Telomerase and Telomeres Biology in Thyroid Cancer


{kind=link}
Abstract
1. Introduction
2. TERT Promoter Mutations Are a Hallmark of TC Aggressiveness
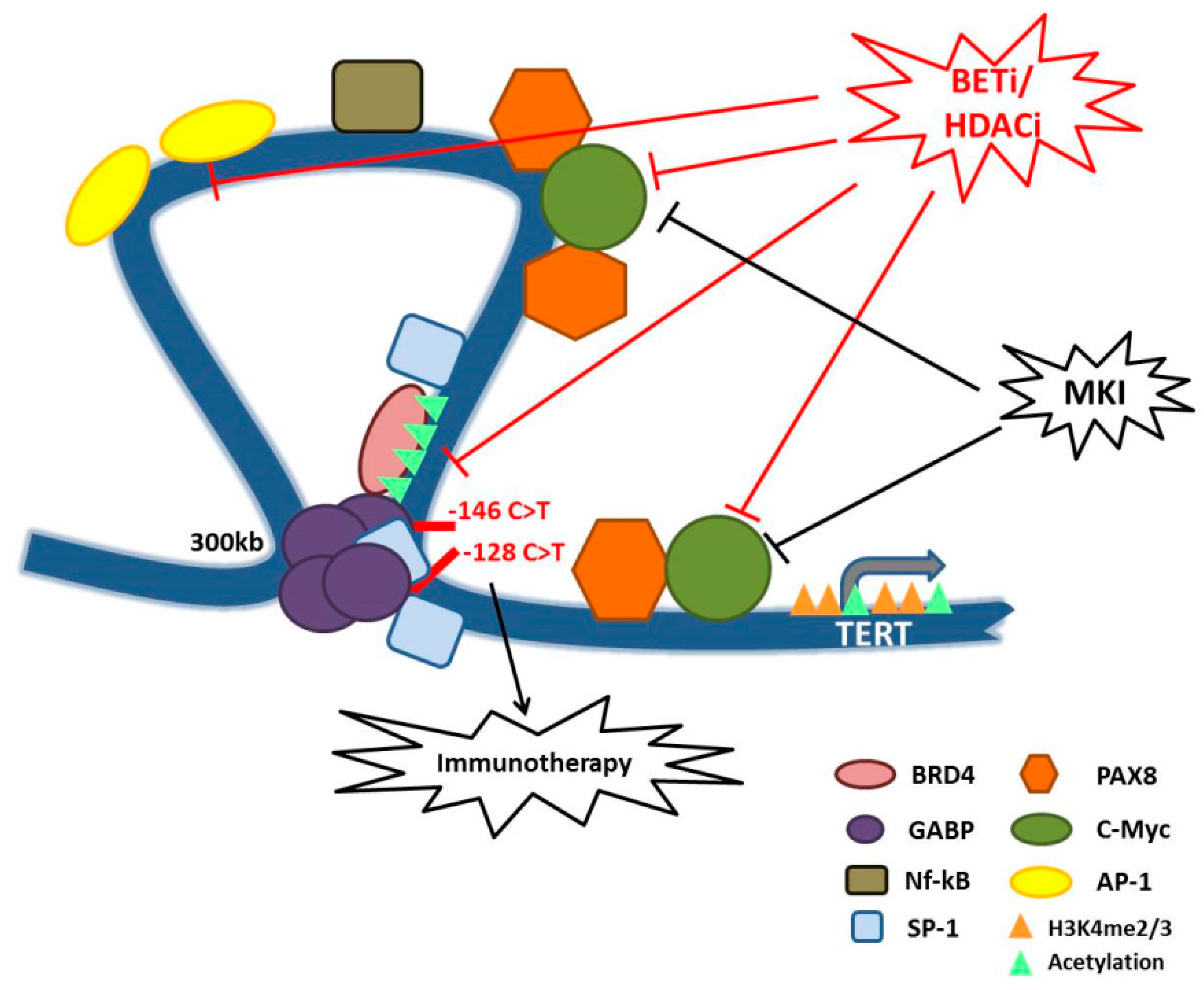
3. Additional Mechanisms of Aberrant TERT Expression Regulation in Cancer
4. Telomere Length Involvement in Thyroid Cancer Development
5. Telomere-Independent Cancer Supportive Mechanisms of TERT
6. Are Telomeres Possible Targets for TC Therapy?
7. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Blackburn, E.H. Structure and function of telomeres. Nature 1991, 350, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Palm, W.; de Lange, T. How shelterin protects mammalian telomeres. Ann. Rev. Genet. 2008, 42, 301–334. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y. The structural biology of the shelterin complex. Biol. Chem. 2019, 400, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K. How long does telomerase extend telomeres? Regulation of telomerase release and telomere length homeostasis. Curr. Genet. 2018, 64, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- de Lange, T. Shelterin-Mediated Telomere Protection. Ann. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef] [PubMed]
- De Vitis, M.; Berardinelli, F.; Sgura, A. Telomere Length Maintenance in Cancer: At the Crossroad between Telomerase and Alternative Lengthening of Telomeres (ALT). Int. J. Mol. Sci. 2018, 19, 606. [Google Scholar] [CrossRef]
- Shay, J.W.; Wright, W.E. Role of telomeres and telomerase in cancer. Semin. Cancer Biol. 2011, 21, 349–353. [Google Scholar] [CrossRef]
- Cacchione, S.; Biroccio, A.; Rizzo, A. Emerging roles of telomeric chromatin alterations in cancer. J. Exp. Clin. Cancer Res. 2019, 38, 21. [Google Scholar] [CrossRef]
- Lovejoy, C.A.; Li, W.; Reisenweber, S.; Thongthip, S.; Bruno, J.; de Lange, T.; De, S.; Petrini, J.H.; Sung, P.A.; Jasin, M.; et al. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet. 2012, 8, e1002772. [Google Scholar] [CrossRef]
- Kim, J.Y.; Brosnan-Cashman, J.A.; An, S.; Kim, S.J.; Song, K.B.; Kim, M.S.; Kim, M.J.; Hwang, D.W.; Meeker, A.K.; Yu, E.; et al. Alternative Lengthening of Telomeres in Primary Pancreatic Neuroendocrine Tumors Is Associated with Aggressive Clinical Behavior and Poor Survival. Clin. Cancer Res. 2017, 23, 1598–1606. [Google Scholar] [CrossRef]
- Lee, M.; Teber, E.T.; Holmes, O.; Nones, K.; Patch, A.M.; Dagg, R.A.; Lau, L.M.S.; Lee, J.H.; Napier, C.E.; Arthur, J.W.; et al. Telomere sequence content can be used to determine ALT activity in tumours. Nucleic Acids Res. 2018, 46, 4903–4918. [Google Scholar] [CrossRef] [PubMed]
- Heaphy, C.M.; de Wilde, R.F.; Jiao, Y.; Klein, A.P.; Edil, B.H.; Shi, C.; Bettegowda, C.; Rodriguez, F.J.; Eberhart, C.G.; Hebbar, S.; et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science 2011, 333, 425. [Google Scholar] [CrossRef] [PubMed]
- Cerone, M.A.; Londono-Vallejo, J.A.; Bacchetti, S. Telomere maintenance by telomerase and by recombination can coexist in human cells. Hum. Mol. Genet. 2001, 10, 1945–1952. [Google Scholar] [CrossRef] [PubMed]
- Elisei, R.; Molinaro, E.; Agate, L.; Bottici, V.; Masserini, L.; Ceccarelli, C.; Lippi, F.; Grasso, L.; Basolo, F.; Bevilacqua, G.; et al. Are the clinical and pathological features of differentiated thyroid carcinoma really changed over the last 35 years? Study on 4187 patients from a single Italian institution to answer this question. J. Clin. Endocrinol. Metabol. 2010, 95, 1516–1527. [Google Scholar] [CrossRef] [PubMed]
- Gandolfi, G.; Ragazzi, M.; de Biase, D.; Visani, M.; Zanetti, E.; Torricelli, F.; Sancisi, V.; Gugnoni, M.; Manzotti, G.; Braglia, L.; et al. Genome-wide profiling identifies the THYT1 signature as a distinctive feature of widely metastatic Papillary Thyroid Carcinomas. Oncotarget 2018, 9, 1813–1825. [Google Scholar] [CrossRef] [PubMed]
- Matrone, A.; Valerio, L.; Pieruzzi, L.; Giani, C.; Cappagli, V.; Lorusso, L.; Agate, L.; Puleo, L.; Viola, D.; Bottici, V.; et al. Protein kinase inhibitors for the treatment of advanced and progressive radiorefractory thyroid tumors: From the clinical trials to the real life. Best Pract. Res. Clin. Endocrinol. Metabol. 2017, 31, 319–334. [Google Scholar] [CrossRef]
- Liu, R.; Xing, M. TERT promoter mutations in thyroid cancer. Endocr. Relat. Cancer 2016, 23, R143–R155. [Google Scholar] [CrossRef]
- Liu, X.; Qu, S.; Liu, R.; Sheng, C.; Shi, X.; Zhu, G.; Murugan, A.K.; Guan, H.; Yu, H.; Wang, Y.; et al. TERT promoter mutations and their association with BRAF V600E mutation and aggressive clinicopathological characteristics of thyroid cancer. J. Clin. Endocrinol. Metabol. 2014, 99, E1130–E1136. [Google Scholar] [CrossRef]
- Landa, I.; Ganly, I.; Chan, T.A.; Mitsutake, N.; Matsuse, M.; Ibrahimpasic, T.; Ghossein, R.A.; Fagin, J.A. Frequent somatic TERT promoter mutations in thyroid cancer: Higher prevalence in advanced forms of the disease. J. Clin. Endocrinol. Metabol. 2013, 98, E1562–E1566. [Google Scholar] [CrossRef]
- Shi, X.; Liu, R.; Qu, S.; Zhu, G.; Bishop, J.; Liu, X.; Sun, H.; Shan, Z.; Wang, E.; Luo, Y.; et al. Association of TERT promoter mutation 1,295,228 C>T with BRAF V600E mutation, older patient age, and distant metastasis in anaplastic thyroid cancer. J. Clin. Endocrinol. Metabol. 2015, 100, E632–E637. [Google Scholar] [CrossRef]
- Vinagre, J.; Almeida, A.; Populo, H.; Batista, R.; Lyra, J.; Pinto, V.; Coelho, R.; Celestino, R.; Prazeres, H.; Lima, L.; et al. Frequency of TERT promoter mutations in human cancers. Nat. Commun. 2013, 4, 2185. [Google Scholar] [CrossRef] [PubMed]
- Muzza, M.; Colombo, C.; Rossi, S.; Tosi, D.; Cirello, V.; Perrino, M.; De Leo, S.; Magnani, E.; Pignatti, E.; Vigo, B.; et al. Telomerase in differentiated thyroid cancer: Promoter mutations, expression and localization. Mol. Cell. Endocrinol. 2015, 399, 288–295. [Google Scholar] [CrossRef]
- Gandolfi, G.; Ragazzi, M.; Frasoldati, A.; Piana, S.; Ciarrocchi, A.; Sancisi, V. TERT promoter mutations are associated with distant metastases in papillary thyroid carcinoma. Eur. J. Endocrinol. 2015, 172, 403–413. [Google Scholar] [CrossRef]
- Chien, M.N.; Yang, P.S.; Hsu, Y.C.; Liu, T.P.; Lee, J.J.; Cheng, S.P. Transcriptome analysis of papillary thyroid cancer harboring telomerase reverse transcriptase promoter mutation. Head Neck 2018, 40, 2528–2537. [Google Scholar] [CrossRef] [PubMed]
- de Biase, D.; Gandolfi, G.; Ragazzi, M.; Eszlinger, M.; Sancisi, V.; Gugnoni, M.; Visani, M.; Pession, A.; Casadei, G.; Durante, C.; et al. TERT Promoter Mutations in Papillary Thyroid Microcarcinomas. Thyroid 2015, 25, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- de Biase, D.; Torricelli, F.; Ragazzi, M.; Donati, B.; Khun, E.; Visani, M.; Acquaviva, G.; Pession, A.; Tallini, G.; Piana, S.; et al. Not the same thing: Metastatic PTCs have a different background than ATCs. Endocr. Connect. 2018, 7, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.; Liu, R.; Liu, X.; Murugan, A.K.; Zhu, G.; Zeiger, M.A.; Pai, S.; Bishop, J. BRAF V600E and TERT promoter mutations cooperatively identify the most aggressive papillary thyroid cancer with highest recurrence. J. Clin. Oncol. 2014, 32, 2718–2726. [Google Scholar] [CrossRef]
- Bell, R.J.; Rube, H.T.; Kreig, A.; Mancini, A.; Fouse, S.D.; Nagarajan, R.P.; Choi, S.; Hong, C.; He, D.; Pekmezci, M.; et al. Cancer. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science 2015, 348, 1036–1039. [Google Scholar] [CrossRef]
- Hollenhorst, P.C.; Ferris, M.W.; Hull, M.A.; Chae, H.; Kim, S.; Graves, B.J. Oncogenic ETS proteins mimic activated RAS/MAPK signaling in prostate cells. Genes Dev. 2011, 25, 2147–2157. [Google Scholar] [CrossRef]
- Yuan, X.; Mu, N.; Wang, N.; Strååt, K.; Sofiadis, A.; Guo, Y.; Stenman, A.; Li, K.; Cheng, G.; Zhang, L.; et al. GABPA inhibits invasion/metastasis in papillary thyroid carcinoma by regulating DICER1 expression. Oncogene 2019, 38, 965–979. [Google Scholar] [CrossRef]
- Stern, J.L.; Paucek, R.D.; Huang, F.W.; Ghandi, M.; Nwumeh, R.; Costello, J.C.; Cech, T.R. Allele-Specific DNA Methylation and Its Interplay with Repressive Histone Marks at Promoter-Mutant TERT Genes. Cell Rep. 2017, 21, 3700–3707. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhang, T.; Zhu, G.; Xing, M. Regulation of mutant TERT by BRAF V600E/MAP kinase pathway through FOS/GABP in human cancer. Nat. Commun. 2018, 9, 579. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.S.; Yoo, S.K.; Kim, H.H.; Jung, G.; Oh, A.R.; Cha, J.Y.; Kim, S.J.; Cho, S.W.; Lee, K.E.; Seo, J.S.; et al. Interaction of BRAF-induced ETS factors with mutant TERT promoter in papillary thyroid cancer. Endor. Relat. Cancer 2019, 26, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Akincilar, S.C.; Khattar, E.; Boon, P.L.; Unal, B.; Fullwood, M.J.; Tergaonkar, V. Long-Range Chromatin Interactions Drive Mutant TERT Promoter Activation. Cancer Discov. 2016, 6, 1276–1291. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, W.; Sun, H. Bromodomaincontaining protein 4 is critical for the antiproliferative and proapoptotic effects of gambogic acid in anaplastic thyroid cancer. Int. J. Mol. Med. 2018, 42, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Enomoto, K.; Zhao, L.; Zhu, Y.J.; Willingham, M.C.; Meltzer, P.; Qi, J.; Cheng, S.Y. Bromodomain and Extraterminal Protein Inhibitor JQ1 Suppresses Thyroid Tumor Growth in a Mouse Model. Clin. Cancer Res. 2017, 23, 430–440. [Google Scholar] [CrossRef]
- Wang, S.; Pike, A.M.; Lee, S.S.; Strong, M.A.; Connelly, C.J.; Greider, C.W. BRD4 inhibitors block telomere elongation. Nucleic Acids Res. 2017, 45, 8403–8410. [Google Scholar] [CrossRef]
- Wu, K.J.; Grandori, C.; Amacker, M.; Simon-Vermot, N.; Polack, A.; Lingner, J.; Dalla-Favera, R. Direct activation of TERT transcription by c-MYC. Nat. Genet. 1999, 21, 220–224. [Google Scholar] [CrossRef]
- Sakr, H.I.; Chute, D.J.; Nasr, C.; Sturgis, C.D. cMYC expression in thyroid follicular cell-derived carcinomas: A role in thyroid tumorigenesis. Diagn. Pathol. 2017, 12, 71. [Google Scholar] [CrossRef][Green Version]
- Hu, Y.J.; Luo, X.Y.; Yang, Y.; Chen, C.Y.; Zhang, Z.Y.; Guo, X. Characterization and significance of MUC1 and c-myc expression in elderly patients with papillary thyroid carcinoma. Genet. Res. 2015, 14, 15325–15330. [Google Scholar] [CrossRef]
- Abruzzese, M.P.; Bilotta, M.T.; Fionda, C.; Zingoni, A.; Soriani, A.; Vulpis, E.; Borrelli, C.; Zitti, B.; Petrucci, M.T.; Ricciardi, M.R.; et al. Inhibition of bromodomain and extra-terminal (BET) proteins increases NKG2D ligand MICA expression and sensitivity to NK cell-mediated cytotoxicity in multiple myeloma cells: Role of cMYC-IRF4-miR-125b interplay. J. Hematol. Oncol. 2016, 9, 134. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Willingham, M.C.; Qi, J.; Cheng, S.Y. Metformin and JQ1 synergistically inhibit obesity-activated thyroid cancer. Endocr. Relat. Cancer 2018, 25, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Sinha-Datta, U.; Horikawa, I.; Michishita, E.; Datta, A.; Sigler-Nicot, J.C.; Brown, M.; Kazanji, M.; Barrett, J.C.; Nicot, C. Transcriptional activation of hTERT through the NF-kappaB pathway in HTLV-I-transformed cells. Blood 2004, 104, 2523–2531. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.Y.; Chen, Y.R.; Wang, T.C. A major role of PKC theta and NFkappaB in the regulation of hTERT in human T lymphocytes. FEBS Lett. 2006, 580, 6819–6824. [Google Scholar] [CrossRef] [PubMed]
- Ramlee, M.K.; Wang, J.; Toh, W.X.; Li, S. Transcription Regulation of the Human Telomerase Reverse Transcriptase (hTERT) Gene. Genes 2016, 7, 50. [Google Scholar] [CrossRef]
- Bauerle, K.T.; Schweppe, R.E.; Haugen, B.R. Inhibition of nuclear factor-kappa B differentially affects thyroid cancer cell growth, apoptosis, and invasion. Mol. Cancer 2010, 9, 117. [Google Scholar] [CrossRef]
- Pozdeyev, N.; Berlinberg, A.; Zhou, Q.; Wuensch, K.; Shibata, H.; Wood, W.M.; Haugen, B.R. Targeting the NF-kappaB Pathway as a Combination Therapy for Advanced Thyroid Cancer. PLoS ONE 2015, 10, e0134901. [Google Scholar] [CrossRef]
- Trovato, M.; Grosso, M.; Vitarelli, E.; Ruggeri, R.M.; Alesci, S.; Trimarchi, F.; Barresi, G.; Benvenga, S. Distinctive expression of STAT3 in papillary thyroid carcinomas and a subset of follicular adenomas. Histol. Histopathol. 2003, 18, 393–399. [Google Scholar] [CrossRef]
- Couto, J.P.; Daly, L.; Almeida, A.; Knauf, J.A.; Fagin, J.A.; Sobrinho-Simoes, M.; Lima, J.; Maximo, V.; Soares, P.; Lyden, D.; et al. STAT3 negatively regulates thyroid tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, E2361–E2370. [Google Scholar] [CrossRef]
- Kim, W.G.; Choi, H.J.; Kim, W.B.; Kim, E.Y.; Yim, J.H.; Kim, T.Y.; Gong, G.; Kim, S.Y.; Chung, N.; Shong, Y.K. Basal STAT3 activities are negatively correlated with tumor size in papillary thyroid carcinomas. J. Endocrinol. Investig. 2012, 35, 413–418. [Google Scholar] [CrossRef]
- Sosonkina, N.; Starenki, D.; Park, J.I. The Role of STAT3 in Thyroid Cancer. Cancers 2014, 6, 526–544. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, E.; Ragazzi, M.; Ciarrocchi, A.; Torricelli, F.; de Biase, D. Angiosarcoma and anaplastic carcinoma of the thyroid are two distinct entities: A morphologic, immunohistochemical, and genetic study. Mod. Pathol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Campbell, H.G.; Wiles, A.K.; Eccles, M.R.; Reddel, R.R.; Braithwaite, A.W.; Royds, J.A. PAX8 regulates telomerase reverse transcriptase and telomerase RNA component in glioma. Cancer Res. 2008, 68, 5724–5732. [Google Scholar] [CrossRef] [PubMed]
- Sancisi, V.; Manzotti, G.; Gugnoni, M.; Rossi, T.; Gandolfi, G.; Gobbi, G.; Torricelli, F.; Catellani, F.; Faria do Valle, I.; Remondini, D.; et al. RUNX2 expression in thyroid and breast cancer requires the cooperation of three non-redundant enhancers under the control of BRD4 and c-JUN. Nucleic Acids Res. 2017, 45, 11249–11267. [Google Scholar] [CrossRef] [PubMed]
- Takakura, M.; Kyo, S.; Inoue, M.; Wright, W.E.; Shay, J.W. Function of AP-1 in transcription of the telomerase reverse transcriptase gene (TERT) in human and mouse cells. Mol. Cell. Biol. 2005, 25, 8037–8043. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wu, W.; Chen, X.; Gong, X. Roles of phosphatidylinositol 3-kinase regulatory subunit alpha, activator protein-1, and programmed cell death 4 in diagnosis of papillary thyroid carcinoma. Tumor Biol. 2016, 37, 6519–6526. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Huang, Y.; Gao, Q.; Feng, Z.; Li, Q.; Liu, Z. Expression of activator protein-1 in papillary thyroid carcinoma and its clinical significance. World J. Surg. Oncol. 2019, 17, 25. [Google Scholar] [CrossRef]
- Capezzone, M.; Marchisotta, S.; Cantara, S.; Busonero, G.; Brilli, L.; Pazaitou-Panayiotou, K.; Carli, A.F.; Caruso, G.; Toti, P.; Capitani, S.; et al. Familial non-medullary thyroid carcinoma displays the features of clinical anticipation suggestive of a distinct biological entity. Endocr. Relat. Cancer 2008, 15, 1075–1081. [Google Scholar] [CrossRef]
- Bonora, E.; Tallini, G.; Romeo, G. Genetic Predisposition to Familial Nonmedullary Thyroid Cancer: An Update of Molecular Findings and State-of-the-Art Studies. J. Oncol. 2010, 2010, 385206. [Google Scholar] [CrossRef]
- Tavarelli, M.; Russo, M.; Terranova, R.; Scollo, C.; Spadaro, A.; Sapuppo, G.; Malandrino, P.; Masucci, R.; Squatrito, S.; Pellegriti, G. Familial Non-Medullary Thyroid Cancer Represents an Independent Risk Factor for Increased Cancer Aggressiveness: A Retrospective Analysis of 74 Families. Front. Endocrinol. 2015, 6, 117. [Google Scholar] [CrossRef]
- Calado, R.T.; Young, N.S. Telomere diseases. N. Engl. J. Med. 2009, 361, 2353–2365. [Google Scholar] [CrossRef]
- Murnane, J.P. Telomere loss as a mechanism for chromosome instability in human cancer. Cancer Res. 2010, 70, 4255–4259. [Google Scholar] [CrossRef] [PubMed]
- Capezzone, M.; Cantara, S.; Marchisotta, S.; Filetti, S.; De Santi, M.M.; Rossi, B.; Ronga, G.; Durante, C.; Pacini, F. Short telomeres, telomerase reverse transcriptase gene amplification, and increased telomerase activity in the blood of familial papillary thyroid cancer patients. J. Clin. Endocrinol. Metabol. 2008, 93, 3950–3957. [Google Scholar] [CrossRef]
- Shen, J.; Terry, M.B.; Gurvich, I.; Liao, Y.; Senie, R.T.; Santella, R.M. Short telomere length and breast cancer risk: A study in sister sets. Cancer Res. 2007, 67, 5538–5544. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; An, C.; Zheng, H.; Lei, T.; Zhang, N.; Zheng, Y.; Yang, M. Leukocyte telomere length and risk of papillary thyroid carcinoma. J. Clin. Endocrinol. Metabol. 2019, 104, 2712–2718. [Google Scholar] [CrossRef] [PubMed]
- Cantara, S.; Pisu, M.; Frau, D.V.; Caria, P.; Dettori, T.; Capezzone, M.; Capuano, S.; Vanni, R.; Pacini, F. Telomere abnormalities and chromosome fragility in patients affected by familial papillary thyroid cancer. J. Clin. Endocrinol. Metabol. 2012, 97, E1327–E1331. [Google Scholar] [CrossRef] [PubMed]
- Ceja-Rangel, H.A.; Sanchez-Suarez, P.; Castellanos-Juarez, E.; Penaroja-Flores, R.; Arenas-Aranda, D.J.; Gariglio, P.; Benitez-Bribiesca, L. Shorter telomeres and high telomerase activity correlate with a highly aggressive phenotype in breast cancer cell lines. Tumor Biol. 2016, 37, 11917–11926. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Meeker, A.K.; Kowalski, J.; Tsai, H.L.; Somervell, H.; Heaphy, C.; Sangenario, L.E.; Prasad, N.; Westra, W.H.; Zeiger, M.A.; et al. Telomere length is related to alternative splice patterns of telomerase in thyroid tumors. Am. J. Pathol. 2011, 179, 1415–1424. [Google Scholar] [CrossRef]
- Caria, P.; Pillai, R.; Dettori, T.; Frau, D.V.; Zavattari, P.; Riva, G.; Romano, G.; Pani, F.; Bentivegna, A.; Giovannoni, R.; et al. Thyrospheres from B-CPAP Cell Line with BRAF and TERT Promoter Mutations have Different Functional and Molecular Features than Parental Cells. J. Cancer 2017, 8, 1629–1639. [Google Scholar] [CrossRef]
- Caria, P.; Cantara, S.; Frau, D.V.; Pacini, F.; Vanni, R.; Dettori, T. Genetic Heterogeneity of HER2 Amplification and Telomere Shortening in Papillary Thyroid Carcinoma. Int. J. Mol. Sci. 2016, 17, 1759. [Google Scholar] [CrossRef] [PubMed]
- Masutomi, K.; Possemato, R.; Wong, J.M.; Currier, J.L.; Tothova, Z.; Manola, J.B.; Ganesan, S.; Lansdorp, P.M.; Collins, K.; Hahn, W.C. The telomerase reverse transcriptase regulates chromatin state and DNA damage responses. Proc. Natl. Acad. Sci. USA 2005, 102, 8222–8227. [Google Scholar] [CrossRef] [PubMed]
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Muller, F.L.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Khattar, E.; Kumar, P.; Liu, C.Y.; Akincilar, S.C.; Raju, A.; Lakshmanan, M.; Maury, J.J.; Qiang, Y.; Li, S.; Tan, E.Y.; et al. Telomerase reverse transcriptase promotes cancer cell proliferation by augmenting tRNA expression. J. Clin. Investig. 2016, 126, 4045–4060. [Google Scholar] [CrossRef] [PubMed]
- Mazzolini, R.; Gonzalez, N.; Garcia-Garijo, A.; Millanes-Romero, A.; Peiro, S.; Smith, S.; Garcia de Herreros, A.; Canudas, S. Snail1 transcription factor controls telomere transcription and integrity. Nucleic Acids Res. 2018, 46, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, Q.; Li, K.; Chen, L.; Li, W.; Hou, M.; Liu, T.; Yang, J.; Lindvall, C.; Bjorkholm, M.; et al. Telomerase reverse transcriptase promotes epithelial-mesenchymal transition and stem cell-like traits in cancer cells. Oncogene 2013, 32, 4203–4213. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Gugnoni, M.; Ciarrocchi, A. Long Noncoding RNA and Epithelial Mesenchymal Transition in Cancer. Int. J. Mol. Sci. 2019, 20, 1924. [Google Scholar] [CrossRef] [PubMed]
- Gugnoni, M.; Sancisi, V.; Gandolfi, G.; Manzotti, G.; Ragazzi, M.; Giordano, D.; Tamagnini, I.; Tigano, M.; Frasoldati, A.; Piana, S.; et al. Cadherin-6 promotes EMT and cancer metastasis by restraining autophagy. Oncogene 2017, 36, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Ciarrocchi, A.; Piana, S.; Valcavi, R.; Gardini, G.; Casali, B. Inhibitor of DNA binding-1 induces mesenchymal features and promotes invasiveness in thyroid tumour cells. Eur. J. Cancer 2011, 47, 934–945. [Google Scholar] [CrossRef]
- Sancisi, V.; Gandolfi, G.; Ragazzi, M.; Nicoli, D.; Tamagnini, I.; Piana, S.; Ciarrocchi, A. Cadherin 6 is a new RUNX2 target in TGF-beta signalling pathway. PLoS ONE 2013, 8, e75489. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Tang, B.; Hu, C.J.; Xiao, Y.F.; Xie, R.; Yong, X.; Wu, Y.Y.; Dong, H.; Yang, S.M. An hTERT/ZEB1 complex directly regulates E-cadherin to promote epithelial-to-mesenchymal transition (EMT) in colorectal cancer. Oncotarget 2016, 7, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Bettin, N.; Oss Pegorar, C.; Cusanelli, E. The Emerging Roles of TERRA in Telomere Maintenance and Genome Stability. Cells 2019, 8, 246. [Google Scholar] [CrossRef] [PubMed]
- Pacini, F.; Castagna, M.G.; Brilli, L.; Pentheroudakis, G. Thyroid cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2012, 23, vii110–vii119. [Google Scholar] [CrossRef] [PubMed]
- Naoum, G.E.; Morkos, M.; Kim, B.; Arafat, W. Novel targeted therapies and immunotherapy for advanced thyroid cancers. Mol. Cancer 2018, 17, 51. [Google Scholar] [CrossRef] [PubMed]
- Brose, M.S.; Cabanillas, M.E.; Cohen, E.E.; Wirth, L.J.; Riehl, T.; Yue, H.; Sherman, S.I.; Sherman, E.J. Vemurafenib in patients with BRAF(V600E)-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: A non-randomised, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 1272–1282. [Google Scholar] [CrossRef]
- Falchook, G.S.; Millward, M.; Hong, D.; Naing, A.; Piha-Paul, S.; Waguespack, S.G.; Cabanillas, M.E.; Sherman, S.I.; Ma, B.; Curtis, M.; et al. BRAF inhibitor dabrafenib in patients with metastatic BRAF-mutant thyroid cancer. Thyroid 2015, 25, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Kurzrock, R.; Wheler, J.J.; Naing, A.; Falchook, G.S.; Fu, S.; Kim, K.B.; Davies, M.A.; Nguyen, L.M.; George, G.C.; et al. Phase I Dose-Escalation Study of the Multikinase Inhibitor Lenvatinib in Patients with Advanced Solid Tumors and in an Expanded Cohort of Patients with Melanoma. Clin. Cancer Res. 2015, 21, 4801–4810. [Google Scholar] [CrossRef] [PubMed]
- Brose, M.S.; Worden, F.P.; Newbold, K.L.; Guo, M.; Hurria, A. Effect of Age on the Efficacy and Safety of Lenvatinib in Radioiodine-Refractory Differentiated Thyroid Cancer in the Phase III SELECT Trial. J. Clin. Oncol. 2017, 35, 2692–2699. [Google Scholar] [CrossRef]
- Kim, M.J.; Kim, S.M.; Lee, E.K.; Hwangbo, Y.; Lee, Y.J.; Cho, S.W.; Park, D.J.; Lee, Y.; Park, Y.J. Tumor doubling time predicts response to sorafenib in radioactive iodine-refractory differentiated thyroid cancer. Endocr. J. 2019. [Google Scholar] [CrossRef]
- Yu, S.T.; Ge, J.N.; Luo, J.Y.; Wei, Z.G.; Sun, B.H.; Lei, S.T. Treatment-related adverse effects with TKIs in patients with advanced or radioiodine refractory differentiated thyroid carcinoma: A systematic review and meta-analysis. Cancer Manag. Res. 2019, 11, 1525–1532. [Google Scholar] [CrossRef] [PubMed]
- Schlumberger, M.; Tahara, M.; Wirth, L.J.; Robinson, B.; Brose, M.S.; Elisei, R.; Habra, M.A.; Newbold, K.; Shah, M.H.; Hoff, A.O.; et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N. Engl. J. Med. 2015, 372, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 1995, 270, 16483–16486. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Bang, Y.J.; Piha-Paul, S.A.; Razak, A.R.A.; Bennouna, J.; Soria, J.C.; Rugo, H.S.; Cohen, R.B.; O’Neil, B.H.; Mehnert, J.M.; et al. T-Cell-Inflamed Gene-Expression Profile, Programmed Death Ligand 1 Expression, and Tumor Mutational Burden Predict Efficacy in Patients Treated with Pembrolizumab Across 20 Cancers: KEYNOTE-028. J. Clin. Oncol. 2019, 37, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Mehnert, J.M.; Varga, A.; Brose, M.S.; Aggarwal, R.R.; Lin, C.C.; Prawira, A.; de Braud, F.; Tamura, K.; Doi, T.; Piha-Paul, S.A.; et al. Safety and antitumor activity of the anti-PD-1 antibody pembrolizumab in patients with advanced, PD-L1-positive papillary or follicular thyroid cancer. BMC Cancer 2019, 19, 196. [Google Scholar] [CrossRef] [PubMed]
- Cantara, S.; Bertelli, E.; Occhini, R.; Regoli, M.; Brilli, L.; Pacini, F.; Castagna, M.G.; Toti, P. Blockade of the programmed death ligand 1 (PD-L1) as potential therapy for anaplastic thyroid cancer. Endocrine 2019, 64, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Pfister, S.X.; Ashworth, A. Marked for death: Targeting epigenetic changes in cancer. Nat. Rev. Drug Discov. 2017, 16, 241–263. [Google Scholar] [CrossRef] [PubMed]
- Hogg, S.J.; Vervoort, S.J.; Deswal, S.; Ott, C.J.; Li, J.; Cluse, L.A.; Beavis, P.A.; Darcy, P.K.; Martin, B.P.; Spencer, A.; et al. BET-Bromodomain Inhibitors Engage the Host Immune System and Regulate Expression of the Immune Checkpoint Ligand PD-L1. Cell Rep. 2017, 18, 2162–2174. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed]
- Vetsika, E.K.; Papadimitraki, E.; Aggouraki, D.; Konsolakis, G.; Mela, M.E.; Kotsakis, A.; Christou, S.; Patramani, S.; Alefantinou, M.; Kaskara, A.; et al. Sequential administration of the native TERT572 cryptic peptide enhances the immune response initiated by its optimized variant TERT(572Y) in cancer patients. J. Immunother. 2011, 34, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Vetsika, E.K.; Konsolakis, G.; Aggouraki, D.; Kotsakis, A.; Papadimitraki, E.; Christou, S.; Menez-Jamet, J.; Kosmatopoulos, K.; Georgoulias, V.; Mavroudis, D. Immunological responses in cancer patients after vaccination with the therapeutic telomerase-specific vaccine Vx-001. Cancer Immunol. Immunother. 2012, 61, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Mavroudis, D.; Bolonakis, I.; Cornet, S.; Myllaki, G.; Kanellou, P.; Kotsakis, A.; Galanis, A.; Nikoloudi, I.; Spyropoulou, M.; Menez, J.; et al. A phase I study of the optimized cryptic peptide TERT(572y) in patients with advanced malignancies. Oncology 2006, 70, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Fenoglio, D.; Traverso, P.; Parodi, A.; Tomasello, L.; Negrini, S.; Kalli, F.; Battaglia, F.; Ferrera, F.; Sciallero, S.; Murdaca, G.; et al. A multi-peptide, dual-adjuvant telomerase vaccine (GX301) is highly immunogenic in patients with prostate and renal cancer. Cancer Immunol. Immunother. 2013, 62, 1041–1052. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Dannull, J.; Yang, B.K.; Dahm, P.; Coleman, D.; Yancey, D.; Sichi, S.; Niedzwiecki, D.; Boczkowski, D.; Gilboa, E.; et al. Telomerase mRNA-transfected dendritic cells stimulate antigen-specific CD8+ and CD4+ T cell responses in patients with metastatic prostate cancer. J. Immunol. 2005, 174, 3798–3807. [Google Scholar] [CrossRef] [PubMed]
- Khoury, H.J.; Collins, R.H., Jr.; Blum, W.; Stiff, P.S.; Elias, L.; Lebkowski, J.S.; Reddy, A.; Nishimoto, K.P.; Sen, D.; Wirth, E.D., 3rd; et al. Immune responses and long-term disease recurrence status after telomerase-based dendritic cell immunotherapy in patients with acute myeloid leukemia. Cancer 2017, 123, 3061–3072. [Google Scholar] [CrossRef] [PubMed]
- Soares, K.C.; Rucki, A.A.; Wu, A.A.; Olino, K.; Xiao, Q.; Chai, Y.; Wamwea, A.; Bigelow, E.; Lutz, E.; Liu, L.; et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J. Immunother. 2015, 38, 1–11. [Google Scholar] [CrossRef]
- Chiappori, A.A.; Kolevska, T.; Spigel, D.R.; Hager, S.; Rarick, M.; Gadgeel, S.; Blais, N.; Von Pawel, J.; Hart, L.; Reck, M.; et al. A randomized phase II study of the telomerase inhibitor imetelstat as maintenance therapy for advanced non-small-cell lung cancer. Ann. Oncol. 2015, 26, 354–362. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Tong, A.W.; Nemunaitis, M.; Senzer, N.; Phadke, A.P.; Bedell, C.; Adams, N.; Zhang, Y.A.; Maples, P.B.; Chen, S.; et al. A phase I study of telomerase-specific replication competent oncolytic adenovirus (telomelysin) for various solid tumors. Mol. Ther. 2010, 18, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Goldkorn, A. Telomere and Telomerase Therapeutics in Cancer. Genes 2016, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Mender, I.; Senturk, S.; Ozgunes, N.; Akcali, K.C.; Kletsas, D.; Gryaznov, S.; Can, A.; Shay, J.W.; Dikmen, Z.G. Imetelstat (a telomerase antagonist) exerts offtarget effects on the cytoskeleton. Int. J. Oncol. 2013, 42, 1709–1715. [Google Scholar] [CrossRef] [PubMed]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and Cancer: Going beyond transcriptional regulation. Mol. Cancer 2018, 17, 164. [Google Scholar] [CrossRef] [PubMed]
- Nilubol, N.; Merkel, R.; Yang, L.; Patel, D.; Reynolds, J.C.; Sadowski, S.M.; Neychev, V.; Kebebew, E. A phase II trial of valproic acid in patients with advanced, radioiodine-resistant thyroid cancers of follicular cell origin. Clin. Endocrinol. 2017, 86, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Catalano, M.G.; Pugliese, M.; Gallo, M.; Brignardello, E.; Milla, P.; Orlandi, F.; Limone, P.P.; Arvat, E.; Boccuzzi, G.; Piovesan, A. Valproic Acid, a Histone Deacetylase Inhibitor, in Combination with Paclitaxel for Anaplastic Thyroid Cancer: Results of a Multicenter Randomized Controlled Phase II/III Trial. Int. J. Endocrinol. 2016, 2016, 2930414. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, Y.; Shan, W.; Hu, Z.; Yuan, J. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Barthel, F.P.; Wei, W.; Tang, M. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef]
- Cantara, S.; Capuano, S.; Capezzone, M.; Benigni, M.; Pisu, M.; Marchisotta, S.; Pacini, F. Lack of mutations of the telomerase RNA component in familial papillary thyroid cancer with short telomeres. Thyroid 2012, 22, 363–368. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donati, B.; Ciarrocchi, A. Telomerase and Telomeres Biology in Thyroid Cancer. Int. J. Mol. Sci. 2019, 20, 2887. https://doi.org/10.3390/ijms20122887
Donati B, Ciarrocchi A. Telomerase and Telomeres Biology in Thyroid Cancer. International Journal of Molecular Sciences. 2019; 20(12):2887. https://doi.org/10.3390/ijms20122887
Chicago/Turabian StyleDonati, Benedetta, and Alessia Ciarrocchi. 2019. "Telomerase and Telomeres Biology in Thyroid Cancer" International Journal of Molecular Sciences 20, no. 12: 2887. https://doi.org/10.3390/ijms20122887
APA StyleDonati, B., & Ciarrocchi, A. (2019). Telomerase and Telomeres Biology in Thyroid Cancer. International Journal of Molecular Sciences, 20(12), 2887. https://doi.org/10.3390/ijms20122887