The Synergistic Effects of APOE Genotype and Obesity on Alzheimer’s Disease Risk


{kind=link}
Abstract
1. Alzheimer’s Disease
2. APOE4 and AD
3. Obesity and AD
4. Obesity and APOE
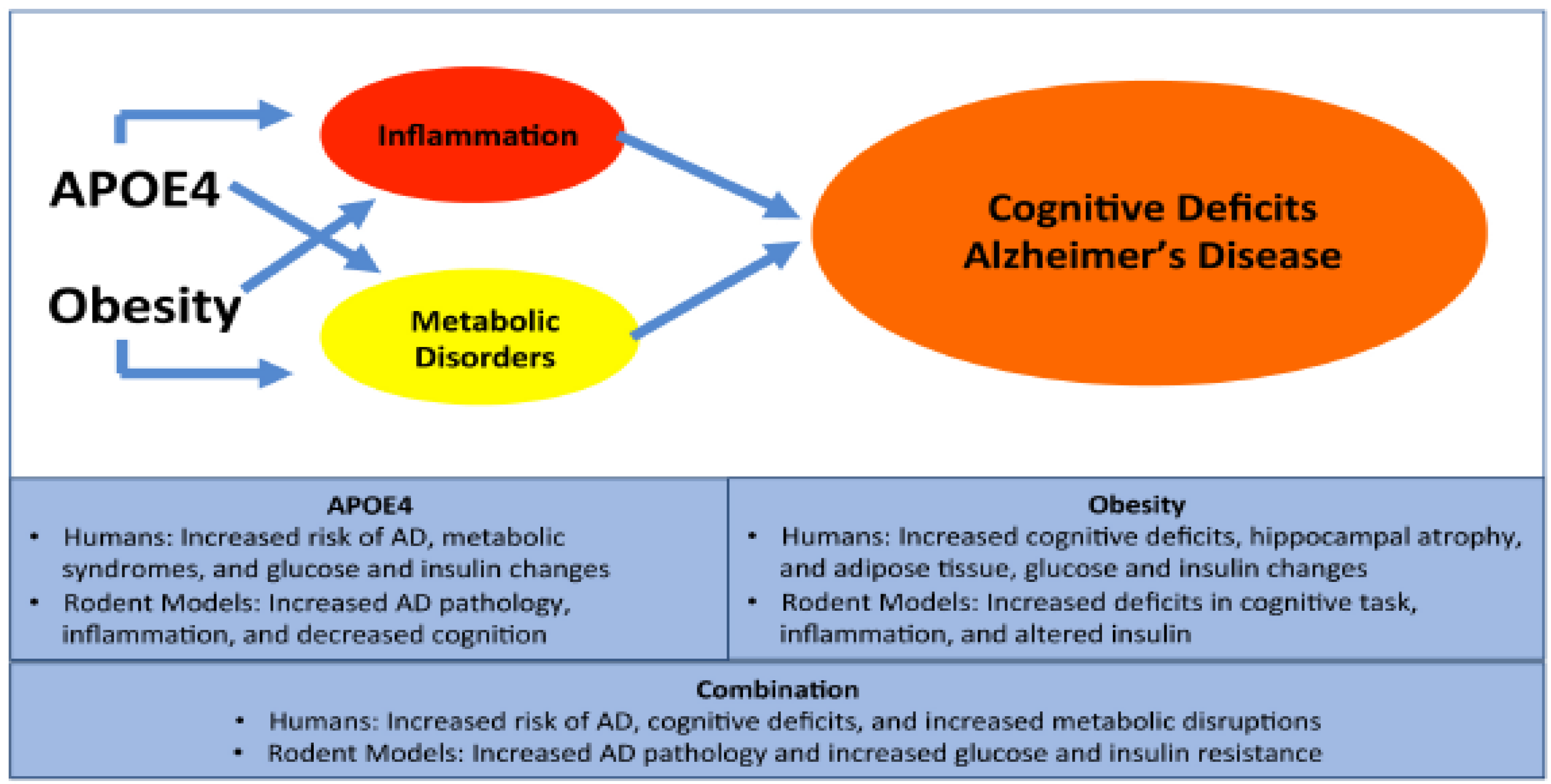
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Jansen, W.J.; Ossenkoppele, R.; Knol, D.L.; Tijms, B.M.; Scheltens, P.; Verhey, F.R.; Visser, P.J.; Aalten, P.; Aarsland, D.; Alcolea, D.; et al. Prevalence of cerebral amyloid pathology in persons without dementia: A meta-analysis. JAMA 2015, 313, 1924–1938. [Google Scholar] [CrossRef]
- Huang, Y.; Mahley, R.W. Apolipoprotein E: Structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis. 2014, 72, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- El-Lebedy, D.; Raslan, H.M.; Mohammed, A.M. Apolipoprotein E gene polymorphism and risk of type 2 diabetes and cardiovascular disease. Cardiovasc. Diabetol. 2016, 15, 12. [Google Scholar] [CrossRef] [PubMed]
- Ungar, L.; Altmann, A.; Greicius, M.D. Apolipoprotein E, gender, and Alzheimer’s disease: An overlooked, but potent and promising interaction. Brain Imaging Behav. 2014, 8, 262–273. [Google Scholar] [CrossRef]
- Bu, G. Apolipoprotein E and its receptors in Alzheimer’s disease: Pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 2009, 10, 333–344. [Google Scholar] [CrossRef]
- Berteau-Pavy, F.; Park, B.; Raber, J. Effects of sex and APOE epsilon4 on object recognition and spatial navigation in the elderly. Neuroscience 2007, 147, 6–17. [Google Scholar] [CrossRef]
- Dumanis, S.B.; Tesoriero, J.A.; Babus, L.W.; Nguyen, M.T.; Trotter, J.H.; Ladu, M.J.; Weeber, E.J.; Turner, R.S.; Xu, B.; Rebeck, G.W.; et al. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J. Neurosci. 2009, 29, 15317–15322. [Google Scholar] [CrossRef]
- Wang, R.; Fratiglioni, L.; Laukka, E.J.; Lovden, M.; Kalpouzos, G.; Keller, L.; Graff, C.; Salami, A.; Backman, L.; Qiu, C. Effects of vascular risk factors and APOE epsilon4 on white matter integrity and cognitive decline. Neurology 2015, 84, 1128–1135. [Google Scholar] [CrossRef]
- Liu, C.C.; Hu, J.; Tsai, C.W.; Yue, M.; Melrose, H.L.; Kanekiyo, T.; Bu, G. Neuronal LRP1 regulates glucose metabolism and insulin signaling in the brain. J. Neurosci. 2015, 35, 5851–5859. [Google Scholar] [CrossRef]
- Beydoun, M.A.; Boueiz, A.; Abougergi, M.S.; Kitner-Triolo, M.H.; Beydoun, H.A.; Resnick, S.M.; O’Brien, R.; Zonderman, A.B. Sex differences in the association of the apolipoprotein E epsilon 4 allele with incidence of dementia, cognitive impairment, and decline. Neurobiol. Aging 2012, 33, 720–731. [Google Scholar] [CrossRef]
- Mortensen, E.L.; Hogh, P. A gender difference in the association between APOE genotype and age-related cognitive decline. Neurology 2001, 57, 89–95. [Google Scholar] [CrossRef]
- Fleisher, A.; Grundman, M.; Jack, C.R., Jr.; Petersen, R.C.; Taylor, C.; Kim, H.T.; Schiller, D.H.; Bagwell, V.; Sencakova, D.; Weiner, M.F.; et al. Alzheimer’s Disease Cooperative, S. Sex, apolipoprotein E epsilon 4 status, and hippocampal volume in mild cognitive impairment. Arch. Neurol. 2005, 62, 953–957. [Google Scholar] [CrossRef]
- Liraz, O.; Boehm-Cagan, A.; Michaelson, D.M. ApoE4 induces Abeta42, tau, and neuronal pathology in the hippocampus of young targeted replacement apoE4 mice. Mol. Neurodegener. 2013, 8, 16. [Google Scholar] [CrossRef]
- Klein, R.C.; Mace, B.E.; Moore, S.D.; Sullivan, P.M. Progressive loss of synaptic integrity in human apolipoprotein E4 targeted replacement mice and attenuation by apolipoprotein E2. Neuroscience 2010, 171, 1265–1272. [Google Scholar] [CrossRef]
- Neustadtl, A.L.; Winston, C.N.; Parsadanian, M.; Main, B.S.; Villapol, S.; Burns, M.P. Reduced cortical excitatory synapse number in APOE4 mice is associated with increased calcineurin activity. Neuroreport 2017, 28, 618–624. [Google Scholar] [CrossRef]
- Bales, K.R. Brain lipid metabolism, apolipoprotein E and the pathophysiology of Alzheimer’s disease. Neuropharmacology 2010, 59, 295–302. [Google Scholar] [CrossRef]
- Rodriguez, G.A.; Burns, M.P.; Weeber, E.J.; Rebeck, G.W. Young APOE4 targeted replacement mice exhibit poor spatial learning and memory, with reduced dendritic spine density in the medial entorhinal cortex. Learn. Mem. 2013, 20, 256–266. [Google Scholar] [CrossRef]
- Tai, L.M.; Balu, D.; Avila-Munoz, E.; Abdullah, L.; Thomas, R.; Collins, N.; Valencia-Olvera, A.C.; LaDu, M.J. EFAD transgenic mice as a human APOE relevant preclinical model of Alzheimer’s disease. J. Lipid Res. 2017, 58, 1733–1755. [Google Scholar] [CrossRef]
- Naderali, E.K.; Ratcliffe, S.H.; Dale, M.C. Obesity and Alzheimer’s disease: A link between body weight and cognitive function in old age. Am. J. Alzheimers Dis. 2009, 24, 445–449. [Google Scholar] [CrossRef]
- Bloor, I.D.; Symonds, M.E. Sexual dimorphism in white and brown adipose tissue with obesity and inflammation. Horm. Behav. 2014, 66, 95–103. [Google Scholar] [CrossRef]
- Whitmer, R.A.; Gustafson, D.R.; Barrett-Connor, E.; Haan, M.N.; Gunderson, E.P.; Yaffe, K. Central obesity and increased risk of dementia more than three decades later. Neurology 2008, 71, 1057–1064. [Google Scholar] [CrossRef]
- Gustafson, D.R.; Backman, K.; Waern, M.; Ostling, S.; Guo, X.; Zandi, P.; Mielke, M.M.; Bengtsson, C.; Skoog, I. Adiposity indicators and dementia over 32 years in Sweden. Neurology 2009, 73, 1559–1566. [Google Scholar] [CrossRef]
- Isaac, V.; Sim, S.; Zheng, H.; Zagorodnov, V.; Tai, E.S.; Chee, M. Adverse Associations between Visceral Adiposity, Brain Structure, and Cognitive Performance in Healthy Elderly. Front. Aging Neurosci. 2011, 3, 12. [Google Scholar] [CrossRef]
- Mathieu, P.; Poirier, P.; Pibarot, P.; Lemieux, I.; Despres, J.P. Visceral obesity: The link among inflammation, hypertension, and cardiovascular disease. Hypertension 2009, 53, 577–584. [Google Scholar] [CrossRef]
- Neth, B.J.; Craft, S. Insulin Resistance and Alzheimer’s Disease: Bioenergetic Linkages. Front. Aging Neurosci. 2017, 9, 345. [Google Scholar] [CrossRef]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Litwin, S.E. Normal weight obesity: Is bigger really badder? Circ. Cardiovasc. Imaging 2012, 5, 286–288. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Arner, P.; Caro, J.F.; Atkinson, R.L.; Spiegelman, B.M. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Investig. 1995, 95, 2409–2415. [Google Scholar] [CrossRef] [PubMed]
- Moser, V.A.; Pike, C.J. Obesity and sex interact in the regulation of Alzheimer’s disease. Neurosci. Biobehav. Rev. 2016, 67, 102–118. [Google Scholar] [CrossRef] [PubMed]
- Solfrizzi, V.; Panza, F.; Colacicco, A.M.; D’Introno, A.; Capurso, C.; Torres, F.; Grigoletto, F.; Maggi, S.; Del Parigi, A.; Reiman, E.M.; et al. Italian Longitudinal Study on Aging Working, G. Vascular risk factors, incidence of MCI, and rates of progression to dementia. Neurology 2004, 63, 1882–1891. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, D.R.; Backman, K.; Joas, E.; Waern, M.; Ostling, S.; Guo, X.; Skoog, I. 37 years of body mass index and dementia: Observations from the prospective population study of women in Gothenburg, Sweden. J. Alzheimers Dis. 2012, 28, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Besser, L.M.; Gill, D.P.; Monsell, S.E.; Brenowitz, W.; Meranus, D.H.; Kukull, W.; Gustafson, D.R. Body mass index, weight change, and clinical progression in mild cognitive impairment and Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2014, 28, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Cournot, M.; Marquie, J.C.; Ansiau, D.; Martinaud, C.; Fonds, H.; Ferrieres, J.; Ruidavets, J.B. Relation between body mass index and cognitive function in healthy middle-aged men and women. Neurology 2006, 67, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Gunstad, J.; Paul, R.H.; Cohen, R.A.; Tate, D.F.; Spitznagel, M.B.; Gordon, E. Elevated body mass index is associated with executive dysfunction in otherwise healthy adults. Compr. Psychiatry 2007, 48, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Enzinger, C.; Fazekas, F.; Matthews, P.M.; Ropele, S.; Schmidt, H.; Smith, S.; Schmidt, R. Risk factors for progression of brain atrophy in aging: Six-year follow-up of normal subjects. Neurology 2005, 64, 1704–1711. [Google Scholar] [CrossRef] [PubMed]
- Siervo, M.; Arnold, R.; Wells, J.C.; Tagliabue, A.; Colantuoni, A.; Albanese, E.; Brayne, C.; Stephan, B.C. Intentional weight loss in overweight and obese individuals and cognitive function: A systematic review and meta-analysis. Obes. Rev. 2011, 12, 968–983. [Google Scholar] [CrossRef]
- Espeland, M.A.; Rapp, S.R.; Bray, G.A.; Houston, D.K.; Johnson, K.C.; Kitabchi, A.E.; Hergenroeder, A.L.; Williamson, J.; Jakicic, J.M.; van Dorsten, B.; et al. Long-term impact of behavioral weight loss intervention on cognitive function. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1101–1108. [Google Scholar] [CrossRef]
- Bruehl, H.; Sweat, V.; Tirsi, A.; Shah, B.; Convit, A. Obese Adolescents with Type 2 Diabetes Mellitus Have Hippocampal and Frontal Lobe Volume Reductions. Neurosci. Med. 2011, 2, 34–42. [Google Scholar] [CrossRef]
- Yau, P.L.; Castro, M.G.; Tagani, A.; Tsui, W.H.; Convit, A. Obesity and metabolic syndrome and functional and structural brain impairments in adolescence. Pediatrics 2012, 130, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Bruehl, H.; Wolf, O.T.; Sweat, V.; Tirsi, A.; Richardson, S.; Convit, A. Modifiers of cognitive function and brain structure in middle-aged and elderly individuals with type 2 diabetes mellitus. Brain Res. 2009, 1280, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Cherbuin, N.; Sargent-Cox, K.; Fraser, M.; Sachdev, P.; Anstey, K.J. Being overweight is associated with hippocampal atrophy: The PATH Through Life Study. Int. J. Obes. 2015, 39, 1509–1514. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, D.R.; Karlsson, C.; Skoog, I.; Rosengren, L.; Lissner, L.; Blennow, K. Mid-life adiposity factors relate to blood-brain barrier integrity in late life. J. Int. Med. 2007, 262, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Thirumangalakudi, L.; Prakasam, A.; Zhang, R.; Bimonte-Nelson, H.; Sambamurti, K.; Kindy, M.S.; Bhat, N.R. High cholesterol-induced neuroinflammation and amyloid precursor protein processing correlate with loss of working memory in mice. J. Neurochem. 2008, 106, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Puig, K.L.; Floden, A.M.; Adhikari, R.; Golovko, M.Y.; Combs, C.K. Amyloid precursor protein and proinflammatory changes are regulated in brain and adipose tissue in a murine model of high fat diet-induced obesity. PLoS ONE 2012, 7, e30378. [Google Scholar] [CrossRef] [PubMed]
- Koga, S.; Kojima, A.; Kuwabara, S.; Yoshiyama, Y. Immunohistochemical analysis of tau phosphorylation and astroglial activation with enhanced leptin receptor expression in diet-induced obesity mouse hippocampus. Neurosci. Lett. 2014, 571, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Heikkila, O.; Lundbom, N.; Timonen, M.; Groop, P.H.; Heikkinen, S.; Makimattila, S. Evidence for abnormal glucose uptake or metabolism in thalamus during acute hyperglycaemia in type 1 diabetes—A 1H MRS study. Metab. Brain Dis. 2010, 25, 227–234. [Google Scholar] [CrossRef]
- Seaquist, E.R.; Tkac, I.; Damberg, G.; Thomas, W.; Gruetter, R. Brain glucose concentrations in poorly controlled diabetes mellitus as measured by high-field magnetic resonance spectroscopy. Metabolism 2005, 54, 1008–1013. [Google Scholar] [CrossRef]
- Hayden, K.M.; Zandi, P.P.; Lyketsos, C.G.; Khachaturian, A.S.; Bastian, L.A.; Charoonruk, G.; Tschanz, J.T.; Norton, M.C.; Pieper, C.F.; Munger, R.G.; et al. Vascular risk factors for incident Alzheimer disease and vascular dementia: The Cache County study. Alzheimer Dis. Assoc. Disord. 2006, 20, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Ahonen, T.; Vanhala, M.; Kautiainen, H.; Kumpusalo, E.; Saltevo, J. Sex differences in the association of adiponectin and low-grade inflammation with changes in the body mass index from youth to middle age. Gend. Med. 2012, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, M.G.; Agellon, L.B. Sex differences in lipid metabolism and metabolic disease risk. Biochem. Cell Biol. 2012, 90, 124–141. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.R.; Clegg, D.J.; Prossnitz, E.R.; Barton, M. Obesity, insulin resistance and diabetes: Sex differences and role of oestrogen receptors. Acta Physiol. 2011, 203, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Charlat, O.; Tartaglia, L.A.; Woolf, E.A.; Weng, X.; Ellis, S.J.; Lakey, N.D.; Culpepper, J.; Moore, K.J.; Breitbart, R.E.; et al. Evidence that the diabetes gene encodes the leptin receptor: Identification of a mutation in the leptin receptor gene in db/db mice. Cell 1996, 84, 491–495. [Google Scholar] [CrossRef]
- Hummel, K.P.; Dickie, M.M.; Coleman, D.L. Diabetes, a new mutation in the mouse. Science 1966, 153, 1127–1128. [Google Scholar] [CrossRef] [PubMed]
- Zucker, L.M.; Antoniades, H.N. Insulin and obesity in the Zucker genetically obese rat “fatty”. Endocrinology 1972, 90, 1320–1330. [Google Scholar] [CrossRef]
- Campfield, L.A.; Smith, F.J.; Guisez, Y.; Devos, R.; Burn, P. Recombinant mouse OB protein: Evidence for a peripheral signal linking adiposity and central neural networks. Science 1995, 269, 546–549. [Google Scholar] [CrossRef]
- Gratuze, M.; El Khoury, N.B.; Turgeon, A.; Julien, C.; Marcouiller, F.; Morin, F.; Whittington, R.A.; Marette, A.; Calon, F.; Planel, E. Tau hyperphosphorylation in the brain of ob/ob mice is due to hypothermia: Importance of thermoregulation in linking diabetes and Alzheimer’s disease. Neurobiol. Dis. 2017, 98, 1–8. [Google Scholar] [CrossRef]
- Finger, B.C.; Dinan, T.G.; Cryan, J.F. Leptin-deficient mice retain normal appetitive spatial learning yet exhibit marked increases in anxiety-related behaviours. Psychopharmacology 2010, 210, 559–568. [Google Scholar] [CrossRef]
- Dinel, A.L.; Andre, C.; Aubert, A.; Ferreira, G.; Laye, S.; Castanon, N. Cognitive and emotional alterations are related to hippocampal inflammation in a mouse model of metabolic syndrome. PLoS ONE 2011, 6, e24325. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Aou, S.; Oomura, Y.; Hori, N.; Fukunaga, K.; Hori, T. Impairment of long-term potentiation and spatial memory in leptin receptor-deficient rodents. Neuroscience 2002, 113, 607–615. [Google Scholar] [CrossRef]
- Lin, B.; Hasegawa, Y.; Takane, K.; Koibuchi, N.; Cao, C.; Kim-Mitsuyama, S. High-Fat-Diet Intake Enhances Cerebral Amyloid Angiopathy and Cognitive Impairment in a Mouse Model of Alzheimer’s Disease, Independently of Metabolic Disorders. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Knight, E.M.; Martins, I.V.; Gumusgoz, S.; Allan, S.M.; Lawrence, C.B. High-fat diet-induced memory impairment in triple-transgenic Alzheimer’s disease (3xTgAD) mice is independent of changes in amyloid and tau pathology. Neurobiol. Aging 2014, 35, 1821–1832. [Google Scholar] [CrossRef] [PubMed]
- Kesby, J.P.; Kim, J.J.; Scadeng, M.; Woods, G.; Kado, D.M.; Olefsky, J.M.; Jeste, D.V.; Achim, C.L.; Semenova, S. Spatial Cognition in Adult and Aged Mice Exposed to High-Fat Diet. PLoS ONE 2015, 10, e0140034. [Google Scholar] [CrossRef] [PubMed]
- Baumgarner, K.M.; Setti, S.; Diaz, C.; Littlefield, A.; Jones, A.; Kohman, R.A. Diet-induced obesity attenuates cytokine production following an immune challenge. Behav. Brain Res. 2014, 267, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; Zuloaga, K.L.; Kugelman, T.L.; Mader, K.S.; Morre, J.T.; Zuloaga, D.G.; Weber, S.; Marzulla, T.; Mulford, A.; Button, D.; et al. Amelioration of Metabolic Syndrome-Associated Cognitive Impairments in Mice via a Reduction in Dietary Fat Content or Infusion of Non-Diabetic Plasma. EBioMedicine 2016, 3, 26–42. [Google Scholar] [CrossRef]
- Kanoski, S.E.; Zhang, Y.; Zheng, W.; Davidson, T.L. The effects of a high-energy diet on hippocampal function and blood-brain barrier integrity in the rat. J. Alzheimers Dis. 2010, 21, 207–219. [Google Scholar] [CrossRef]
- Julien, C.; Tremblay, C.; Phivilay, A.; Berthiaume, L.; Emond, V.; Julien, P.; Calon, F. High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse model. Neurobiol. Aging 2010, 31, 1516–1531. [Google Scholar] [CrossRef]
- Barron, A.M.; Rosario, E.R.; Elteriefi, R.; Pike, C.J. Sex-specific effects of high fat diet on indices of metabolic syndrome in 3xTg-AD mice: Implications for Alzheimer’s disease. PLoS ONE 2013, 8, e78554. [Google Scholar] [CrossRef]
- Hwang, L.L.; Wang, C.H.; Li, T.L.; Chang, S.D.; Lin, L.C.; Chen, C.P.; Chen, C.T.; Liang, K.C.; Ho, I.K.; Yang, W.S.; et al. Sex differences in high-fat diet-induced obesity, metabolic alterations and learning, and synaptic plasticity deficits in mice. Obesity 2010, 18, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, A.; Mohapel, P.; Bouter, B.; Frielingsdorf, H.; Pizzo, D.; Brundin, P.; Erlanson-Albertsson, C. High-fat diet impairs hippocampal neurogenesis in male rats. Eur. J. Neurol. 2006, 13, 1385–1388. [Google Scholar] [CrossRef] [PubMed]
- Macotela, Y.; Boucher, J.; Tran, T.T.; Kahn, C.R. Sex and depot differences in adipocyte insulin sensitivity and glucose metabolism. Diabetes 2009, 58, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Torres-Perez, E.; Ledesma, M.; Garcia-Sobreviela, M.P.; Leon-Latre, M.; Arbones-Mainar, J.M. Apolipoprotein E4 association with metabolic syndrome depends on body fatness. Atherosclerosis 2016, 245, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Arbones-Mainar, J.M.; Johnson, L.A.; Altenburg, M.K.; Maeda, N. Differential modulation of diet-induced obesity and adipocyte functionality by human apolipoprotein E3 and E4 in mice. Int. J. Obes. 2008, 32, 1595–1605. [Google Scholar] [CrossRef] [PubMed]
- Tejedor, M.T.; Garcia-Sobreviela, M.P.; Ledesma, M.; Arbones-Mainar, J.M. The apolipoprotein E polymorphism rs7412 associates with body fatness independently of plasma lipids in middle aged men. PLoS ONE 2014, 9, e108605. [Google Scholar] [CrossRef] [PubMed]
- Volcik, K.A.; Barkley, R.A.; Hutchinson, R.G.; Mosley, T.H.; Heiss, G.; Sharrett, A.R.; Ballantyne, C.M.; Boerwinkle, E. Apolipoprotein E polymorphisms predict low density lipoprotein cholesterol levels and carotid artery wall thickness but not incident coronary heart disease in 12,491 ARIC study participants. Am. J. Epidemiol. 2006, 164, 342–348. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef]
- Sun, Y.; Wei, R.; Yan, D.; Xu, F.; Zhang, X.; Zhang, B.; Yimiti, D.; Li, H.; Sun, H.; Hu, C.; et al. Association between APOE polymorphism and metabolic syndrome in Uyghur ethnic men. BMJ Open 2016, 6, e010049. [Google Scholar] [CrossRef]
- Fallaize, R.; Carvalho-Wells, A.L.; Tierney, A.C.; Marin, C.; Kiec-Wilk, B.; Dembinska-Kiec, A.; Drevon, C.A.; DeFoort, C.; Lopez-Miranda, J.; Riserus, U.; et al. APOE genotype influences insulin resistance, apolipoprotein CII and CIII according to plasma fatty acid profile in the Metabolic Syndrome. Sci. Rep. 2017, 7, 6274. [Google Scholar] [CrossRef]
- Bennet, A.M.; Di Angelantonio, E.; Ye, Z.; Wensley, F.; Dahlin, A.; Ahlbom, A.; Keavney, B.; Collins, R.; Wiman, B.; de Faire, U.; et al. Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA 2007, 298, 1300–1311. [Google Scholar] [CrossRef] [PubMed]
- Stiefel, P.; Montilla, C.; Muniz-Grijalvo, O.; Garcia-Lozano, R.; Alonso, A.; Miranda, M.L.; Pamies, E.; Villar, J. Apolipoprotein E gene polymorphism is related to metabolic abnormalities, but does not influence erythrocyte membrane lipid composition or sodium-lithium countertransport activity in essential hypertension. Metabolism 2001, 50, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Elosua, R.; Demissie, S.; Cupples, L.A.; Meigs, J.B.; Wilson, P.W.; Schaefer, E.J.; Corella, D.; Ordovas, J.M. Obesity modulates the association among APOE genotype, insulin, and glucose in men. Obes. Res. 2003, 11, 1502–1508. [Google Scholar] [CrossRef] [PubMed]
- Ghebranious, N.; Mukesh, B.; Giampietro, P.F.; Glurich, I.; Mickel, S.F.; Waring, S.C.; McCarty, C.A. A pilot study of gene/gene and gene/environment interactions in Alzheimer disease. Clin. Med. Res. 2011, 9, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Zade, D.; Beiser, A.; McGlinchey, R.; Au, R.; Seshadri, S.; Palumbo, C.; Wolf, P.A.; DeCarli, C.; Milberg, W. Apolipoprotein epsilon 4 allele modifies waist-to-hip ratio effects on cognition and brain structure. J. Stroke Cereb. Dis. 2013, 22, 119–125. [Google Scholar] [CrossRef][Green Version]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Cherrier, M.M.; Schellenberg, G.D.; Frey, W.H.; et al. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J. Alzheimers Dis. 2008, 13, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Asthana, S.; Schellenberg, G.; Baker, L.; Cherrier, M.; Boyt, A.A.; Martins, R.N.; Raskind, M.; Peskind, E.; Plymate, S. Insulin effects on glucose metabolism, memory, and plasma amyloid precursor protein in Alzheimer’s disease differ according to apolipoprotein-E genotype. Ann. N. Y. Acad. Sci. 2000, 903, 222–228. [Google Scholar] [CrossRef]
- Arbones-Mainar, J.M.; Johnson, L.A.; Torres-Perez, E.; Garcia, A.E.; Perez-Diaz, S.; Raber, J.; Maeda, N. Metabolic shifts toward fatty-acid usage and increased thermogenesis are associated with impaired adipogenesis in mice expressing human APOE4. Int. J. Obes. 2016, 40, 1574–1581. [Google Scholar] [CrossRef]
- Karagiannides, I.; Abdou, R.; Tzortzopoulou, A.; Voshol, P.J.; Kypreos, K.E. Apolipoprotein E predisposes to obesity and related metabolic dysfunctions in mice. FEBS J. 2008, 275, 4796–4809. [Google Scholar] [CrossRef]
- Lo Sasso, G.; Schlage, W.K.; Boue, S.; Veljkovic, E.; Peitsch, M.C.; Hoeng, J. The Apoe(-/-) mouse model: A suitable model to study cardiovascular and respiratory diseases in the context of cigarette smoke exposure and harm reduction. J. Transl. Med. 2016, 14, 146. [Google Scholar] [CrossRef]
- Huang, Z.H.; Reardon, C.A.; Mazzone, T. Endogenous ApoE expression modulates adipocyte triglyceride content and turnover. Diabetes 2006, 55, 3394–3402. [Google Scholar] [CrossRef] [PubMed]
- Huebbe, P.; Dose, J.; Schloesser, A.; Campbell, G.; Gluer, C.C.; Gupta, Y.; Ibrahim, S.; Minihane, A.M.; Baines, J.F.; Nebel, A.; et al. Apolipoprotein E (APOE) genotype regulates body weight and fatty acid utilization-Studies in gene-targeted replacement mice. Mol. Nutr. Food Res. 2015, 59, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; Torres, E.R.; Impey, S.; Stevens, J.F.; Raber, J. Apolipoprotein E4 and Insulin Resistance Interact to Impair Cognition and Alter the Epigenome and Metabolome. Sci. Rep. 2017, 7, 43701. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; Torres, E.R.; Weber Boutros, S.; Patel, E.; Akinyeke, T.; Alkayed, N.J.; Raber, J. Apolipoprotein E4 mediates insulin resistance-associated cerebrovascular dysfunction and the post-prandial response. J. Cerebr. Blood Flow Metabol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Moser, V.A.; Pike, C.J. Obesity Accelerates Alzheimer-Related Pathology in APOE4 but not APOE3 Mice. eNeuro 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Janssen, C.I.; Jansen, D.; Mutsaers, M.P.; Dederen, P.J.; Geenen, B.; Mulder, M.T.; Kiliaan, A.J. The Effect of a High-Fat Diet on Brain Plasticity, Inflammation and Cognition in Female ApoE4-Knockin and ApoE-Knockout Mice. PLoS ONE 2016, 11, e0155307. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E.; Lucki, I.; Brookshire, B.R.; Carlson, G.C.; Browne, C.A.; Kazi, H.; Bang, S.; Choi, B.R.; Chen, Y.; McMullen, M.F.; et al. High fat diet produces brain insulin resistance, synaptodendritic abnormalities and altered behavior in mice. Neurobiol. Dis. 2014, 67, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Kothari, V.; Luo, Y.; Tornabene, T.; O’Neill, A.M.; Greene, M.W.; Geetha, T.; Babu, J.R. High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 499–508. [Google Scholar] [CrossRef]
- Varatharaj, A.; Galea, I. The blood-brain barrier in systemic inflammation. Brain Behav. Immunol. 2017, 60, 1–12. [Google Scholar] [CrossRef]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jones, N.S.; Rebeck, G.W. The Synergistic Effects of APOE Genotype and Obesity on Alzheimer’s Disease Risk. Int. J. Mol. Sci. 2019, 20, 63. https://doi.org/10.3390/ijms20010063
Jones NS, Rebeck GW. The Synergistic Effects of APOE Genotype and Obesity on Alzheimer’s Disease Risk. International Journal of Molecular Sciences. 2019; 20(1):63. https://doi.org/10.3390/ijms20010063
Chicago/Turabian StyleJones, Nahdia S., and G. William Rebeck. 2019. "The Synergistic Effects of APOE Genotype and Obesity on Alzheimer’s Disease Risk" International Journal of Molecular Sciences 20, no. 1: 63. https://doi.org/10.3390/ijms20010063
APA StyleJones, N. S., & Rebeck, G. W. (2019). The Synergistic Effects of APOE Genotype and Obesity on Alzheimer’s Disease Risk. International Journal of Molecular Sciences, 20(1), 63. https://doi.org/10.3390/ijms20010063