Auraptene Mitigates Parkinson’s Disease-Like Behavior by Protecting Inhibition of Mitochondrial Respiration and Scavenging Reactive Oxygen Species

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
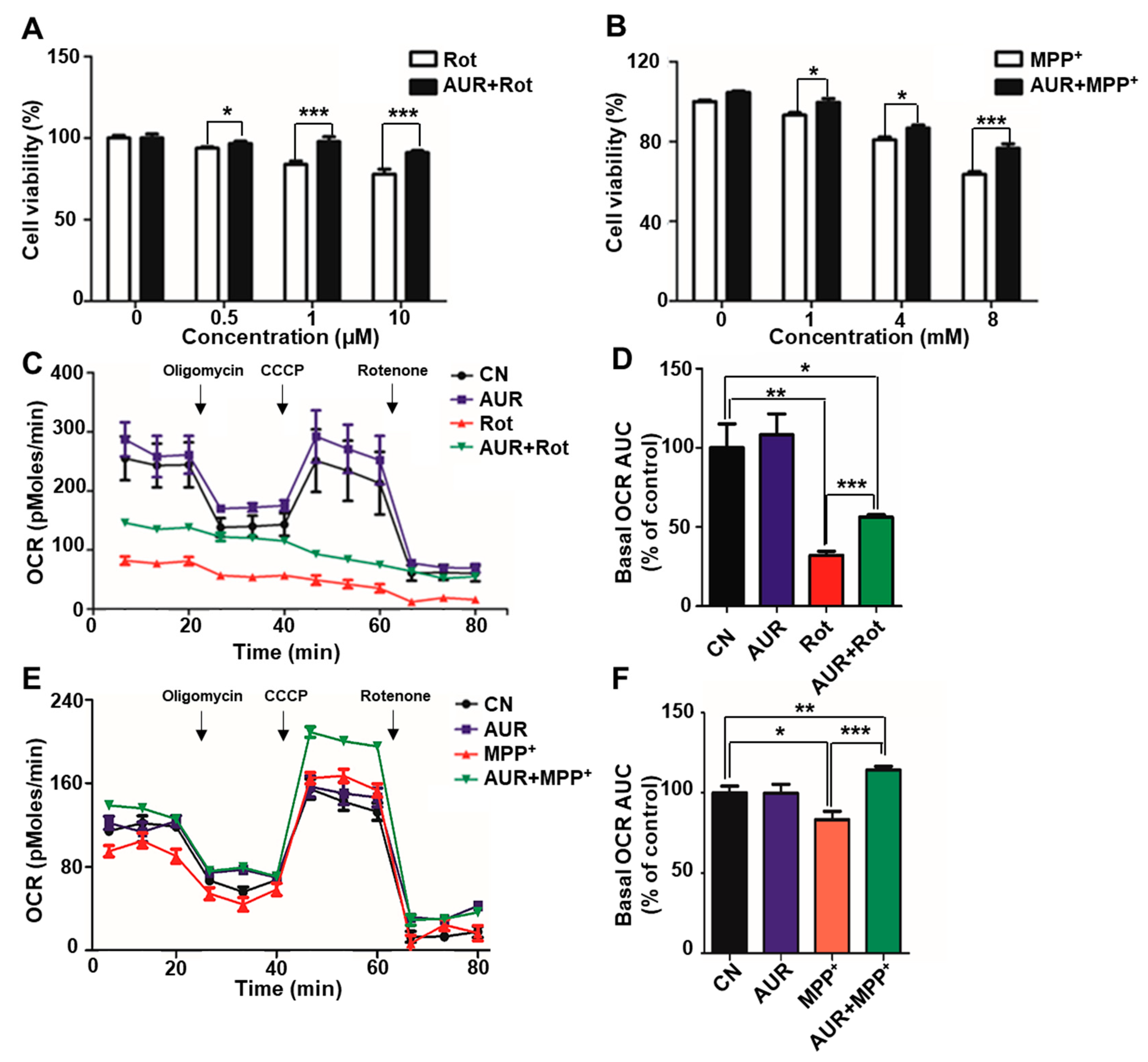
2.1. AUR Increases Cell Viability and Protects Against Neurotoxin-Induced Inhibition of Mitochondrial Respiration
2.2. AUR Induces Antioxidant Enzyme Expression in a Rotenone-Treated Cell Model
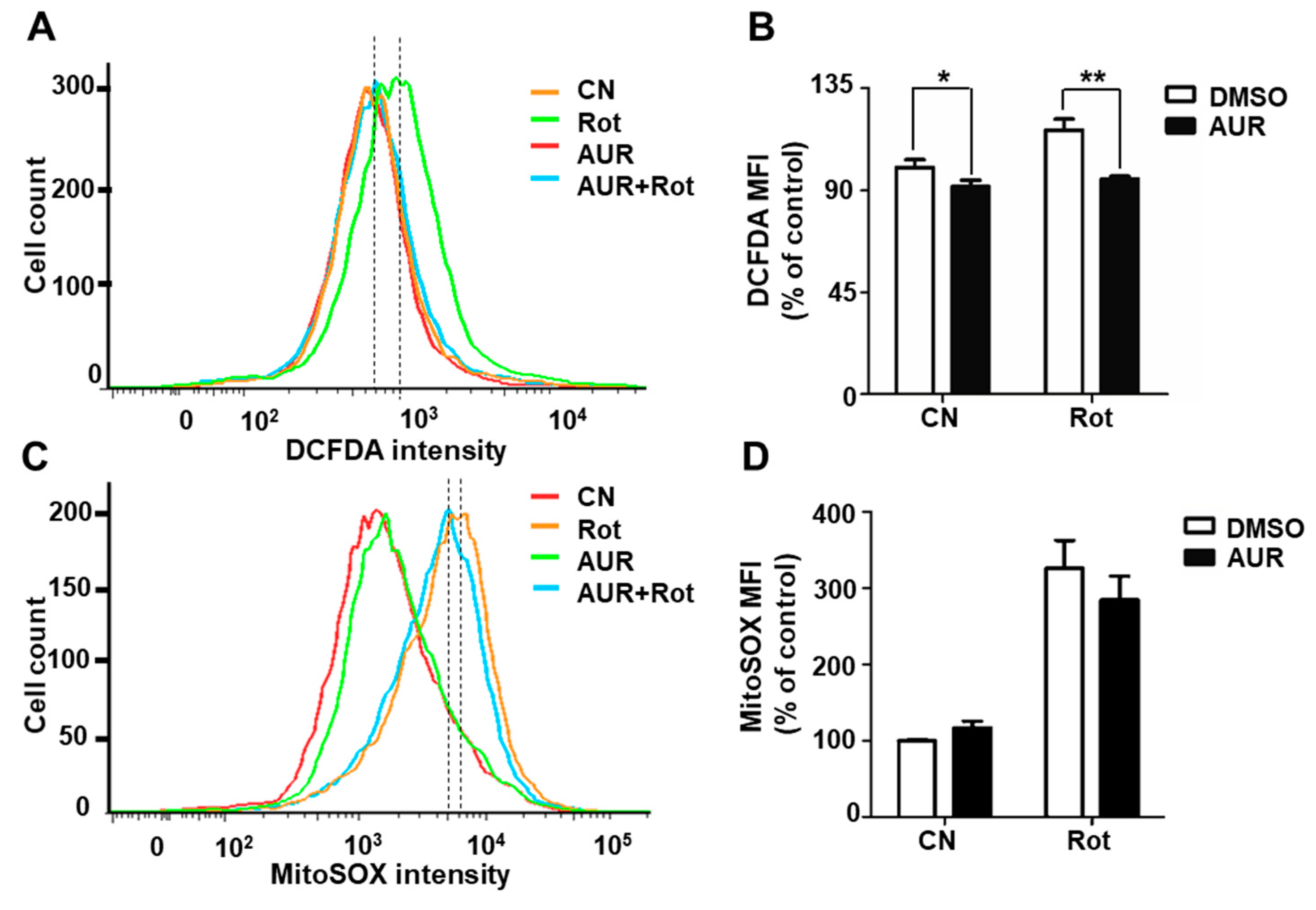
2.3. AUR Inhibits Rotenone-Induced Cytosolic ROS Production
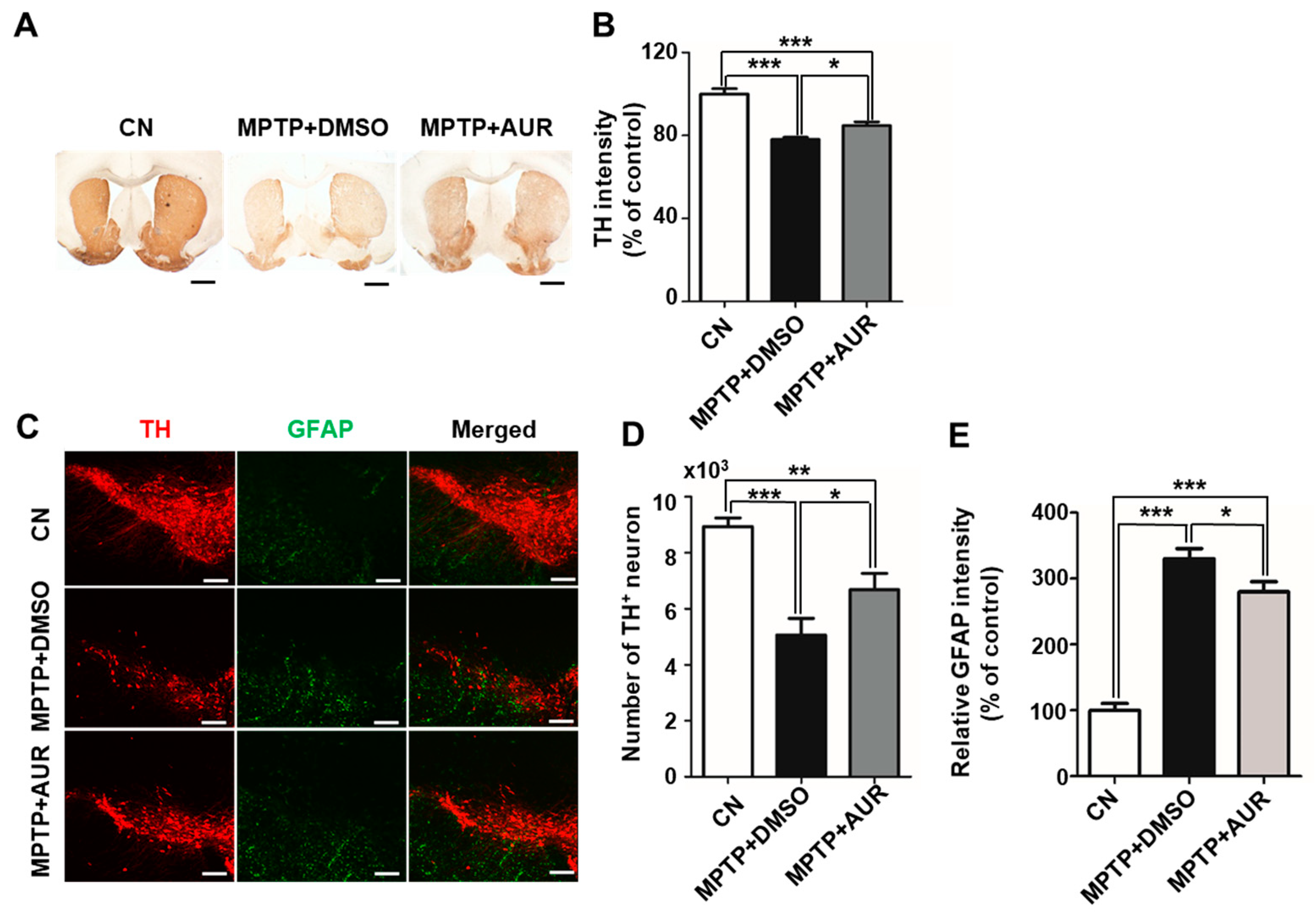
2.4. AUR Protects Neurotoxin-Induced Loss of Tyrosine Hydroxylase Expression
2.5. AUR Ameliorates MPTP-Induced Motor Deficits
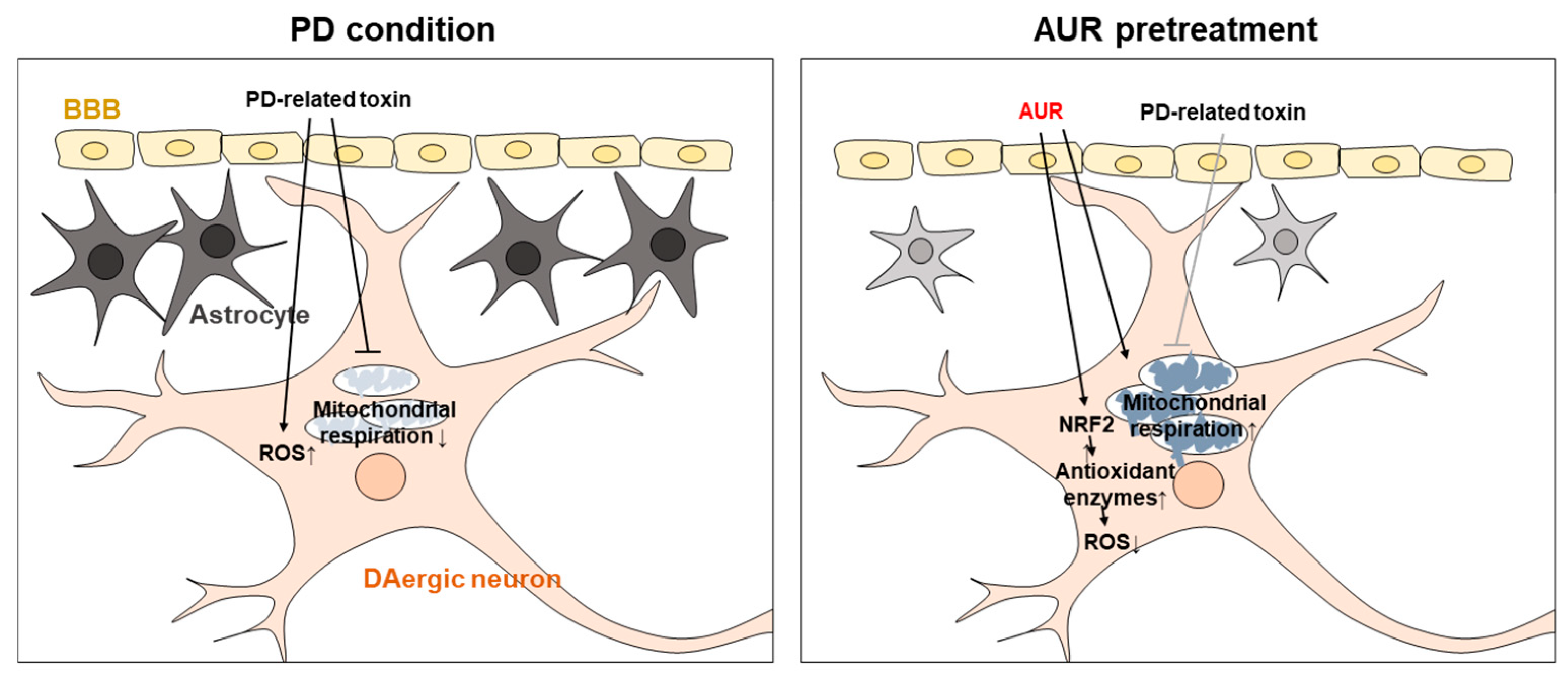
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Measurement of Cell Viability
4.3. Flow Cytometry
4.4. Measurement of Oxygen Consumption Rate (OCR)
4.5. RNA Isolation and Real Time PCR
4.6. Animal Experiments
4.7. Immunofluorescence Staining and Immunohistochemistry
4.8. Protein Isolation and Western Blotting
4.9. Behavior Test
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kalia, L.V.; Lang, A.E. Parkinson disease in 2015: Evolving basic, pathological and clinical concepts in PD. Nat. Rev. Neurol. 2016, 12, 65. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Berg, D.; Adler, C.H.; Bloem, B.R.; Chan, P.; Deuschl, G.; Gasser, T.; Goetz, C.G.; Halliday, G.; Joseph, L. The new definition and diagnostic criteria of Parkinson’s disease. Lancet Neurol. 2016, 15, 546–548. [Google Scholar] [CrossRef]
- Dal Ben, M.; Bongiovanni, R.; Tuniz, S.; Fioriti, E.; Tiribelli, C.; Moretti, R.; Gazzin, S. Earliest Mechanisms of Dopaminergic Neurons Sufferance in a Novel Slow Progressing Ex Vivo Model of Parkinson Disease in Rat Organotypic Cultures of Substantia Nigra. Int. J. Mol. Sci. 2019, 20, 2224. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Perier, C.; Vila, M. Mitochondrial biology and Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009332. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, G.; Chakrabarti, S.; Chatterjee, U.; Saso, L. Proteinopathy, oxidative stress and mitochondrial dysfunction: Cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Des. Dev. Ther. 2017, 11, 797. [Google Scholar] [CrossRef]
- Li, J.; Mahdi, F.; Du, L.; Jekabsons, M.B.; Zhou, Y.-D.; Nagle, D.G. Semisynthetic studies identify mitochondria poisons from botanical dietary supplements—Geranyloxycoumarins from Aegle marmelos. Bioorg. Med. Chem. 2013, 21, 1795–1803. [Google Scholar] [CrossRef]
- Ghanbarabadi, M.; Iranshahi, M.; Amoueian, S.; Mehri, S.; Motamedshariaty, V.S.; Mohajeri, S.A. Neuroprotective and memory enhancing effects of auraptene in a rat model of vascular dementia: Experimental study and histopathological evaluation. Neurosci. Lett. 2016, 623, 13–21. [Google Scholar] [CrossRef]
- Okuyama, S.; Morita, M.; Kaji, M.; Amakura, Y.; Yoshimura, M.; Shimamoto, K.; Ookido, Y.; Nakajima, M.; Furukawa, Y. Auraptene acts as an anti-inflammatory agent in the mouse brain. Molecules 2015, 20, 20230–20239. [Google Scholar] [CrossRef]
- Jang, Y.; Han, J.; Kim, S.J.; Kim, J.; Lee, M.J.; Jeong, S.; Ryu, M.J.; Seo, K.-S.; Choi, S.-Y.; Shong, M. Suppression of mitochondrial respiration with auraptene inhibits the progression of renal cell carcinoma: Involvement of HIF-1α degradation. Oncotarget 2015, 6, 38127. [Google Scholar] [CrossRef] [PubMed]
- Epifano, F.; Molinaro, G.; Genovese, S.; Ngomba, R.T.; Nicoletti, F.; Curini, M. Neuroprotective effect of prenyloxycoumarins from edible vegetables. Neurosci. Lett. 2008, 443, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, S.; Minami, S.; Shimada, N.; Makihata, N.; Nakajima, M.; Furukawa, Y. Anti-inflammatory and neuroprotective effects of auraptene, a citrus coumarin, following cerebral global ischemia in mice. Eur. J. Pharmacol. 2013, 699, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, S.; Semba, T.; Toyoda, N.; Epifano, F.; Genovese, S.; Fiorito, S.; Taddeo, V.A.; Sawamoto, A.; Nakajima, M.; Furukawa, Y. Auraptene and other prenyloxyphenylpropanoids suppress microglial activation and dopaminergic neuronal cell death in a lipopolysaccharide-induced model of Parkinson’s disease. Int. J. Mol. Sci. 2016, 17, 1716. [Google Scholar] [CrossRef] [PubMed]
- Tabrizian, K.; Yaghoobi, N.S.; Iranshahi, M.; Shahraki, J.; Rezaee, R.; Hashemzaei, M. Auraptene consolidates memory, reverses scopolamine-disrupted memory in passive avoidance task, and ameliorates retention deficits in mice. Iran. J. Basic Med. Sci. 2015, 18, 1014. [Google Scholar] [PubMed]
- Bove, J.; Perier, C. Neurotoxin-based models of Parkinson’s disease. Neuroscience 2012, 211, 51–76. [Google Scholar] [CrossRef] [PubMed]
- Martinez, T.N.; Greenamyre, J.T. Toxin models of mitochondrial dysfunction in Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 920–934. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, S.; Guillemin, G.J.; Abiramasundari, R.S.; Essa, M.M.; Akbar, M.; Akbar, M.D. The role of reactive oxygen species in the pathogenesis of Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease: A mini review. Oxid. Med. Cell. Longev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Perry, V.H. Microglial physiology: Unique stimuli, specialized responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Campolo, M.; Casili, G.; Biundo, F.; Crupi, R.; Cordaro, M.; Cuzzocrea, S.; Esposito, E. The neuroprotective effect of dimethyl fumarate in an MPTP-mouse model of Parkinson’s disease: Involvement of reactive oxygen species/nuclear factor-κB/nuclear transcription factor related to NF-E2. Antioxid. Redox Signal. 2017, 27, 453–471. [Google Scholar] [CrossRef]
- Zhang, Y.; Dawson, V.L.; Dawson, T.M. Oxidative stress and genetics in the pathogenesis of Parkinson’s disease. Neurobiol. Dis. 2000, 7, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Orth, M.; Schapira, A. Mitochondrial involvement in Parkinson’s disease. Neurochem. Int. 2002, 40, 533–541. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931. [Google Scholar] [CrossRef] [PubMed]
- Soltani, F.; Mosaffa, F.; Iranshahi, M.; Karimi, G.; Malekaneh, M.; Haghighi, F.; Behravan, J. Auraptene from Ferula szowitsiana protects human peripheral lymphocytes against oxidative stress. Phytother. Res. Int. J. Devoted Pharmacol. Toxicol. Eval. Nat. Prod. Deriv. 2010, 24, 85–89. [Google Scholar]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Mates, J. Effects of antioxidant enzymes in the molecular control of reactive oxygen species toxicology. Toxicology 2000, 153, 83–104. [Google Scholar] [CrossRef]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sánchez-Pérez, P.; Cadenas, S.; Lamas, S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef]
- Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J.A.; Robinson, J.P. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem. 2003, 278, 8516–8525. [Google Scholar] [CrossRef]
- Cheng, H.C.; Ulane, C.M.; Burke, R.E. Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 2010, 67, 715–725. [Google Scholar] [CrossRef]
- Meredith, G.E.; Rademacher, D.J. MPTP mouse models of Parkinson’s disease: An update. J. Parkinsons Dis. 2011, 1, 19–33. [Google Scholar]
- Ghosh, A.; Kanthasamy, A.; Joseph, J.; Anantharam, V.; Srivastava, P.; Dranka, B.P.; Kalyanaraman, B.; Kanthasamy, A.G. Anti-inflammatory and neuroprotective effects of an orally active apocynin derivative in pre-clinical models of Parkinson’s disease. J. Neuroinflamm. 2012, 9, 241. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Ryu, M.J.; Han, J.; Jang, Y.; Lee, M.J.; Ju, X.; Ryu, I.; Lee, Y.L.; Oh, E.; Chung, W.; et al. Non-cell autonomous modulation of tyrosine hydroxylase by HMGB1 released from astrocytes in an acute MPTP-induced Parkinsonian mouse model. Lab. Investig. 2019. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.; Lee, M.J.; Han, J.; Kim, S.J.; Ryu, I.; Ju, X.; Ryu, M.J.; Chung, W.; Oh, E.; Kweon, G.R.; et al. A High-fat Diet Induces a Loss of Midbrain Dopaminergic Neuronal Function That Underlies Motor Abnormalities. Exp. Neurobiol. 2017, 26, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Korchounov, A.; Meyer, M.F.; Krasnianski, M. Postsynaptic nigrostriatal dopamine receptors and their role in movement regulation. J. Neural Transm. 2010, 117, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Son, H.J.; Choi, J.H.; Ji, I.J.; Hwang, O. Vertical grid test and modified horizontal grid test are sensitive methods for evaluating motor dysfunctions in the MPTP mouse model of Parkinson’s disease. Brain Res. 2010, 1306, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Connolly, B.S.; Lang, A.E. Pharmacological treatment of Parkinson disease: A review. JAMA 2014, 311, 1670–1683. [Google Scholar] [CrossRef] [PubMed]
- Schirinzi, T.; Martella, G.; Imbriani, P.; Di Lazzaro, G.; Franco, D.; Colona, V.L.; Alwardat, M.; Sinibaldi Salimei, P.; Mercuri, N.B.; Pierantozzi, M.; et al. Dietary Vitamin E as a Protective Factor for Parkinson’s Disease: Clinical and Experimental Evidence. Front. Neurol. 2019, 10, 148. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, O.; Mandel, S.; Youdim, M.B. Gene and protein expression profiles of anti-and pro-apoptotic actions of dopamine, R-apomorphine, green tea polyphenol (−)-epigallocatechine-3-gallate, and melatonin. Ann. N. Y. Acad. Sci. 2003, 993, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Briffa, M.; Ghio, S.; Neuner, J.; Gauci, A.J.; Cacciottolo, R.; Marchal, C.; Caruana, M.; Cullin, C.; Vassallo, N.; Cauchi, R.J. Extracts from two ubiquitous Mediterranean plants ameliorate cellular and animal models of neurodegenerative proteinopathies. Neurosci. Lett. 2017, 638, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Bao, Y.; Korthuis, R.J. Mitochondrial reactive oxygen species: A double edged sword in ischemia/reperfusion vs. preconditioning. Redox Biol. 2014, 2, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B.; Liby, K.T. NRF2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer 2012, 12, 564. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Alcivar, A.L.; Ma, J.; Foo, T.K.; Zywea, S.; Mahdi, A.; Huo, Y.; Kensler, T.W.; Gatza, M.L.; Xia, B. NRF2 Induction Supporting Breast Cancer Cell Survival Is Enabled by Oxidative Stress-Induced DPP3-KEAP1 Interaction. Cancer Res. 2017, 77, 2881–2892. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, Y.; Choo, H.; Lee, M.J.; Han, J.; Kim, S.J.; Ju, X.; Cui, J.; Lee, Y.L.; Ryu, M.J.; Oh, E.S.; et al. Auraptene Mitigates Parkinson’s Disease-Like Behavior by Protecting Inhibition of Mitochondrial Respiration and Scavenging Reactive Oxygen Species. Int. J. Mol. Sci. 2019, 20, 3409. https://doi.org/10.3390/ijms20143409
Jang Y, Choo H, Lee MJ, Han J, Kim SJ, Ju X, Cui J, Lee YL, Ryu MJ, Oh ES, et al. Auraptene Mitigates Parkinson’s Disease-Like Behavior by Protecting Inhibition of Mitochondrial Respiration and Scavenging Reactive Oxygen Species. International Journal of Molecular Sciences. 2019; 20(14):3409. https://doi.org/10.3390/ijms20143409
Chicago/Turabian StyleJang, Yunseon, Hyosun Choo, Min Joung Lee, Jeongsu Han, Soo Jeong Kim, Xianshu Ju, Jianchen Cui, Yu Lim Lee, Min Jeong Ryu, Eung Seok Oh, and et al. 2019. "Auraptene Mitigates Parkinson’s Disease-Like Behavior by Protecting Inhibition of Mitochondrial Respiration and Scavenging Reactive Oxygen Species" International Journal of Molecular Sciences 20, no. 14: 3409. https://doi.org/10.3390/ijms20143409
APA StyleJang, Y., Choo, H., Lee, M. J., Han, J., Kim, S. J., Ju, X., Cui, J., Lee, Y. L., Ryu, M. J., Oh, E. S., Choi, S.-Y., Chung, W., Kweon, G. R., & Heo, J. Y. (2019). Auraptene Mitigates Parkinson’s Disease-Like Behavior by Protecting Inhibition of Mitochondrial Respiration and Scavenging Reactive Oxygen Species. International Journal of Molecular Sciences, 20(14), 3409. https://doi.org/10.3390/ijms20143409