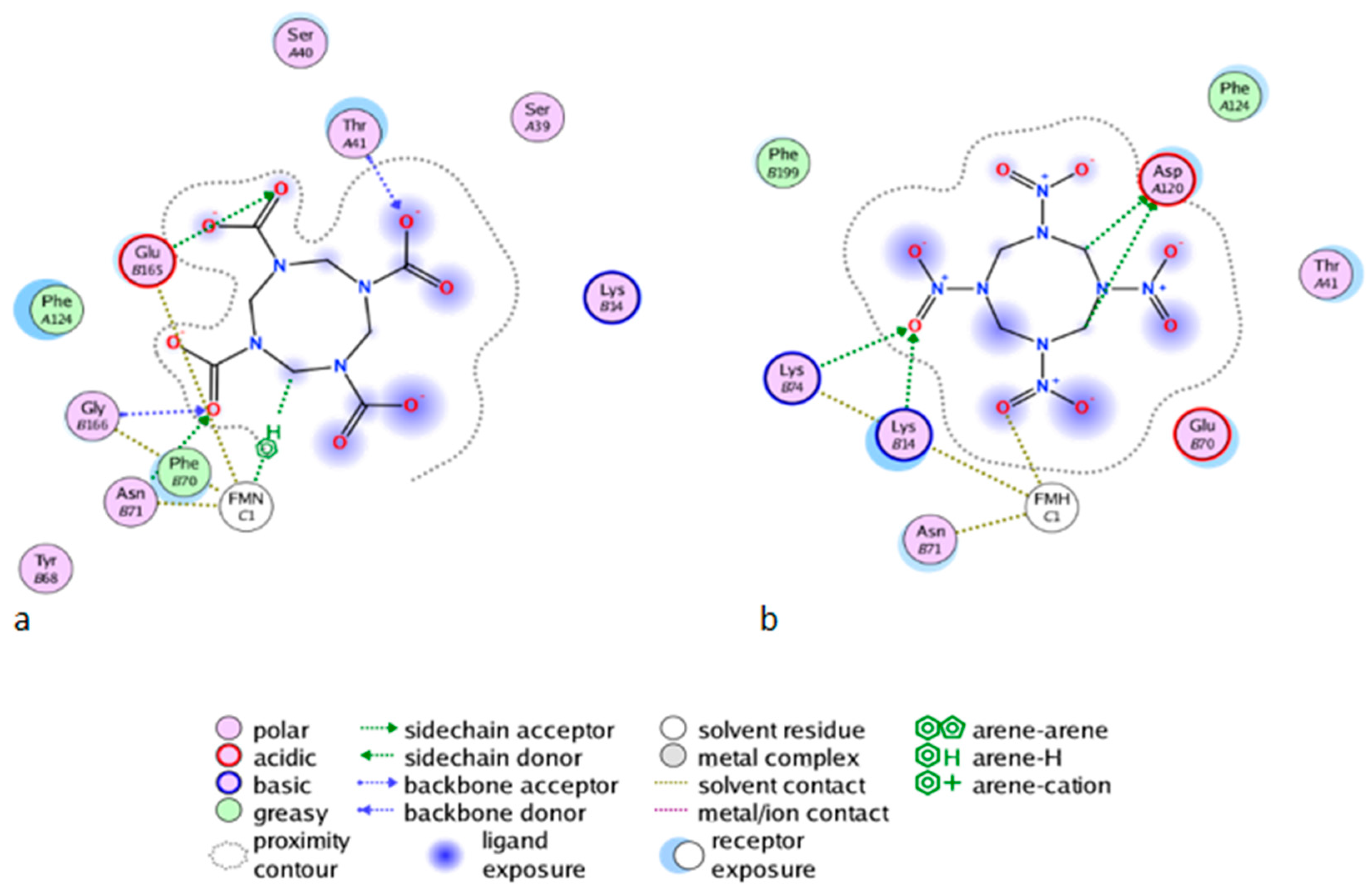
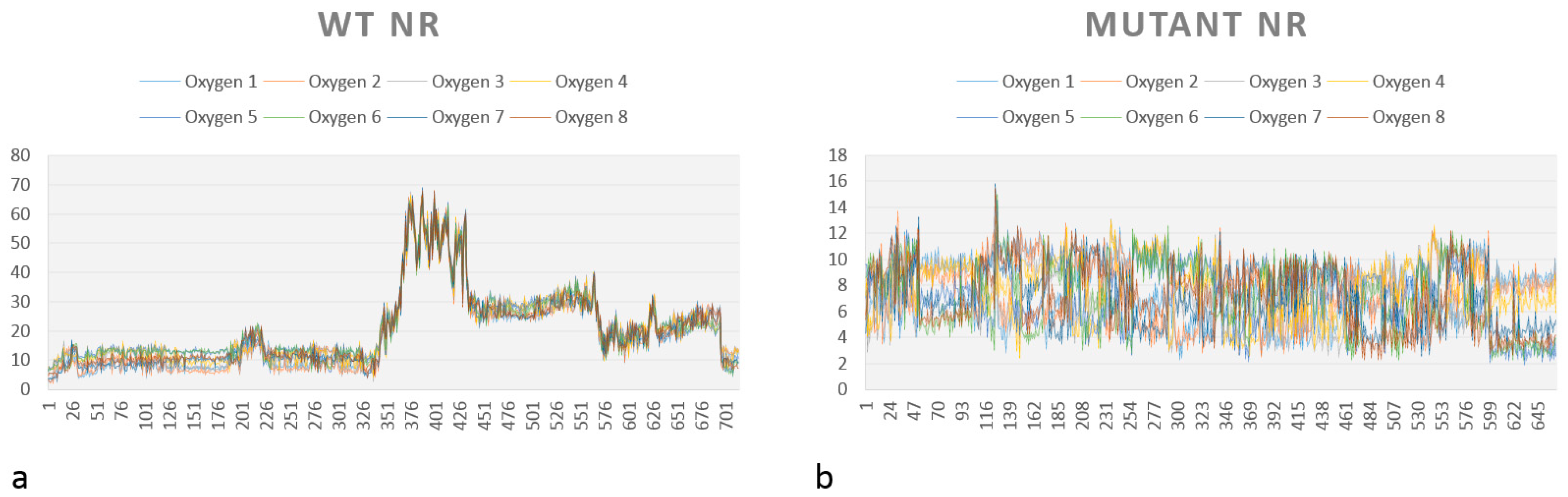
2.2.3. Affinity Studies
2.2.4. HMX Behavior during the Simulation
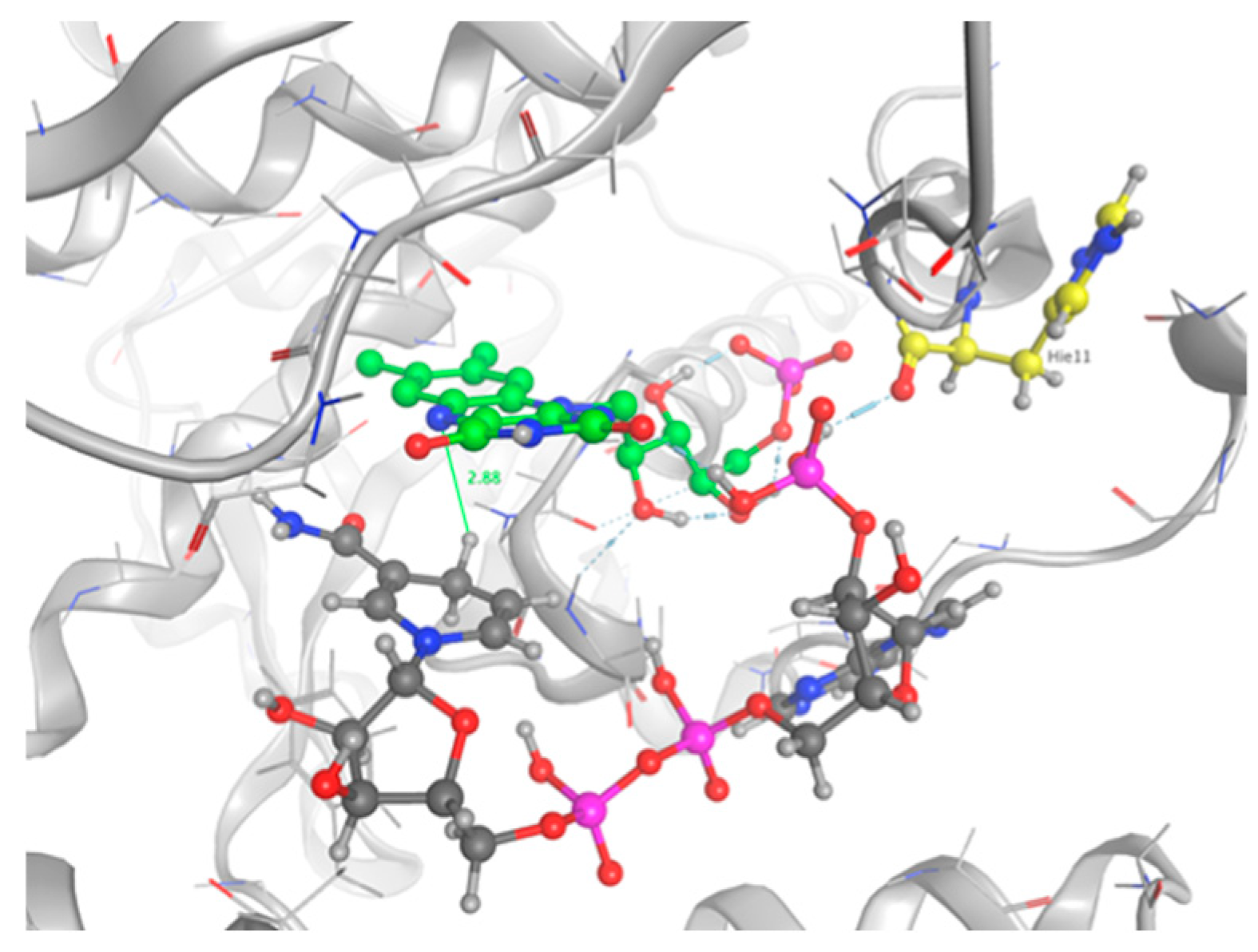
2.3. Docking Studies
3. Discussion
4. Materials and Methods
4.1. Workflow
4.2. Computational protein engineering
4.2.1. Protein Preparation
4.2.2. Cofactor Preparation
4.2.3. Ligand Preparation
4.2.4. Generation of HMX Rotamer Library
4.2.5. Resfile Generation
4.2.6. Design Method
4.2.7. Model Evaluation (Data Analysis)
4.3. Molecular Dynamics Simulations
4.3.1. MD Methods
4.3.2. Trajectory Analysis
4.3.3. Binding Free Energy Calculation
4.4. Docking Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HEM | high energy molecule |
| WWII | world war II |
| NAD(P) | oxidized form of nicotinamide adenine dinucleotide phosphate |
| NAD(P)H | reduced form of nicotinamide adenine dinucleotide phosphate |
| NR | nitroreductase |
| FMN | oxidized form of flavin mononucleotide |
| FMNH2 | reduced form of flavin mononucleotide |
| TNT | 2,4,6-TriNitroToluene |
| RDX | hexahydro-1,3,5-trinitro-1,3,5-triazine |
| HMX | octahydro-1,3,5,7-tetranitro- 1,3,5,7-tetrazocine |
| p-NBA | acid para nitro benzoic |
| NAAD | nicotinic acid adenine dinucleotide |
| MD | molecular dynamics |
| MMGBSA | molecular mechanics energies combined with the generalized born and surface area continuum solvation |
| QM/MM | quantum-mechanics/molecular-mechanics |
| RMSD | root-mean-square deviation |
| WT | wild type |
| DEE | dead end elimination |
| ALLAA | All amino acids |
| PDB | Protein data bank |
| MOE | molecular operating environment |
| AMBER | Assisted Model Building with Energy Refinement |
References
- Stierstorfer, J.; Klapötke, T.M. High Energy Materials. Propellants, Explosives and Pyrotechnics. By Jai Prakash Agrawal. Angew. Chem. Int. Ed. 2010, 49, 6253. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1002/anie.201003666 (accessed on 7 August 2019). [CrossRef]
- Chatterjee, S.; Deb, U.; Datta, S.; Walther, C.; Gupta, D.K. Common explosives (TNT, RDX, HMX) and their fate in the environment: Emphasizing bioremediation. Chemosphere 2017, 184, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Rylott, E.L.; Bruce, N.C. Right on target: Using plants and microbes to remediate explosives. Int. J. Phytoremediation 2019, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lewis, T.A.; Newcombe, D.A.; Crawford, R.L. Bioremediation of soils contaminated with explosives. J. Environ. Manag. 2004, 70, 291–307. [Google Scholar] [CrossRef]
- UN Environment. The Fog of War; UN Environment: Nairobi, Kenya, 2017; Available online: http://www.unenvironment.org/news-and-stories/story/fog-war (accessed on 19 August 2019).
- Singh, S.N. (Ed.) Biological Remediation of Explosive Residues; Springer International Publishing: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Zhang, L.; Rylott, E.L.; Bruce, N.C.; Strand, S.E. Phytodetoxification of TNT by transplastomic tobacco (Nicotiana tabacum) expressing a bacterial nitroreductase. Plant Mol. Biol. 2017, 95, 99–109. [Google Scholar] [CrossRef]
- Zhu, B.; Han, H.; Fu, X.; Li, Z.; Gao, J.; Yao, Q. Degradation of trinitrotoluene by transgenic nitroreductase in Arabidopsis plants. Plant Soil Environ. 2018, 64, 379–385. [Google Scholar] [CrossRef]
- Zhang, L.; Rylott, E.L.; Bruce, N.C.; Strand, S.E. Genetic modification of western wheatgrass (Pascopyrum smithii) for the phytoremediation of RDX and TNT. Planta 2019, 249, 1007–1015. [Google Scholar] [CrossRef]
- Beck, A.J.; Gledhill, M.; Schlosser, C.; Stamer, B.; Böttcher, C.; Sternheim, J.; Greinert, J.; Achterberg, E.P. Spread, Behavior, and Ecosystem Consequences of Conventional Munitions Compounds in Coastal Marine Waters. Front. Mar. Sci. 2018, 5. [Google Scholar] [CrossRef]
- Nagar, S.; Shaw, A.K.; Anand, S.; Celin, S.M.; Rai, P.K. Aerobic biodegradation of HMX by Planomicrobium flavidum. 3 Biotech 2018, 8, 455. [Google Scholar] [CrossRef]
- Caballero, A.; Lazaro, J.J.; Ramos, J.L.; Esteve-Nunez, A. PnrA, a new nitroreductase-family enzyme in the TNT-degrading strain Pseudomonas putida JLR11. Environ. Microbiol. 2005, 7, 1211–1219. [Google Scholar] [CrossRef]
- Kitts, C.L.; Green, C.E.; Otley, R.A.; Alvarez, M.A.; Unkefer, P.J. Type I nitroreductases in soil enterobacteria reduce TNT (2,4,6-trinitrotoluene) and RDX (hexahydro-1,3,5-trinitro-1,3,5-triazine). Can. J. Microbiol. 2000, 46, 8. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pitsawong, W.; Hoben, J.P.; Miller, A.-F. Understanding the Broad Substrate Repertoire of Nitroreductase Based on Its Kinetic Mechanism. J. Biol. Chem. 2014, 289, 15203–15214. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.A.; Koder, R.L.; Miller, A.-F.; Rodgers, D.W. Structures of nitroreductase in three states: Effects of inhibitor binding and reduction. J. Biol. Chem. 2002, 277, 11513–11520. [Google Scholar] [CrossRef] [PubMed]
- Fagan, R.L.; Palfey, B.A. 7.03—Flavin-Dependent Enzymes. In Comprehensive Natural Products II; Liu, H.-W., Mander, L., Eds.; Elsevier: Oxford, UK, 2010; pp. 37–113. [Google Scholar] [CrossRef]
- Isayev, O.; Crespo-Hernández, C.E.; Gorb, L.; Hill, F.C.; Leszczynski, J. In silico structure–function analysis of E. cloacae nitroreductase. Proteins Struct. Funct. Bioinform. 2012, 80, 2728–2741. [Google Scholar] [CrossRef]
- Fraaije, M.W.; Mattevi, A. Flavoenzymes: Diverse catalysts with recurrent features. Trends Biochem. Sci. 2000, 25, 126–132. [Google Scholar] [CrossRef]
- Iuliano, J.N.; French, J.B.; Tonge, P.J. Chapter Eight—Vibrational spectroscopy of flavoproteins. In Methods in Enzymology; Palfey, B.A., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 189–214. [Google Scholar] [CrossRef]
- Koder, R.L.; Miller, A.-F. Steady-state kinetic mechanism, stereospecificity, substrate and inhibitor specificity of Enterobacter cloacae nitroreductase1This work was supported by PRF Grant ACS-PRF 28379 (A.F.M.) and a National Science Foundation Graduate Research Fellowship (R.L.K.).1. Biochim. Biophys. Acta (BBA) Protein Struct. Mol. Enzymol. 1998, 1387, 395–405. [Google Scholar] [CrossRef]
- Race, P.R.; Lovering, A.L.; Green, R.M.; Ossor, A.; White, S.A.; Searle, P.F.; Wrighton, C.J.; Hyde, E.I. Structural and Mechanistic Studies of Escherichia coli Nitroreductase with the Antibiotic Nitrofurazone REVERSED BINDING ORIENTATIONS IN DIFFERENT REDOX STATES OF THE ENZYME. J. Biol. Chem. 2005, 280, 13256–13264. [Google Scholar] [CrossRef]
- Chaignon, P.; Cortial, S.; Ventura, A.P.; Lopes, P.; Halgand, F.; Laprevote, O.; Ouazzani, J. Purification and identification of a Bacillus nitroreductase: Potential use in 3,5-DNBTF biosensoring system. Enzym. Microb. Technol. 2006, 39, 1499–1506. [Google Scholar] [CrossRef]
- Yang, Y.; Lin, J.; Wei, D. Heterologous Overexpression and Biochemical Characterization of a Nitroreductase from Gluconobacter oxydans 621H. Mol. Biotechnol. 2016, 58, 428–440. [Google Scholar] [CrossRef]
- Miller, A.-F.; Park, J.T.; Ferguson, K.L.; Pitsawong, W.; Bommarius, A.S. Informing Efforts to Develop Nitroreductase for Amine Production. Molecules 2018, 23, 211. [Google Scholar] [CrossRef]
- Bryant, C.; DeLuca, M. Purification and characterization of an oxygen-insensitive NAD(P)H nitroreductase from Enterobacter cloacae. J. Biol. Chem. 1991, 266, 4119–4125. [Google Scholar] [PubMed]
- Pitsawong, W.; Haynes, C.A.; Koder, R.L.; Rodgers, D.W.; Miller, A.-F. Mechanism-Informed Refinement Reveals Altered Substrate-Binding Mode for Catalytically Competent Nitroreductase. Structure 2017, 25, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Driggers, C.M.; Dayal, P.V.; Ellis, H.R.; Karplus, P.A. Crystal structure of Escherichia coli SsuE: Defining a general catalytic cycle for FMN reductases of the flavodoxin-like superfamily. Biochemistry 2014, 53, 3509–3519. [Google Scholar] [CrossRef] [PubMed]
- Lovering, A.L.; Hyde, E.I.; Searle, P.F.; White, S.A. The structure of Escherichia coli nitroreductase complexed with nicotinic acid: Three crystal forms at 1.7 A, 1.8 A and 2.4 A resolution. J. Mol. Biol. 2001, 309, 203–213. [Google Scholar] [CrossRef]
- Grove, J.I.; Lovering, A.L.; Guise, C.; Race, P.R.; Wrighton, C.J.; White, S.A.; Hyde, E.I.; Searle, P.F. Generation of Escherichia Coli Nitroreductase Mutants Conferring Improved Cell Sensitization to the Prodrug CB1954. Cancer Res. 2003, 63, 5532. [Google Scholar]
- Johansson, E.; Parkinson, G.N.; Denny, W.A.; Neidle, S. Studies on the Nitroreductase Prodrug-Activating System. Crystal Structures of Complexes with the Inhibitor Dicoumarol and Dinitrobenzamide Prodrugs and of the Enzyme Active Form. J. Med. Chem. 2003, 46, 4009–4020. [Google Scholar] [CrossRef]
- Ndibe, T.O.; Benjamin, B.; Eugene, W.C.; Usman, J.J. A Review on Biodegradation and Biotransformation of Explosive Chemicals. Eur. J. Eng. Res. Sci. 2018, 3, 58–65. [Google Scholar] [CrossRef]
- Berrisford, J.M.; Sazanov, L.A. Structural basis for the mechanism of respiratory complex I. J. Biol. Chem. 2009, 284, 29773–29783. [Google Scholar] [CrossRef]
- Pai, E.F.; Karplus, P.A.; Schulz, G.E. Crystallographic analysis of the binding of NADPH, NADPH fragments, and NADPH analogues to glutathione reductase. Biochemistry 1988, 27, 4465–4474. [Google Scholar] [CrossRef]
- Zenno, S.; Koike, H.; Kumar, A.N.; Jayaraman, R.; Tanokura, M.; Saigo, K. Biochemical characterization of NfsA, the Escherichia coli major nitroreductase exhibiting a high amino acid sequence homology to Frp, a Vibrio harveyi flavin oxidoreductase. J. Bacteriol. 1996, 178, 4508–4514. [Google Scholar] [CrossRef]
- McCormick, N.G.; Feeherry, F.E.; Levinson, H.S. Microbial transformation of 2,4,6-trinitrotoluene and other nitroaromatic compounds. Appl. Environ. Microbiol. 1976, 31, 949–958. [Google Scholar] [PubMed]
- Corbett, M.D.; Corbett, B.R. Bioorganic Chemistry of the Arylhydroxylamine and Nitrosoarene Functional Groups. In Biodegradation of Nitroaromatic Compounds; Spain, J.C., Ed.; Springer: Boston, MA, USA, 1995; pp. 151–182. [Google Scholar] [CrossRef]
- Kufareva, I.; Abagyan, R. Methods of protein structure comparison. Methods Mol. Biol. 2012, 857, 231–257. [Google Scholar] [CrossRef] [PubMed]
- Looger, L.L.; Dwyer, M.A.; Smith, J.J.; Hellinga, H.W. Computational design of receptor and sensor proteins with novel functions. Nature 2003, 423, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Announcing the worldwide Protein Data Bank|Nature Structural & Molecular Biology. Available online: https://www.nature.com/articles/nsb1203-980 (accessed on 7 August 2019).
- Chemical Computing Group. Molecular Operating Environment (MOE); GTF Databanks Bulletin: Montreal, QC, Canada, 1994. [Google Scholar]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- ChemSpider|Search and Share Chemistry. Available online: http://www.chemspider.com/ (accessed on 14 August 2019).
- Cambridge Structural Database (CSD). Available online: www.ccdc.cam.ac.uk (accessed on 4 March 2019).
- Ollikainen, N.; de Jong, R.M.; Kortemme, T. Coupling Protein Side-Chain and Backbone Flexibility Improves the Re-design of Protein-Ligand Specificity. PLoS Comput. Biol. 2015, 11, e1004335. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. Available online: https://www.ncbi.nlm.nih.gov/pubmed/16458552 (accessed on 7 August 2019). [CrossRef] [PubMed]
- VMD—Visual Molecular Dynamics. Available online: https://www.ks.uiuc.edu/Research/vmd/ (accessed on 14 August 2019).
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex NR/HMX | mmGBSA (kcal/mol) |
|---|---|
| Wild Type NR/p-NBA | −15.2117 |
| Wild Type NR/HMX | −11.8261 |
| Mutant NR/HMX | −29.0704 |
| HMX Derivative Molecule | Name |
|---|---|
 | 1,3,5,7-tetranitro-1,3,5,7-tetrazocane |
 | 1,3,5-trinitro-7-nitroso-1,3,5,7-tetrazocane |
 | 1,3-ditrinitro-5,7-dinitroso-1,3,5,7-tetrazocane |
 | 1,5-ditrinitro-3,7-dinitroso-1,3,5,7-tetrazocane |
 | 1-Nitro-3,5,7-trinitroso-1,3,5,7-tetrazocane |
 | 1,3,5,7-tetranitroso-1,3,5,7-tetrazocane |
 | N-(3,5,7-trinitroso-1,3,5,7-tetrazocan-1-yl) hydroxylamine |
 | N-[3-(hydroxyamino)-5,7-dinitroso-1,3,5,7-tetrazocan-1-yl] hydroxylamine |
 | N-[5-(hydroxyamino)-3,7-dinitroso-1,3,5,7-tetrazocan-1-yl] hydroxylamine |
 | N-[(3,5-bis(hydroxyamino)-7-nitroso-1,3,5,7-tetrazocan-1-yl] hydroxylamine |
 | N-[3,5,7-tris(hydroxyamino)-1,3,5,7-tetrazocan-1-yl] hydroxylamine |
 | N1,N3,N5-trihydroxy-1,3,5,7-tetrazocane-1,3,5,7-tetramine |
 | N1,N3-dihydroxy-1,3,5,7-tetrazocane-1,3,5,7-tetramine |
 | N1,N5-dihydroxy-1,3,5,7-tetrazocane-1,3,5,7-tetramine |
 | N1-hydroxy-1,3,5,7-tetrazocane-1,3,5,7-tetramine |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguero, S.; Terreux, R. Degradation of High Energy Materials Using Biological Reduction: A Rational Way to Reach Bioremediation. Int. J. Mol. Sci. 2019, 20, 5556. https://doi.org/10.3390/ijms20225556
Aguero S, Terreux R. Degradation of High Energy Materials Using Biological Reduction: A Rational Way to Reach Bioremediation. International Journal of Molecular Sciences. 2019; 20(22):5556. https://doi.org/10.3390/ijms20225556
Chicago/Turabian StyleAguero, Stephanie, and Raphaël Terreux. 2019. "Degradation of High Energy Materials Using Biological Reduction: A Rational Way to Reach Bioremediation" International Journal of Molecular Sciences 20, no. 22: 5556. https://doi.org/10.3390/ijms20225556
APA StyleAguero, S., & Terreux, R. (2019). Degradation of High Energy Materials Using Biological Reduction: A Rational Way to Reach Bioremediation. International Journal of Molecular Sciences, 20(22), 5556. https://doi.org/10.3390/ijms20225556