Actin-Myosin Interaction: Structure, Function and Drug Discovery


Abstract
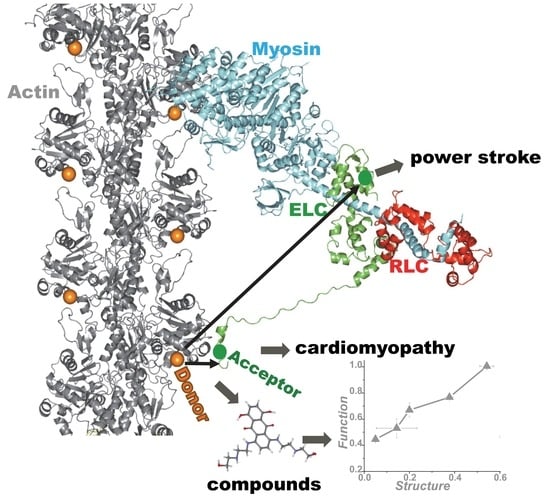
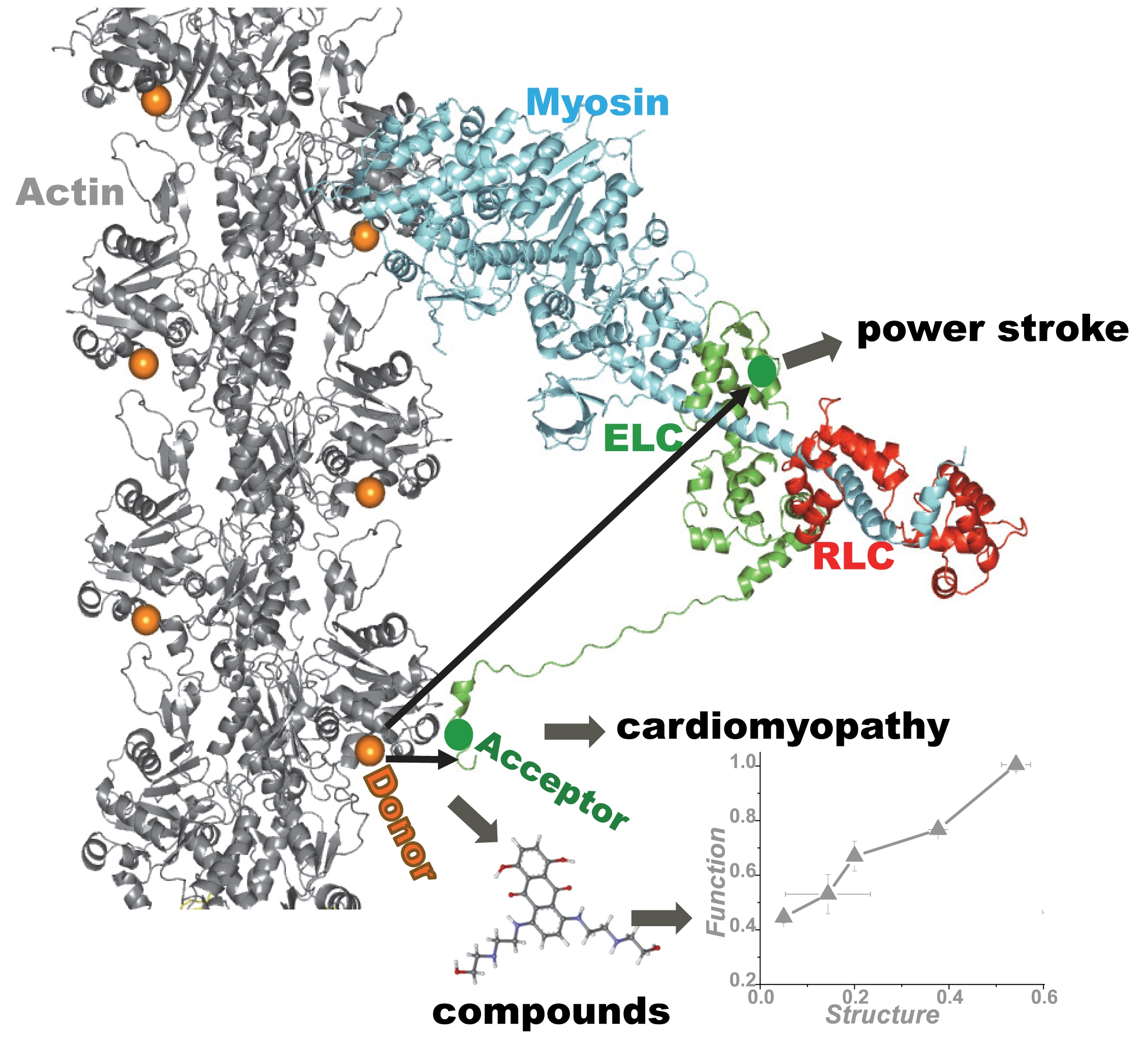{kind=link}
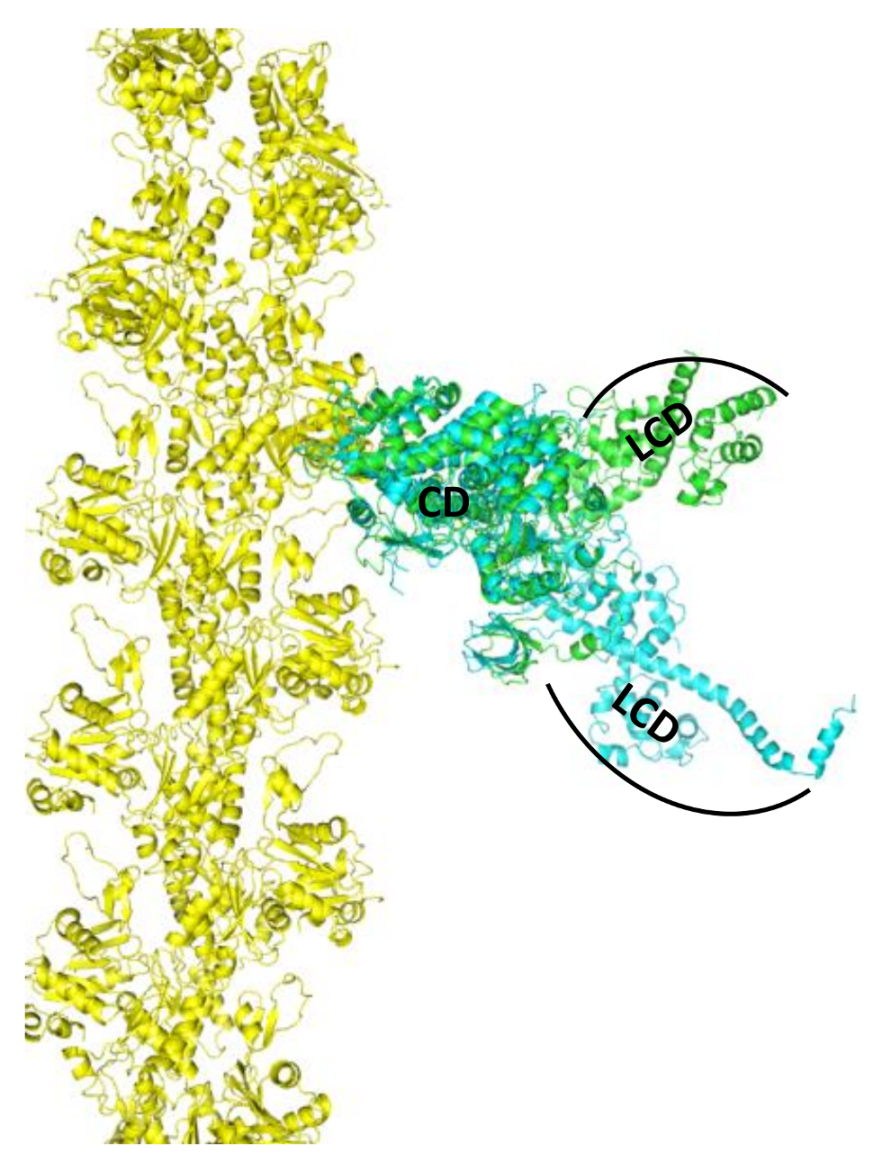{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Skeletal Muscle Actin-Myosin Structural Transition Depends on the Myosin ELC Isoform
3. Cardiac Actin-Myosin Structural Transition Is Affected by a Disease-Causing Mutation
4. Application of TR-FRET for Drug Discovery
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ELC | Essential light chain |
| RLC | Regulatory light chain |
| CD | Catalytic domain |
| LCD | Light chain domain |
| TR-FRET | Time-resolved Fluorescence resonance energy transfer |
| HCM | Hypertrophic cardiomyopathy |
| NCC | National clinical collection |
| FLTPR | Fluorescence lifetime plate reader |
| ANT | Acceptor labeled N-terminal peptide |
References
- Thomas, D.D.; Prochniewicz, E.; Roopnarine, O. Changes in actin and myosin structural dynamics due to their weak and strong interactions. Result. Probl. Cell Differ. 2002, 36, 7–19. [Google Scholar]
- Thomas, D.D.; Kast, D.; Korman, V.L. Site-directed spectroscopic probes of actomyosin structural dynamics. Annu. Rev. Biophys. 2009, 38, 347–369. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.D.; Muretta, J.M.; Colson, B.A.; Mello, R.N.; Kast, D. Spectroscopic Probes of Muscle Proteins. In Comprehensive Biophysics; Edward, H.E., Ed.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 226–250. [Google Scholar]
- Sweeney, H.L.; Houdusse, A. Structural and functional insights into the Myosin motor mechanism. Annu. Rev. Biophys. 2010, 39, 539–557. [Google Scholar] [CrossRef] [PubMed]
- Wulf, S.F.; Ropars, V.; Fujita-Becker, S.; Oster, M.; Hofhaus, G.; Trabuco, L.G.; Pylypenko, O.; Sweeney, H.L.; Houdusse, A.M.; Schröder, R.R. Force-producing ADP state of myosin bound to actin. Proc. Natl. Acad. Sci. USA 2016, 113, E1844–E1852. [Google Scholar] [CrossRef] [PubMed]
- Von der Ecken, J.; Heissler, S.M.; Pathan-Chhatbar, S.; Manstein, D.J.; Raunser, S. Cryo-EM structure of a human cytoplasmic actomyosin complex at near-atomic resolution. Nature 2016, 534, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.D.; dos Remedios, C.G. (Eds.) Molecular Interactions of Actin. Actin-Myosin Interaction and Actin-Based Regulation; Springer: Berlin, Germany, 2002. [Google Scholar]
- Fajer, P.G.; Fajer, E.A.; Brunsvold, N.J.; Thomas, D.D. Effects of AMPPNP on the orientation and rotational dynamics of spin-labeled muscle cross-bridges. Biophys. J. 1988, 53, 513–524. [Google Scholar] [CrossRef]
- Ostap, E.M.; Barnett, V.A.; Thomas, D.D. Resolution of three structural states of spin-labeled myosin in contracting muscle. Biophys. J. 1995, 69, 177–188. [Google Scholar] [CrossRef]
- Suzuki, Y.; Yasunaga, T.; Ohkura, R.; Wakabayashi, T.; Sutoh, K. Swing of the lever arm of a myosin motor at the isomerization and phosphate-release steps. Nature 1998, 396, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Campbell, R.E.; Ting, A.Y.; Tsien, R.Y. Creating new fluorescent probes for cell biology. Nat. Rev. Mol. Cell Biol. 2002, 3, 906–918. [Google Scholar] [CrossRef] [PubMed]
- Gruber, S.J.; Cornea, R.L.; Li, J.; Peterson, K.C.; Schaaf, T.M.; Gillispie, G.D.; Dahl, R.; Zsebo, K.M.; Robia, S.L.; Thomas, D.D. Discovery of enzyme modulators via high-throughput time-resolved FRET in living cells. J. Biomol. Screen. 2014, 19, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Rohde, J.A.; Thomas, D.D.; Muretta, J.M. Heart failure drug changes the mechanoenzymology of the cardiac myosin powerstroke. Proc. Natl. Acad. Sci. USA 2017, 114, E1796–E1804. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, H.L.; Bowman, B.F.; Stull, J.T. Myosin light chain phosphorylation in vertebrate striated muscle: Regulation and function. Am. J. Physiol 1993, 264, C1085–C1095. [Google Scholar] [CrossRef] [PubMed]
- Lowey, S.; Waller, G.S.; Trybus, K.M. Skeletal muscle myosin light chains are essential for physiological speeds of shortening. Nature 1993, 365, 454–456. [Google Scholar] [CrossRef] [PubMed]
- VanBuren, P.; Waller, G.S.; Harris, D.E.; Trybus, K.M.; Warshaw, D.M.; Lowey, S. The essential light chain is required for full force production by skeletal muscle myosin. Proc. Natl. Acad. Sci. USA 1994, 91, 12403–12407. [Google Scholar] [CrossRef] [PubMed]
- Frank, G.; Weeds, A.G. The amino-acid sequence of the alkali light chains of rabbit skeletal-muscle myosin. Eur. J. Biochem. 1974, 44, 317–334. [Google Scholar] [PubMed]
- Trayer, I.P.; Trayer, H.R.; Levine, B.A. Evidence that the N-terminal region of A1-light chain of myosin interacts directly with the C-terminal region of actin. A proton magnetic resonance study. Eur. J. Biochem. 1987, 164, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Timson, D.J.; Trayer, H.R.; Trayer, I.P. The N-terminus of A1-type myosin essential light chains binds actin and modulates myosin motor function. Eur. J. Biochem. 1998, 255, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Lowey, S.; Risby, D. Light chains from fast and slow muscle myosins. Nature 1971, 234, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Morano, I.; Ritter, O.; Bonz, A.; Timek, T.; Vahl, C.F.; Michel, G. Myosin light chain-actin interaction regulates cardiac contractility. Circ. Res. 1995, 76, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Trybus, K.M. Regulation of expressed truncated smooth muscle myosins. Role of the essential light chain and tail length. J. Biol. Chem. 1994, 269, 20819–20822. [Google Scholar] [PubMed]
- Chen, T.L.; Kowalczyk, P.A.; Ho, G.; Chisholm, R.L. Targeted disruption of the Dictyostelium myosin essential light chain gene produces cells defective in cytokinesis and morphogenesis. J. Cell Sci. 1995, 108 Pt 10, 3207–3218. [Google Scholar]
- Wagner, P.D.; Slater, C.S.; Pope, B.; Weeds, A.G. Studies on the actin activation of myosin subfragment-1 isoezymes and the role of myosin light chains. Eur. J. Biochem. 1979, 99, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Lowey, S.; Waller, G.S.; Trybus, K.M. Function of skeletal muscle myosin heavy and light chain isoforms by an in vitro motility assay. J. Biol. Chem. 1993, 268, 20414–20418. [Google Scholar] [PubMed]
- Guhathakurta, P.; Prochniewicz, E.; Thomas, D.D. Amplitude of the actomyosin power stroke depends strongly on the isoform of the myosin essential light chain. Proc. Natl. Acad. Sci. USA 2015, 112, 4660–4665. [Google Scholar] [CrossRef] [PubMed]
- Guhathakurta, P.; Prochniewicz, E.; Roopnarine, O.; Rohde, J.A.; Thomas, D.D. A Cardiomyopathy Mutation in the Myosin Essential Light Chain Alters Actomyosin Structure. Biophys. J. 2017, 113, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Guhathakurta, P.; Prochniewicz, E.; Grant, B.D.; Peterson, K.C.; Thomas, D.D. High-throughput screen, using time-resolved FRET, yields actin-binding compounds that modulate actin-myosin structure and function. J. Biol. Chem. 2018, 293, 12288–12298. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.J.; Peterson, K.C.; Muretta, J.M.; Higgins, S.E.; Gillispie, G.D.; Thomas, D.D. Fluorescence lifetime plate reader: Resolution and precision meet high-throughput. Rev. Sci. Instrum. 2014, 85, 113101. [Google Scholar] [CrossRef] [PubMed]
- Rayment, I.; Holden, H.M.; Whittaker, M.; Yohn, C.B.; Lorenz, M.; Holmes, K.C.; Milligan, R.A. Structure of the actin-myosin complex and its implications for muscle contraction. Science 1993, 261, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, R.; Freyzon, Y.; Trybus, K.M.; Cohen, C. Crystal structure of a vertebrate smooth muscle myosin motor domain and its complex with the essential light chain: Visualization of the pre-power stroke state. Cell 1998, 94, 559–571. [Google Scholar] [CrossRef]
- Llinas, P.; Isabet, T.; Song, L.; Sweeney, H.L.; Houdusse, A. How actin initiates the motor activity of Myosin. Dev. Cell 2015, 33, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Houdusse, A.; Szent-Gyorgyi, A.G.; Cohen, C. Three conformational states of scallop myosin S1. Proc. Natl. Acad. Sci. USA 2000, 97, 11238–11243. [Google Scholar] [CrossRef] [PubMed]
- Volkmann, N.; Liu, H.; Hazelwood, L.; Krementsova, E.B.; Lowey, S.; Trybus, K.M.; Hanein, D. The structural basis of myosin V processive movement as revealed by electron cryomicroscopy. Mol. Cell 2005, 19, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Refaat, M.M.; Fahed, A.C.; Hassanieh, S.; Hotait, M.; Arabi, M.; Skouri, H.; Seidman, J.G.; Seidman, C.E.; Bitar, F.F.; Nemer, G. The Muscle-Bound Heart. Card. Electrophysiol. Clin. 2016, 8, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Arad, M.; Seidman, J.G.; Seidman, C.E. Phenotypic diversity in hypertrophic cardiomyopathy. Hum. Mol. Genet. 2002, 11, 2499–2506. [Google Scholar] [CrossRef] [PubMed]
- Marsiglia, J.D.; Pereira, A.C. Hypertrophic cardiomyopathy: How do mutations lead to disease? Arq. Bras. Cardiol. 2014, 102, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Spudich, J.A. The myosin mesa and a possible unifying hypothesis for the molecular basis of human hypertrophic cardiomyopathy. Biochem. Soc. Trans. 2015, 43, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, A.S.; Kooiker, K.B.; Sarkar, S.S.; Liu, C.; Bernstein, D.; Spudich, J.A.; Ruppel, K.M. Early-Onset Hypertrophic Cardiomyopathy Mutations Significantly Increase the Velocity, Force, and Actin-Activated ATPase Activity of Human beta-Cardiac Myosin. Cell Rep. 2016, 17, 2857–2864. [Google Scholar] [CrossRef] [PubMed]
- Spudich, J.A. Hypertrophic and dilated cardiomyopathy: Four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys. J. 2014, 106, 1236–1249. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Szczesna-Cordary, D. Molecular mechanisms of cardiomyopathy phenotypes associated with myosin light chain mutations. J. Muscle Res. Cell Motil. 2015, 36, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Mijailovich, S.M.; Nedic, D.; Svicevic, M.; Stojanovic, B.; Walklate, J.; Ujfalusi, Z.; Geeves, M.A. Modeling the Actin.myosin ATPase Cross-Bridge Cycle for Skeletal and Cardiac Muscle Myosin Isoforms. Biophys. J. 2017, 112, 984–996. [Google Scholar] [CrossRef] [PubMed]
- Spudich, J.A.; Aksel, T.; Bartholomew, S.R.; Nag, S.; Kawana, M.; Yu, E.C.; Sarkar, S.S.; Sung, J.; Sommese, R.F.; Sutton, S.; et al. Effects of hypertrophic and dilated cardiomyopathy mutations on power output by human beta-cardiac myosin. J. Exp. Biol. 2016, 219, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Bonow, R.O. Left ventricular diastolic function in hypertrophic cardiomyopathy. Herz 1991, 16, 13–21. [Google Scholar] [PubMed]
- Schaaf, T.M.; Peterson, K.C.; Grant, B.D.; Bawaskar, P.; Yuen, S.; Li, J.; Muretta, J.M.; Gillispie, G.D.; Thomas, D.D. High-Throughput Spectral and Lifetime-Based FRET Screening in Living Cells to Identify Small-Molecule Effectors of SERCA. SLAS Discov. 2017, 22, 262–273. [Google Scholar] [PubMed]
- Rebbeck, R.T.; Essawy, M.M.; Nitu, F.R.; Grant, B.D.; Gillispie, G.D.; Thomas, D.D.; Bers, D.M.; Cornea, R.L. High-Throughput Screens to Discover Small-Molecule Modulators of Ryanodine Receptor Calcium Release Channels. SLAS Discov. 2017, 22, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Malik, F.I.; Hartman, J.J.; Elias, K.A.; Morgan, B.P.; Rodriguez, H.; Brejc, K.; Anderson, R.L.; Sueoka, S.H.; Lee, K.H.; Finer, J.T. Cardiac myosin activation: A potential therapeutic approach for systolic heart failure. Science 2011, 331, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Green, E.M.; Wakimoto, H.; Anderson, R.L.; Evanchik, M.J.; Gorham, J.M.; Harrison, B.C.; Henze, M.; Kawas, R.; Oslob, J.D.; Rodriguez, H.M.; et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 2016, 351, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Sirigu, S.; Hartman, J.J.; Planelles-Herrero, V.J.; Ropars, V.; Clancy, S.; Wang, X.; Chuang, G.; Qian, X.; Lu, P.P.; Barrett, E.; et al. Highly selective inhibition of myosin motors provides the basis of potential therapeutic application. Proc. Natl. Acad. Sci. USA 2016, 113, E7448–E7455. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guhathakurta, P.; Prochniewicz, E.; Thomas, D.D. Actin-Myosin Interaction: Structure, Function and Drug Discovery. Int. J. Mol. Sci. 2018, 19, 2628. https://doi.org/10.3390/ijms19092628
Guhathakurta P, Prochniewicz E, Thomas DD. Actin-Myosin Interaction: Structure, Function and Drug Discovery. International Journal of Molecular Sciences. 2018; 19(9):2628. https://doi.org/10.3390/ijms19092628
Chicago/Turabian StyleGuhathakurta, Piyali, Ewa Prochniewicz, and David D. Thomas. 2018. "Actin-Myosin Interaction: Structure, Function and Drug Discovery" International Journal of Molecular Sciences 19, no. 9: 2628. https://doi.org/10.3390/ijms19092628
APA StyleGuhathakurta, P., Prochniewicz, E., & Thomas, D. D. (2018). Actin-Myosin Interaction: Structure, Function and Drug Discovery. International Journal of Molecular Sciences, 19(9), 2628. https://doi.org/10.3390/ijms19092628