Cardiomyopathies and Related Changes in Contractility of Human Heart Muscle


Abstract
1. Introduction
2. Approaches and Parameters to Estimate Contractility
3. Contractile Properties of HCM and DCM Hearts
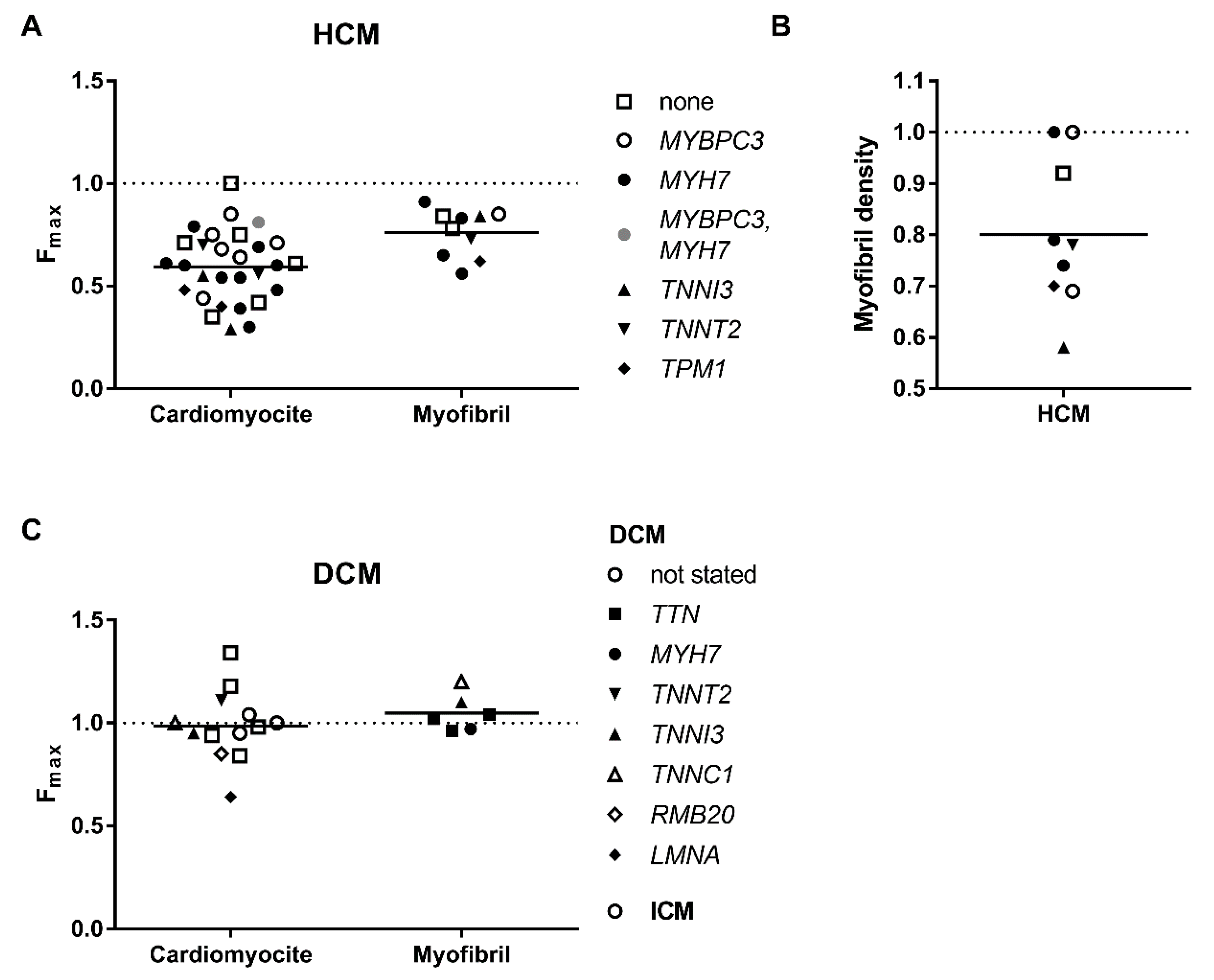
3.1. Decreased Force Capacity
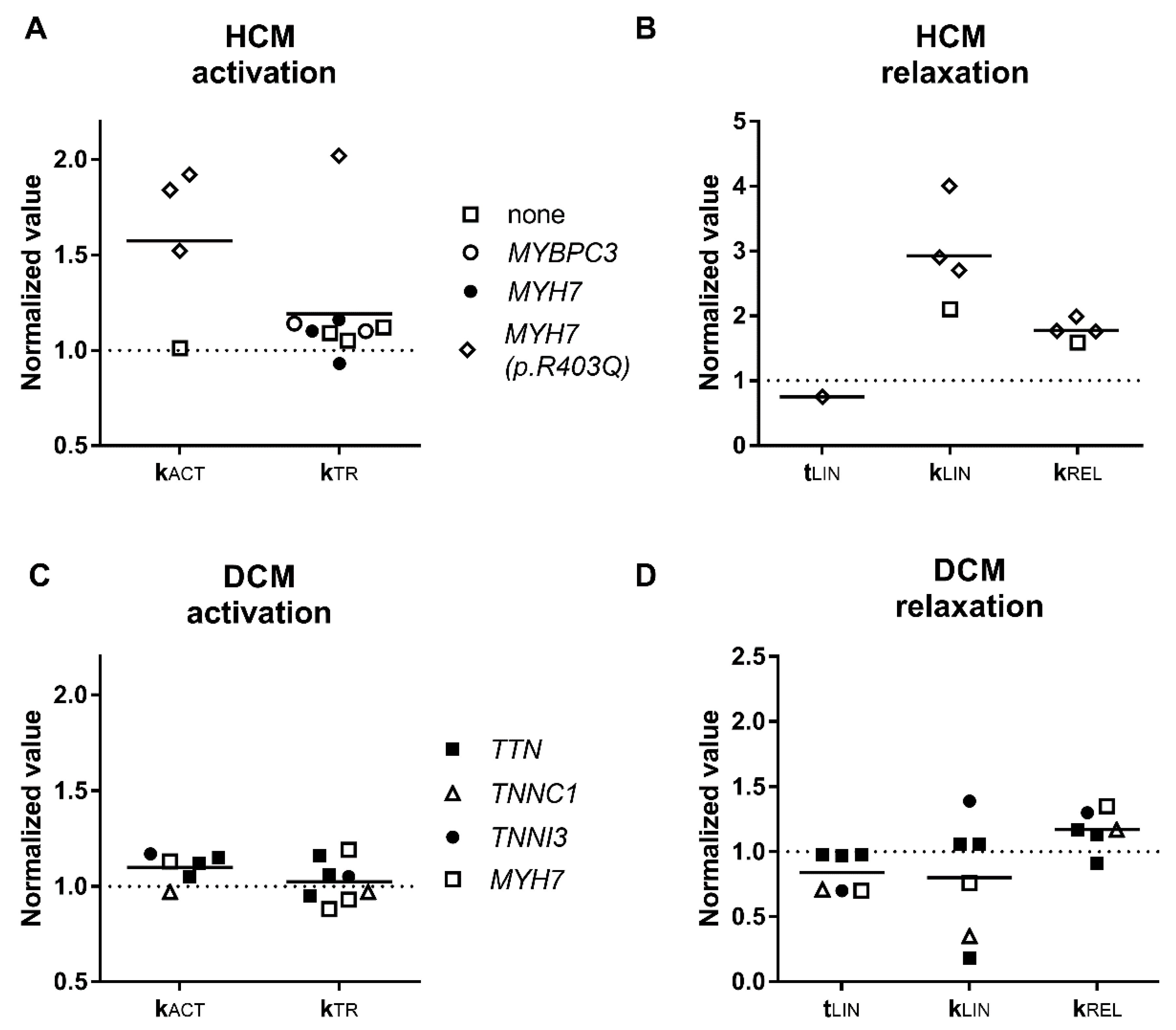
3.2. Activation and Relaxation Kinetics
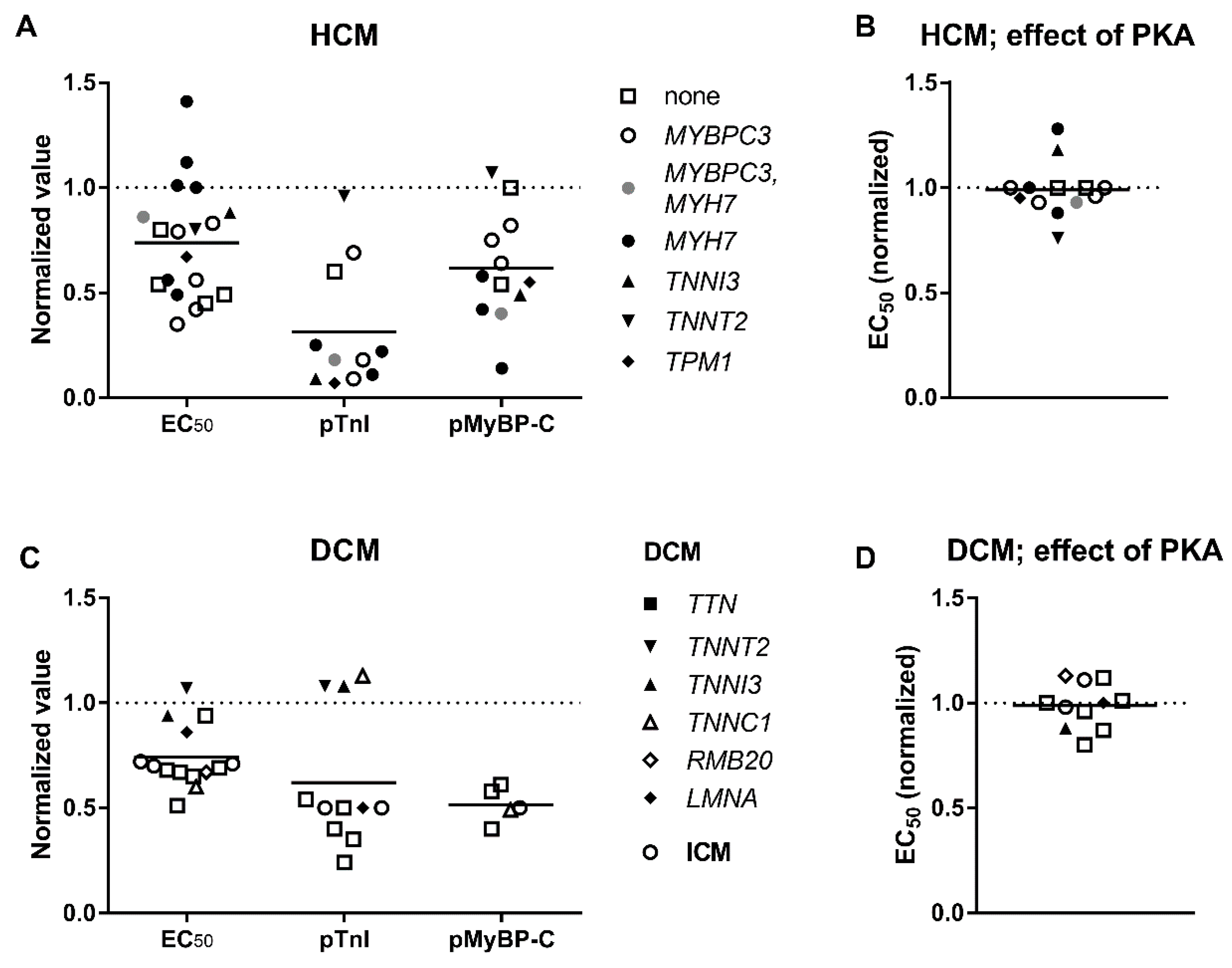
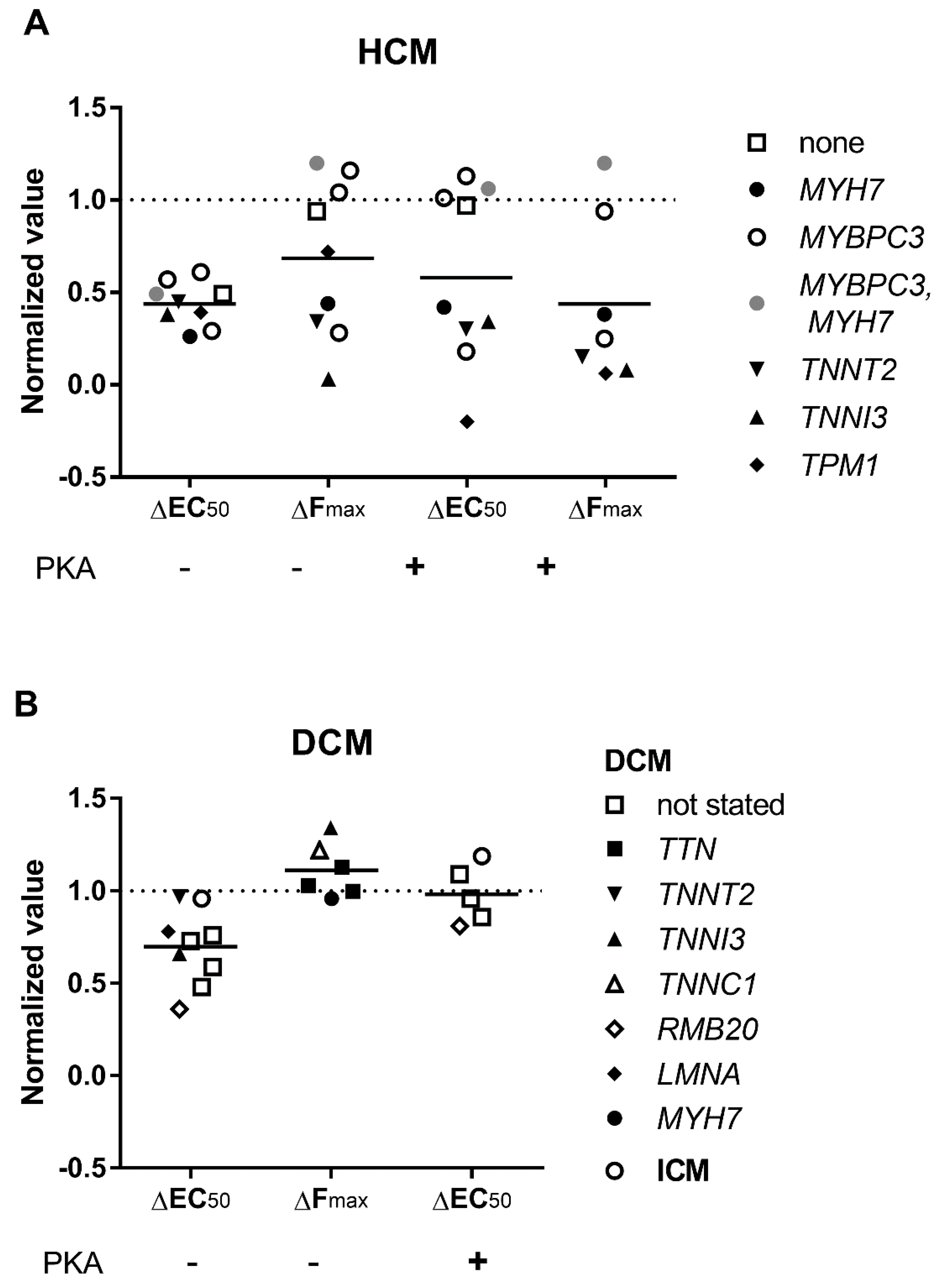
3.3. Elevated Ca2+-Sensitivity
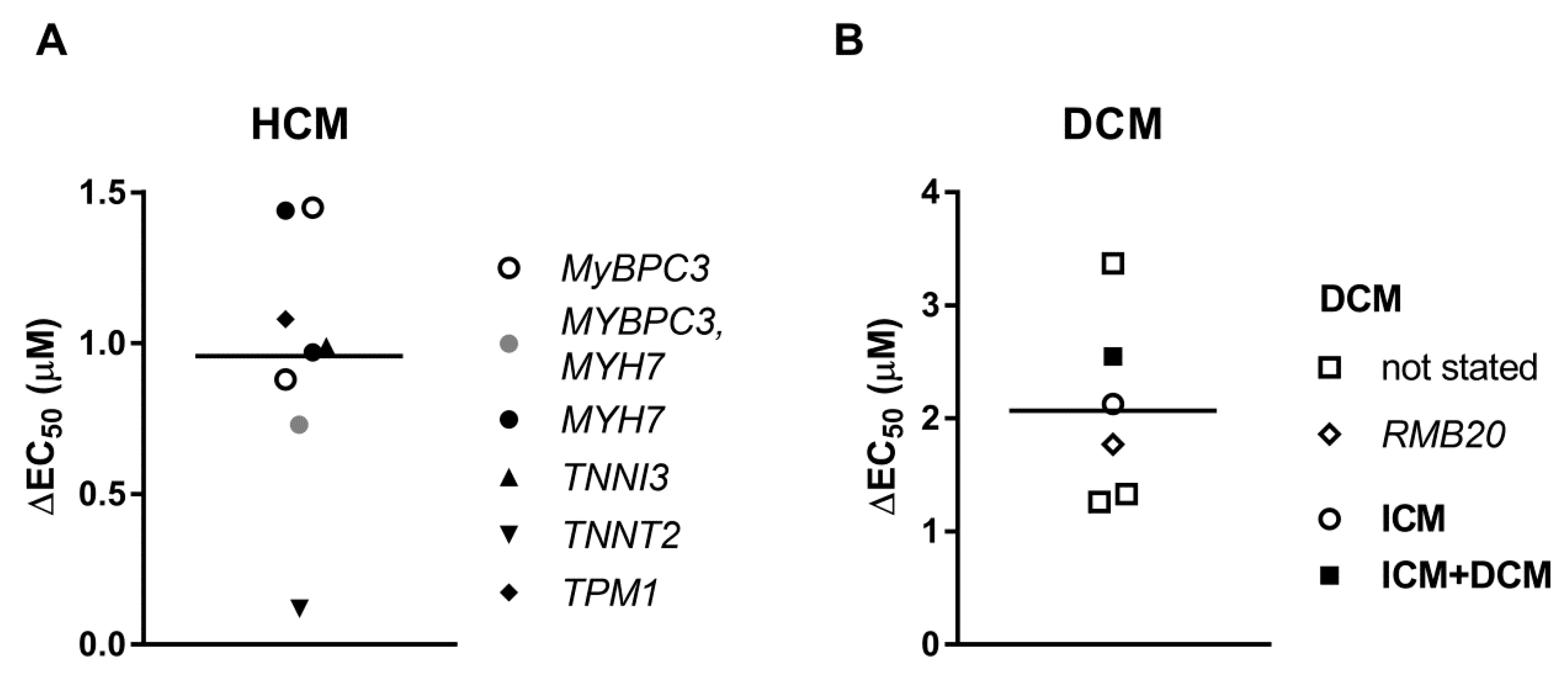
3.4. Uncoupling of TnI Phosphorylation from the Changes in Ca2+-Sensitivity
3.5. Length Dependent Activation
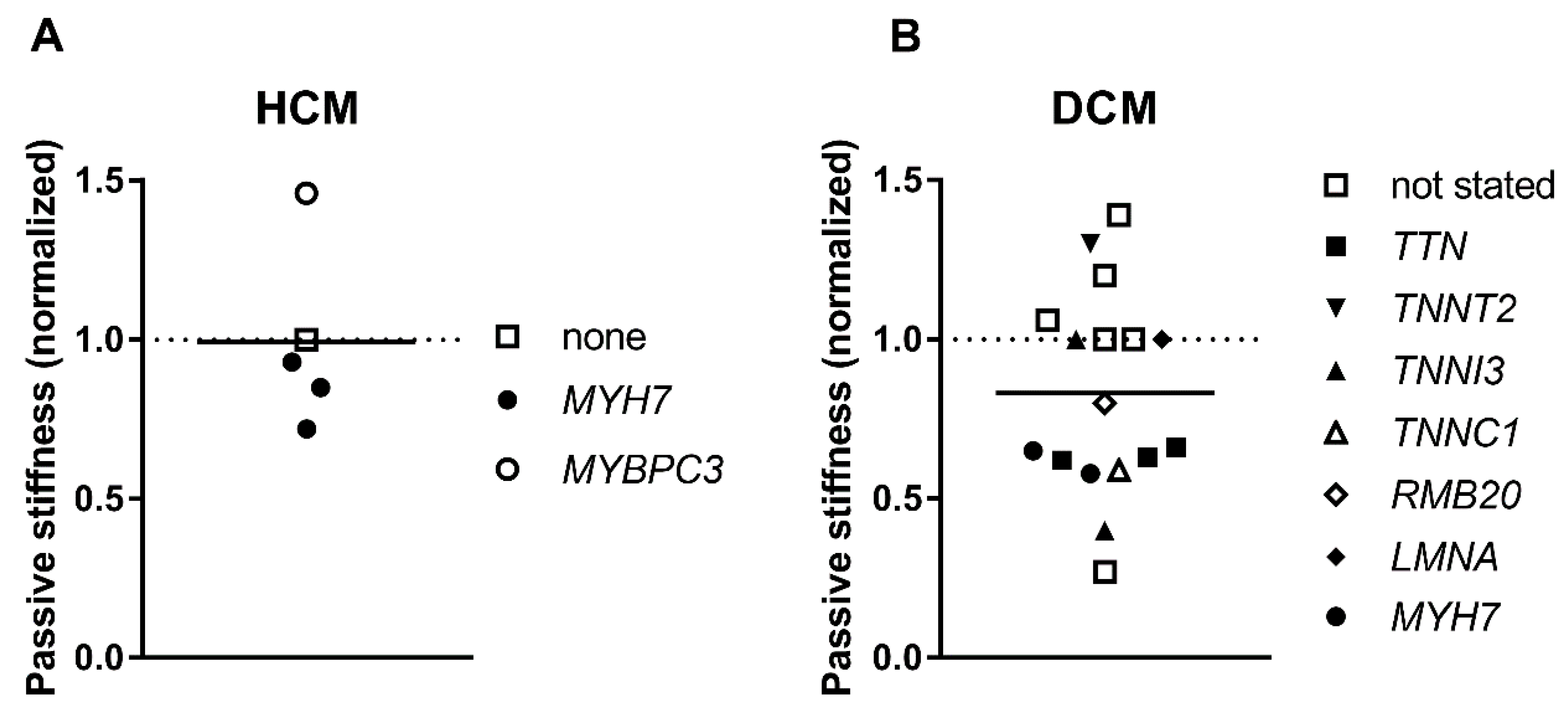
3.6. Passive Stiffness
4. Conclusions
Funding
Conflicts of Interest
References
- Gordan, R.; Gwathmey, J.K.; Xie, L.H. Autonomic and endocrine control of cardiovascular function. World J. Cardiol. 2015, 7, 204–214. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, K.T. Recent advances in understanding cardiac contractility in health and disease. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Eisner, D.A.; Caldwell, J.L.; Kistamas, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.R.; Campbell, S.G.; Lehman, W. Structural determinants of muscle thin filament cooperativity. Arch. Biochem. Biophys. 2016, 594, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Risi, C.; Eisner, J.; Belknap, B.; Heeley, D.H.; White, H.D.; Schroder, G.F.; Galkin, V.E. Ca(2+)-induced movement of tropomyosin on native cardiac thin filaments revealed by cryoelectron microscopy. Proc. Natl. Acad. Sci. USA 2017, 114, 6782–6787. [Google Scholar] [CrossRef] [PubMed]
- Layland, J.; Solaro, R.J.; Shah, A.M. Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovasc. Res. 2005, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Wijnker, P.J.; Murphy, A.M.; Stienen, G.J.; van der Velden, J. Troponin I phosphorylation in human myocardium in health and disease. Neth. Heart J. 2014, 22, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Marston, S.B. Why Is there a Limit to the Changes in Myofilament Ca(2+)-Sensitivity Associated with Myopathy Causing Mutations? Front. Physiol. 2016, 7, 415. [Google Scholar] [CrossRef] [PubMed]
- Craig, R.; Lee, K.H.; Mun, J.Y.; Torre, I.; Luther, P.K. Structure, sarcomeric organization, and thin filament binding of cardiac myosin-binding protein-C. Pflugers Arch. 2014, 466, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Szczesna-Cordary, D. Pseudophosphorylation of cardiac myosin regulatory light chain: A promising new tool for treatment of cardiomyopathy. Biophys. Rev. 2017, 9, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Kampourakis, T.; Yan, Z.; Gautel, M.; Sun, Y.B.; Irving, M. Myosin binding protein-C activates thin filaments and inhibits thick filaments in heart muscle cells. Proc. Natl. Acad. Sci. USA 2014, 111, 18763–18768. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Govindan, S.; Zhang, M.; Khairallah, R.J.; Martin, J.L.; Sadayappan, S.; de Tombe, P.P. Cardiac Myosin-binding Protein C and Troponin-I Phosphorylation Independently Modulate Myofilament Length-dependent Activation. J. Biol. Chem. 2015, 290, 29241–29249. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, M.; Sharma, S.; Cox, S.; Sheppard, M.N.; Panoulas, V.F.; Behr, E.R. The magnitude of sudden cardiac death in the young: A death certificate-based review in England and Wales. Europace 2009, 11, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.E.; McKenna, W.J. New insights into the pathology of inherited cardiomyopathy. Heart 2005, 91, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Rapezzi, C.; Arbustini, E.; Caforio, A.L.; Charron, P.; Gimeno-Blanes, J.; Helio, T.; Linhart, A.; Mogensen, J.; Pinto, Y.; Ristic, A.; et al. Diagnostic work-up in cardiomyopathies: Bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A.; Bowles, N.E. The failing heart. Nature 2002, 415, 227–233. [Google Scholar] [CrossRef] [PubMed]
- McKenna, W.J.; Maron, B.J.; Thiene, G. Classification, Epidemiology, and Global Burden of Cardiomyopathies. Circ. Res. 2017, 121, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Abelmann, W.H.; Lorell, B.H. The challenge of cardiomyopathy. J. Am. Coll. Cardiol. 1989, 13, 1219–1239. [Google Scholar] [CrossRef]
- Haas, J.; Frese, K.S.; Peil, B.; Kloos, W.; Keller, A.; Nietsch, R.; Feng, Z.; Muller, S.; Kayvanpour, E.; Vogel, B.; et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J. 2015, 36, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.A.; Cook, S.A.; Seidman, J.G.; Seidman, C.E. Clinical and Mechanistic Insights Into the Genetics of Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 68, 2871–2886. [Google Scholar] [CrossRef] [PubMed]
- Deo, R.; Albert, C.M. Epidemiology and genetics of sudden cardiac death. Circulation 2012, 125, 620–637. [Google Scholar] [CrossRef] [PubMed]
- Konstam, M.A.; Abboud, F.M. Ejection Fraction: Misunderstood and Overrated (Changing the Paradigm in Categorizing Heart Failure). Circulation 2017, 135, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.E. The pathology of hypertrophic cardiomyopathy. Histopathology 2004, 44, 412–427. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Burke, M.; Fornes, P.; Gallagher, P.J.; de Gouveia, R.H.; Sheppard, M.; Thiene, G.; van der Wal, A.; Association for European Cardiovascular, Pathology. Guidelines for autopsy investigation of sudden cardiac death. Virchows Arch. 2008, 452, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Mathew, T.; Williams, L.; Navaratnam, G.; Rana, B.; Wheeler, R.; Collins, K.; Harkness, A.; Jones, R.; Knight, D.; O’Gallagher, K.; et al. Diagnosis and assessment of dilated cardiomyopathy: A guideline protocol from the British Society of Echocardiography. Echo Res. Pract. 2017, 4, G1–G13. [Google Scholar] [CrossRef] [PubMed]
- Pantazis, A.; Vischer, A.S.; Perez-Tome, M.C.; Castelletti, S. Diagnosis and management of hypertrophic cardiomyopathy. Echo Res. Pract. 2015, 2, R45–R53. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, R.; Fleming, E.J.; Garratt, C.J. Mimics of Hypertrophic Cardiomyopathy—Diagnostic Clues to Aid Early Identification of Phenocopies. Arrhythm. Electrophysiol. Rev. 2013, 2, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, B.; Colvin, M.; Cook, J.; Cooper, L.T.; Deswal, A.; Fonarow, G.C.; Francis, G.S.; Lenihan, D.; Lewis, E.F.; McNamara, D.M.; et al. Current Diagnostic and Treatment Strategies for Specific Dilated Cardiomyopathies: A Scientific Statement From the American Heart Association. Circulation 2016, 134, e579–e646. [Google Scholar] [CrossRef] [PubMed]
- Japp, A.G.; Gulati, A.; Cook, S.A.; Cowie, M.R.; Prasad, S.K. The Diagnosis and Evaluation of Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2996–3010. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kuhl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Bohm, M.; Duboc, D.; Gimeno, J.; de Groote, P.; Imazio, M.; et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Ware, J.S.; Amor-Salamanca, A.; Tayal, U.; Govind, R.; Serrano, I.; Salazar-Mendiguchia, J.; Garcia-Pinilla, J.M.; Pascual-Figal, D.A.; Nunez, J.; Guzzo-Merello, G.; et al. Genetic Etiology for Alcohol-Induced Cardiac Toxicity. J. Am. Coll. Cardiol. 2018, 71, 2293–2302. [Google Scholar] [CrossRef] [PubMed]
- Linschoten, M.; Teske, A.J.; Baas, A.F.; Vink, A.; Dooijes, D.; Baars, H.F.; Asselbergs, F.W. Truncating Titin (TTN) Variants in Chemotherapy-Induced Cardiomyopathy. J. Card. Fail. 2017, 23, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Bakalakos, A.; Ritsatos, K.; Anastasakis, A. Current perspectives on the diagnosis and management of dilated cardiomyopathy Beyond heart failure: A Cardiomyopathy Clinic Doctor’s point of view. Hellenic J. Cardiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Felker, G.M.; Thompson, R.E.; Hare, J.M.; Hruban, R.H.; Clemetson, D.E.; Howard, D.L.; Baughman, K.L.; Kasper, E.K. Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. N. Engl. J. Med. 2000, 342, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A.; Lowe, A.M.; Colan, S.D.; Sleeper, L.A.; Orav, E.J.; Clunie, S.; Messere, J.; Cox, G.F.; Lurie, P.R.; Hsu, D.; et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA 2006, 296, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Schafer, S.; de Marvao, A.; Adami, E.; Fiedler, L.R.; Ng, B.; Khin, E.; Rackham, O.J.; van Heesch, S.; Pua, C.J.; Kui, M.; et al. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat. Genet. 2017, 49, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Tayal, U.; Newsome, S.; Buchan, R.; Whiffin, N.; Halliday, B.; Lota, A.; Roberts, A.; Baksi, A.J.; Voges, I.; Midwinter, W.; et al. Phenotype and Clinical Outcomes of Titin Cardiomyopathy. J. Am. Coll. Cardiol. 2017, 70, 2264–2274. [Google Scholar] [CrossRef] [PubMed]
- Begay, R.L.; Graw, S.; Sinagra, G.; Merlo, M.; Slavov, D.; Gowan, K.; Jones, K.L.; Barbati, G.; Spezzacatene, A.; Brun, F.; et al. Role of Titin Missense Variants in Dilated Cardiomyopathy. J. Am. Heart Assoc. 2015, 4, e002645. [Google Scholar] [CrossRef] [PubMed]
- Van Velzen, H.G.; Schinkel, A.F.L.; Baart, S.J.; Oldenburg, R.A.; Frohn-Mulder, I.M.E.; van Slegtenhorst, M.A.; Michels, M. Outcomes of Contemporary Family Screening in Hypertrophic Cardiomyopathy. Circ. Genom. Precis. Med. 2018, 11, e001896. [Google Scholar] [PubMed]
- Mathew, J.; Zahavich, L.; Lafreniere-Roula, M.; Wilson, J.; George, K.; Benson, L.; Bowdin, S.; Mital, S. Utility of genetics for risk stratification in pediatric hypertrophic cardiomyopathy. Clin. Genet. 2018, 93, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Lopes, L.R.; Syrris, P.; Guttmann, O.P.; O’Mahony, C.; Tang, H.C.; Dalageorgou, C.; Jenkins, S.; Hubank, M.; Monserrat, L.; McKenna, W.J.; et al. Novel genotype-phenotype associations demonstrated by high-throughput sequencing in patients with hypertrophic cardiomyopathy. Heart 2015, 101, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Van Driest, S.L.; Vasile, V.C.; Ommen, S.R.; Will, M.L.; Tajik, A.J.; Gersh, B.J.; Ackerman, M.J. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2004, 44, 1903–1910. [Google Scholar] [CrossRef] [PubMed]
- Kaski, J.P.; Syrris, P.; Esteban, M.T.; Jenkins, S.; Pantazis, A.; Deanfield, J.E.; McKenna, W.J.; Elliott, P.M. Prevalence of sarcomere protein gene mutations in preadolescent children with hypertrophic cardiomyopathy. Circ. Cardiovasc. Genet. 2009, 2, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Millat, G.; Bouvagnet, P.; Chevalier, P.; Dauphin, C.; Jouk, P.S.; Da Costa, A.; Prieur, F.; Bresson, J.L.; Faivre, L.; Eicher, J.C.; et al. Prevalence and spectrum of mutations in a cohort of 192 unrelated patients with hypertrophic cardiomyopathy. Eur. J. Med. Genet. 2010, 53, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Mazzarotto, F.; Girolami, F.; Boschi, B.; Barlocco, F.; Tomberli, A.; Baldini, K.; Coppini, R.; Tanini, I.; Bardi, S.; Contini, E.; et al. Defining the diagnostic effectiveness of genes for inclusion in panels: The experience of two decades of genetic testing for hypertrophic cardiomyopathy at a single center. Genet. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Authors/Task Force Members; Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef] [PubMed]
- Alfares, A.A.; Kelly, M.A.; McDermott, G.; Funke, B.H.; Lebo, M.S.; Baxter, S.B.; Shen, J.; McLaughlin, H.M.; Clark, E.H.; Babb, L.J.; et al. Results of clinical genetic testing of 2912 probands with hypertrophic cardiomyopathy: Expanded panels offer limited additional sensitivity. Genet. Med. 2015, 17, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Mademont-Soler, I.; Mates, J.; Yotti, R.; Espinosa, M.A.; Perez-Serra, A.; Fernandez-Avila, A.I.; Coll, M.; Mendez, I.; Iglesias, A.; Del Olmo, B.; et al. Additional value of screening for minor genes and copy number variants in hypertrophic cardiomyopathy. PLoS ONE 2017, 12, e0181465. [Google Scholar] [CrossRef] [PubMed]
- Dos Remedios, C.G.; Lal, S.P.; Li, A.; McNamara, J.; Keogh, A.; Macdonald, P.S.; Cooke, R.; Ehler, E.; Knoll, R.; Marston, S.B.; et al. The Sydney Heart Bank: Improving translational research while eliminating or reducing the use of animal models of human heart disease. Biophys. Rev. 2017, 9, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Ait Mou, Y.; Bollensdorff, C.; Cazorla, O.; Magdi, Y.; de Tombe, P.P. Exploring cardiac biophysical properties. Glob. Cardiol. Sci. Pract. 2015, 2015, 10. [Google Scholar] [PubMed]
- Poggesi, C.; Tesi, C.; Stehle, R. Sarcomeric determinants of striated muscle relaxation kinetics. Pflugers Arch. 2005, 449, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Stehle, R.; Solzin, J.; Iorga, B.; Poggesi, C. Insights into the kinetics of Ca2+-regulated contraction and relaxation from myofibril studies. Pflugers Arch. 2009, 458, 337–357. [Google Scholar] [CrossRef] [PubMed]
- Vikhorev, P.G.; Ferenczi, M.A.; Marston, S.B. Instrumentation to study myofibril mechanics from static to artificial simulations of cardiac cycle. MethodsX 2016, 3, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B. Effect of Ca2+ on cross-bridge turnover kinetics in skinned single rabbit psoas fibers: Implications for regulation of muscle contraction. Proc. Natl. Acad. Sci. USA 1988, 85, 3265–3269. [Google Scholar] [CrossRef] [PubMed]
- Witjas-Paalberends, E.R.; Piroddi, N.; Stam, K.; van Dijk, S.J.; Oliviera, V.S.; Ferrara, C.; Scellini, B.; Hazebroek, M.; ten Cate, F.J.; van Slegtenhorst, M.; et al. Mutations in MYH7 reduce the force generating capacity of sarcomeres in human familial hypertrophic cardiomyopathy. Cardiovasc. Res. 2013, 99, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Bollen, I.A.E.; van der Meulen, M.; de Goede, K.; Kuster, D.W.D.; Dalinghaus, M.; van der Velden, J. Cardiomyocyte Hypocontractility and Reduced Myofibril Density in End-Stage Pediatric Cardiomyopathy. Front. Physiol. 2017, 8, 1103. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, G.; Zimmermann, R.; Hess, O.M.; Schneider, J.; Kubler, W.; Krayenbuehl, H.P.; Hagl, S.; Mall, G. Decreased concentration of myofibrils and myofiber hypertrophy are structural determinants of impaired left ventricular function in patients with chronic heart diseases: A multiple logistic regression analysis. J. Am. Coll. Cardiol. 1992, 20, 1135–1142. [Google Scholar] [CrossRef]
- Van Dijk, S.J.; Holewijn, R.A.; Tebeest, A.; Dos Remedios, C.; Stienen, G.J.; van der Velden, J. A piece of the human heart: Variance of protein phosphorylation in left ventricular samples from end-stage primary cardiomyopathy patients. J. Muscle Res. Cell Motil. 2009, 30, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Yan, L.J. Protein Oxidative Modifications: Beneficial Roles in Disease and Health. J. Biochem. Pharmacol. Res. 2013, 1, 15–26. [Google Scholar] [PubMed]
- Wijnker, P.J.M.; Sequeira, V.; Kuster, D.W.D.; Velden, J.V. Hypertrophic Cardiomyopathy: A Vicious Cycle Triggered by Sarcomere Mutations and Secondary Disease Hits. Antioxid. Redox Signal. 2018. [Google Scholar] [CrossRef] [PubMed]
- Predmore, J.M.; Wang, P.; Davis, F.; Bartolone, S.; Westfall, M.V.; Dyke, D.B.; Pagani, F.; Powell, S.R.; Day, S.M. Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation 2010, 121, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Ren, J.; Guo, W. Sarcomeric protein isoform transitions in cardiac muscle: A journey to heart failure. Biochim. Biophys. Acta 2015, 1852, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, A.C.; Jacques, A.; Bardswell, S.C.; McKenna, W.J.; Tsang, V.; dos Remedios, C.G.; Ehler, E.; Adams, K.; Jalilzadeh, S.; Avkiran, M.; et al. Normal passive viscoelasticity but abnormal myofibrillar force generation in human hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2010, 49, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Marston, S.; Copeland, O.; Jacques, A.; Livesey, K.; Tsang, V.; McKenna, W.J.; Jalilzadeh, S.; Carballo, S.; Redwood, C.; Watkins, H. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ. Res. 2009, 105, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, V.; Wijnker, P.J.; Nijenkamp, L.L.; Kuster, D.W.; Najafi, A.; Witjas-Paalberends, E.R.; Regan, J.A.; Boontje, N.; Ten Cate, F.J.; Germans, T.; et al. Perturbed length-dependent activation in human hypertrophic cardiomyopathy with missense sarcomeric gene mutations. Circ. Res. 2013, 112, 1491–1505. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, S.J.; Paalberends, E.R.; Najafi, A.; Michels, M.; Sadayappan, S.; Carrier, L.; Boontje, N.M.; Kuster, D.W.; van Slegtenhorst, M.; Dooijes, D.; et al. Contractile dysfunction irrespective of the mutant protein in human hypertrophic cardiomyopathy with normal systolic function. Circ. Heart Fail. 2012, 5, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Belus, A.; Piroddi, N.; Scellini, B.; Tesi, C.; D’Amati, G.; Girolami, F.; Yacoub, M.; Cecchi, F.; Olivotto, I.; Poggesi, C. The familial hypertrophic cardiomyopathy-associated myosin mutation R403Q accelerates tension generation and relaxation of human cardiac myofibrils. J. Physiol. 2008, 586, 3639–3644. [Google Scholar] [CrossRef] [PubMed]
- Piroddi, N.; Belus, A.; Scellini, B.; Tesi, C.; Giunti, G.; Cerbai, E.; Mugelli, A.; Poggesi, C. Tension generation and relaxation in single myofibrils from human atrial and ventricular myocardium. Pflugers Arch. 2007, 454, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Witjas-Paalberends, E.R.; Ferrara, C.; Scellini, B.; Piroddi, N.; Montag, J.; Tesi, C.; Stienen, G.J.; Michels, M.; Ho, C.Y.; Kraft, T.; et al. Faster cross-bridge detachment and increased tension cost in human hypertrophic cardiomyopathy with the R403Q MYH7 mutation. J. Physiol. 2014, 592, 3257–3272. [Google Scholar] [CrossRef] [PubMed]
- Montag, J.; Kowalski, K.; Makul, M.; Ernstberger, P.; Radocaj, A.; Beck, J.; Becker, E.; Tripathi, S.; Keyser, B.; Muhlfeld, C.; et al. Burst-Like Transcription of Mutant and Wildtype MYH7-Alleles as Possible Origin of Cell-to-Cell Contractile Imbalance in Hypertrophic Cardiomyopathy. Front. Physiol. 2018, 9, 359. [Google Scholar] [CrossRef] [PubMed]
- Kraft, T.; Witjas-Paalberends, E.R.; Boontje, N.M.; Tripathi, S.; Brandis, A.; Montag, J.; Hodgkinson, J.L.; Francino, A.; Navarro-Lopez, F.; Brenner, B.; et al. Familial hypertrophic cardiomyopathy: Functional effects of myosin mutation R723G in cardiomyocytes. J. Mol. Cell. Cardiol. 2013, 57, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Schultz, I.; Becker, E.; Montag, J.; Borchert, B.; Francino, A.; Navarro-Lopez, F.; Perrot, A.; Ozcelik, C.; Osterziel, K.J.; et al. Unequal allelic expression of wild-type and mutated beta-myosin in familial hypertrophic cardiomyopathy. Basic Res. Cardiol. 2011, 106, 1041–1055. [Google Scholar] [CrossRef] [PubMed]
- Montag, J.; Syring, M.; Rose, J.; Weber, A.L.; Ernstberger, P.; Mayer, A.K.; Becker, E.; Keyser, B.; Dos Remedios, C.; Perrot, A.; et al. Intrinsic MYH7 expression regulation contributes to tissue level allelic imbalance in hypertrophic cardiomyopathy. J. Muscle Res. Cell Motil. 2017, 38, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Vikhorev, P.G.; Smoktunowicz, N.; Munster, A.B.; Copeland, O.; Kostin, S.; Montgiraud, C.; Messer, A.E.; Toliat, M.R.; Li, A.; Dos Remedios, C.G.; et al. Abnormal contractility in human heart myofibrils from patients with dilated cardiomyopathy due to mutations in TTN and contractile protein genes. Sci. Rep. 2017, 7, 14829. [Google Scholar] [CrossRef] [PubMed]
- Van Spaendonck-Zwarts, K.Y.; Posafalvi, A.; van den Berg, M.P.; Hilfiker-Kleiner, D.; Bollen, I.A.; Sliwa, K.; Alders, M.; Almomani, R.; van Langen, I.M.; van der Meer, P.; et al. Titin gene mutations are common in families with both peripartum cardiomyopathy and dilated cardiomyopathy. Eur. Heart J. 2014, 35, 2165–2173. [Google Scholar] [CrossRef] [PubMed]
- Bollen, I.A.E.; Schuldt, M.; Harakalova, M.; Vink, A.; Asselbergs, F.W.; Pinto, J.R.; Kruger, M.; Kuster, D.W.D.; van der Velden, J. Genotype-specific pathogenic effects in human dilated cardiomyopathy. J. Physiol. 2017, 595, 4677–4693. [Google Scholar] [CrossRef] [PubMed]
- Hoorntje, E.T.; Bollen, I.A.; Barge-Schaapveld, D.Q.; van Tienen, F.H.; Te Meerman, G.J.; Jansweijer, J.A.; van Essen, A.J.; Volders, P.G.; Constantinescu, A.A.; van den Akker, P.C.; et al. Lamin A/C-Related Cardiac Disease: Late Onset With a Variable and Mild Phenotype in a Large Cohort of Patients With the Lamin A/C p.(Arg331Gln) Founder Mutation. Circ. Cardiovasc. Genet. 2017, 10, e001631. [Google Scholar] [CrossRef] [PubMed]
- Beqqali, A.; Bollen, I.A.; Rasmussen, T.B.; van den Hoogenhof, M.M.; van Deutekom, H.W.; Schafer, S.; Haas, J.; Meder, B.; Sorensen, K.E.; van Oort, R.J.; et al. A mutation in the glutamate-rich region of RNA-binding motif protein 20 causes dilated cardiomyopathy through missplicing of titin and impaired Frank-Starling mechanism. Cardiovasc. Res. 2016, 112, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Dyer, E.C.; Jacques, A.M.; Hoskins, A.C.; Ward, D.G.; Gallon, C.E.; Messer, A.E.; Kaski, J.P.; Burch, M.; Kentish, J.C.; Marston, S.B. Functional analysis of a unique troponin c mutation, GLY159ASP, that causes familial dilated cardiomyopathy, studied in explanted heart muscle. Circ. Heart Fail. 2009, 2, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Vallins, W.J.; Brand, N.J.; Dabhade, N.; Butler-Browne, G.; Yacoub, M.H.; Barton, P.J. Molecular cloning of human cardiac troponin I using polymerase chain reaction. FEBS Lett. 1990, 270, 57–61. [Google Scholar] [CrossRef]
- Chaturvedi, R.R.; Herron, T.; Simmons, R.; Shore, D.; Kumar, P.; Sethia, B.; Chua, F.; Vassiliadis, E.; Kentish, J.C. Passive stiffness of myocardium from congenital heart disease and implications for diastole. Circulation 2010, 121, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Van der Velden, J.; Papp, Z.; Zaremba, R.; Boontje, N.M.; de Jong, J.W.; Owen, V.J.; Burton, P.B.; Goldmann, P.; Jaquet, K.; Stienen, G.J. Increased Ca2+-sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc. Res. 2003, 57, 37–47. [Google Scholar] [CrossRef]
- Bollen, I.A.E.; Ehler, E.; Fleischanderl, K.; Bouwman, F.; Kempers, L.; Ricke-Hoch, M.; Hilfiker-Kleiner, D.; Dos Remedios, C.G.; Kruger, M.; Vink, A.; et al. Myofilament Remodeling and Function Is More Impaired in Peripartum Cardiomyopathy Compared with Dilated Cardiomyopathy and Ischemic Heart Disease. Am. J. Pathol. 2017, 187, 2645–2658. [Google Scholar] [CrossRef] [PubMed]
- Kooij, V.; Saes, M.; Jaquet, K.; Zaremba, R.; Foster, D.B.; Murphy, A.M.; Dos Remedios, C.; van der Velden, J.; Stienen, G.J. Effect of troponin I Ser23/24 phosphorylation on Ca2+-sensitivity in human myocardium depends on the phosphorylation background. J. Mol. Cell. Cardiol. 2010, 48, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Kotter, S.; Gout, L.; Von Frieling-Salewsky, M.; Muller, A.E.; Helling, S.; Marcus, K.; Dos Remedios, C.; Linke, W.A.; Kruger, M. Differential changes in titin domain phosphorylation increase myofilament stiffness in failing human hearts. Cardiovasc. Res. 2013, 99, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Papp, Z.; van der Velden, J.; Borbely, A.; Edes, I.; Stienen, G.J.M. Altered myocardial force generation in end-stage human heart failure. ESC Heart Fail. 2014, 1, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Mamidi, R.; Li, J.; Gresham, K.S.; Verma, S.; Doh, C.Y.; Li, A.; Lal, S.; Dos Remedios, C.G.; Stelzer, J.E. Dose-Dependent Effects of the Myosin Activator Omecamtiv Mecarbil on Cross-Bridge Behavior and Force Generation in Failing Human Myocardium. Circ. Heart Fail. 2017, 10, e004257. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.Y.; Lin, Y.H.; Wennersten, S.A.; Demos-Davies, K.M.; Cavasin, M.A.; Mahaffey, J.H.; Monzani, V.; Saripalli, C.; Mascagni, P.; Reece, T.B.; et al. Histone deacetylase activity governs diastolic dysfunction through a nongenomic mechanism. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, J.; Kawai, M.; Minai, K.; Ogawa, K.; Ogawa, T.; Yoshimura, M. Associations between Left Ventricular Cavity Size and Cardiac Function and Overload Determined by Natriuretic Peptide Levels and a Covariance Structure Analysis. Sci. Rep. 2017, 7, 2037. [Google Scholar] [CrossRef] [PubMed]
- Reinstein, E.; Gutierrez-Fernandez, A.; Tzur, S.; Bormans, C.; Marcu, S.; Tayeb-Fligelman, E.; Vinkler, C.; Raas-Rothschild, A.; Irge, D.; Landau, M.; et al. Congenital dilated cardiomyopathy caused by biallelic mutations in Filamin C. Eur. J. Hum. Genet. 2016, 24, 1792–1796. [Google Scholar] [CrossRef] [PubMed]
- Messer, A.E.; Gallon, C.E.; McKenna, W.J.; Dos Remedios, C.G.; Marston, S.B. The use of phosphate-affinity SDS-PAGE to measure the cardiac troponin I phosphorylation site distribution in human heart muscle. Proteom. Clin. Appl. 2009, 3, 1371–1382. [Google Scholar] [CrossRef] [PubMed]
- Bodor, G.S.; Oakeley, A.E.; Allen, P.D.; Crimmins, D.L.; Ladenson, J.H.; Anderson, P.A. Troponin I phosphorylation in the normal and failing adult human heart. Circulation 1997, 96, 1495–1500. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Dyer, E.; Stuckey, D.J.; Copeland, O.; Leung, M.C.; Bayliss, C.; Messer, A.; Wilkinson, R.; Tremoleda, J.L.; Schneider, M.D.; et al. Molecular mechanism of the E99K mutation in cardiac actin (ACTC Gene) that causes apical hypertrophy in man and mouse. J. Biol. Chem. 2011, 286, 27582–27593. [Google Scholar] [CrossRef] [PubMed]
- Hamdani, N.; Kooij, V.; van Dijk, S.; Merkus, D.; Paulus, W.J.; Remedios, C.D.; Duncker, D.J.; Stienen, G.J.; van der Velden, J. Sarcomeric dysfunction in heart failure. Cardiovasc. Res. 2008, 77, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Copeland, O.; Sadayappan, S.; Messer, A.E.; Steinen, G.J.; van der Velden, J.; Marston, S.B. Analysis of cardiac myosin binding protein-C phosphorylation in human heart muscle. J. Mol. Cell. Cardiol. 2010, 49, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Jacques, A.M.; Briceno, N.; Messer, A.E.; Gallon, C.E.; Jalilzadeh, S.; Garcia, E.; Kikonda-Kanda, G.; Goddard, J.; Harding, S.E.; Watkins, H.; et al. The molecular phenotype of human cardiac myosin associated with hypertrophic obstructive cardiomyopathy. Cardiovasc. Res. 2008, 79, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Papadaki, M.; Vikhorev, P.G.; Marston, S.B.; Messer, A.E. Uncoupling of myofilament Ca2+ sensitivity from troponin I phosphorylation by mutations can be reversed by epigallocatechin-3-gallate. Cardiovasc. Res. 2015, 108, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Messer, A.E.; Bayliss, C.R.; El-Mezgueldi, M.; Redwood, C.S.; Ward, D.G.; Leung, M.C.; Papadaki, M.; Dos Remedios, C.; Marston, S.B. Mutations in troponin T associated with Hypertrophic Cardiomyopathy increase Ca(2+)-sensitivity and suppress the modulation of Ca(2+)-sensitivity by troponin I phosphorylation. Arch. Biochem. Biophys. 2016, 601, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Messer, A.E.; Chan, J.; Daley, A.; Copeland, O.; Marston, S.B.; Connolly, D.J. Investigations into the Sarcomeric Protein and Ca(2+)-Regulation Abnormalities Underlying Hypertrophic Cardiomyopathy in Cats (Felix catus). Front. Physiol. 2017, 8, 348. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, C.R.; Jacques, A.M.; Leung, M.C.; Ward, D.G.; Redwood, C.S.; Gallon, C.E.; Copeland, O.; McKenna, W.J.; Dos Remedios, C.; Marston, S.B.; et al. Myofibrillar Ca(2+) sensitivity is uncoupled from troponin I phosphorylation in hypertrophic obstructive cardiomyopathy due to abnormal troponin T. Cardiovasc. Res. 2013, 97, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Vikhorev, P.G.; Song, W.; Wilkinson, R.; Copeland, O.; Messer, A.E.; Ferenczi, M.A.; Marston, S.B. The dilated cardiomyopathy-causing mutation ACTC E361G in cardiac muscle myofibrils specifically abolishes modulation of Ca(2+) regulation by phosphorylation of troponin I. Biophys. J. 2014, 107, 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Biesiadecki, B.J.; Kobayashi, T.; Walker, J.S.; Solaro, R.J.; de Tombe, P.P. The troponin C G159D mutation blunts myofilament desensitization induced by troponin I Ser23/24 phosphorylation. Circ. Res. 2007, 100, 1486–1493. [Google Scholar] [CrossRef] [PubMed]
- Messer, A.E.; Marston, S.B. Investigating the role of uncoupling of troponin I phosphorylation from changes in myofibrillar Ca(2+)-sensitivity in the pathogenesis of cardiomyopathy. Front. Physiol. 2014, 5, 315. [Google Scholar] [CrossRef] [PubMed]
- Edes, I.F.; Czuriga, D.; Csanyi, G.; Chlopicki, S.; Recchia, F.A.; Borbely, A.; Galajda, Z.; Edes, I.; van der Velden, J.; Stienen, G.J.; et al. Rate of tension redevelopment is not modulated by sarcomere length in permeabilized human, murine, and porcine cardiomyocytes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R20–R29. [Google Scholar] [CrossRef] [PubMed]
- McNamara, J.W.; Li, A.; Lal, S.; Bos, J.M.; Harris, S.P.; van der Velden, J.; Ackerman, M.J.; Cooke, R.; Dos Remedios, C.G. MYBPC3 mutations are associated with a reduced super-relaxed state in patients with hypertrophic cardiomyopathy. PLoS ONE 2017, 12, e0180064. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, S.J.; Dooijes, D.; dos Remedios, C.; Michels, M.; Lamers, J.M.; Winegrad, S.; Schlossarek, S.; Carrier, L.; ten Cate, F.J.; Stienen, G.J.; et al. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: Haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation 2009, 119, 1473–1483. [Google Scholar] [CrossRef] [PubMed]
- Rottbauer, W.; Gautel, M.; Zehelein, J.; Labeit, S.; Franz, W.M.; Fischer, C.; Vollrath, B.; Mall, G.; Dietz, R.; Kubler, W.; et al. Novel splice donor site mutation in the cardiac myosin-binding protein-C gene in familial hypertrophic cardiomyopathy. Characterization Of cardiac transcript and protein. J. Clin. Investig. 1997, 100, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Gilda, J.E.; Gomes, A.V. Proteasome dysfunction in cardiomyopathies. J. Physiol. 2017, 595, 4051–4071. [Google Scholar] [CrossRef] [PubMed]
- Makarenko, I.; Opitz, C.A.; Leake, M.C.; Neagoe, C.; Kulke, M.; Gwathmey, J.K.; del Monte, F.; Hajjar, R.J.; Linke, W.A. Passive stiffness changes caused by upregulation of compliant titin isoforms in human dilated cardiomyopathy hearts. Circ. Res. 2004, 95, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Nagueh, S.F.; Shah, G.; Wu, Y.; Torre-Amione, G.; King, N.M.; Lahmers, S.; Witt, C.C.; Becker, K.; Labeit, S.; Granzier, H.L. Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation 2004, 110, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.M.; Ware, J.S.; Herman, D.S.; Schafer, S.; Baksi, J.; Bick, A.G.; Buchan, R.J.; Walsh, R.; John, S.; Wilkinson, S.; et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci. Transl. Med. 2015, 7, 270ra6. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.J.; Winslow, R.L.; Hunter, W.C. Comparison of putative cooperative mechanisms in cardiac muscle: Length dependence and dynamic responses. Am. J. Physiol. 1999, 276, H1734–H1754. [Google Scholar] [CrossRef] [PubMed]
- Beuckelmann, D.J.; Nabauer, M.; Erdmann, E. Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation 1992, 85, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Gwathmey, J.K.; Slawsky, M.T.; Hajjar, R.J.; Briggs, G.M.; Morgan, J.P. Role of intracellular calcium handling in force-interval relationships of human ventricular myocardium. J. Clin. Investig. 1990, 85, 1599–1613. [Google Scholar] [CrossRef] [PubMed]
- Bayer, J.D.; Narayan, S.M.; Lalani, G.G.; Trayanova, N.A. Rate-dependent action potential alternans in human heart failure implicates abnormal intracellular calcium handling. Heart Rhythm 2010, 7, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Dewey, S.; Xu, Q.; Gomes, A. Static and dynamic properties of the HCM myocardium. J. Mol. Cell. Cardiol. 2010, 49, 715–718. [Google Scholar] [CrossRef] [PubMed]
- Poggesi, C.; Ho, C.Y. Muscle dysfunction in hypertrophic cardiomyopathy: What is needed to move to translation? J. Muscle Res. Cell. Motil. 2014, 35, 37–45. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation, Gene Expression and Mutated (mut) Allele Expression | Phosphorylation Level (Cardiomyopathy/Healthy) | Titin Isoforms and Passive Stiffness (Cardiomyopathy/Healthy) | Maximal Force and Myofibril Density (Cardiomyopathy/Healthy) | Contractile Kinetics Parameters (Cardiomyopathy/Healthy) | Ca2-Sensitivity of Force (Cardiomyopathy/Healthy) | Length Dependent Activation Changes upon Stretch (Cardiomyopathy/Healthy) | Effect PKA on EC50 (in µM) | Patient Information (Sex, Age and Number of Patients) | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Expression | Myocyte | Myofibril | Activation | Relaxation | +PKA | +PKA | ||||||||||||||||||||||||
| Diagnosis | Gene with Mutation | Mutation and Type: Truncated (T) or Not (NT) | Gene | Mut Allele | TnI | MyBP-C | LC2 | N2AB/N2B | Passive Stiffness | Fmax | Myofibril Density | Fmax | kACT | kTR | tLIN | kLIN | kREL | EC50 | nH | EC50 | ΔEC50 | ΔF | ΔEC50 | ΔF | ΔEC50 | Sex M/F | Age | N | Refs | |
| HCM | MYBPC3 | g.T2604A+C del at 2605 | T | 0.64 * | 0.34 * | 0.64 | 1 | 1.14 | 0.42 | M | 42 | 1 | [65,66] | |||||||||||||||||
| HCM | MYBPC3 | T | 0.18 | 0.75 | 0.85 * | 0.79 * | 0.96 | 0.57 * | 1.16 | 1.01 | 0.94 | 0.88 * | M/F | 22–69 | 17 | [67] | ||||||||||||||
| HCM | MYBPC3 | T | 0.67 | 0.69 * | 0.82 | 1.46 | 0.71 * | 0.83 * | 0.84 * | 1 | 0.61 * | 1.04 | 1.13 | M/F | 22–69 | 17 | [68] | |||||||||||||
| HCM | MYBPC3 | T/NT | 0.68 * | 0.69 * | 0.85 * | 1 | 1 | MF | 32–69 | 28 | [57] | |||||||||||||||||||
| HCM | MYBPC3 | p.R502W | NT | 0.8 * | 0.75 | 1.10 | 0.35 | M | 23 | 1 | [65] | |||||||||||||||||||
| HCM | MYBPC3 | NT | 0.09 | 0.64 | 0.44 * | 0.56 * | 0.93 | 0.29 * | 0.28 * | 0.18 * | 0.25 * | 1.45 * | M/F | 31–51 | 4 | [67] | ||||||||||||||
| HCM | MYH7 | p.R719Q | NT | 0.79 | 1.16 | 1.01 | F | 27 | 1 | [65] | ||||||||||||||||||||
| HCM | MYH7 | NT | 0.22 | 0.58 | 0.54 * | 0.56 * | 0.88 * | 0.26 * | 0.44 * | 0.42 * | 0.38 * | 0.97 * | M/F | 25–61 | 4–6 | [67] | ||||||||||||||
| fHCM | MYH7 | p.R403Q | NT | 0.35 * | 0.93 | 0.3 | 0.56 * | 1.92 * | 2.02 * | 0.75 * | 4 * | 1.76 * | M | 24 | 1 | [69,70,71] | ||||||||||||||
| fHCM | MYH7 | p.R403Q | NT | 0.5 | 0.6 | 0.83 | 1.84 | 2.9 | 1.77 | M | 35 | 1 | [69,70,71] | |||||||||||||||||
| fHCM | MYH7 | p.R403Q | NT | 0.45 | 0.6 | 0.91 | 1.52 | 2.7 | 1.99 | F | 39 | 1 | [69,70,71] | |||||||||||||||||
| fHCM | MYH7 | p.R723G | NT | 0.7 | 0.54 * | 1.41 * | M | 38, 55 | 2 | [72] | ||||||||||||||||||||
| fHCM | MYH7 | p.R723G | NT | 0.68 | 0.11 * | 0.42 * | 0.42 * | 0.72 * | 0.69 * | 0.74 * | 0.93 | 1 | 1.28 * | 1.44 * | M/F | 53, 55 | 2 | [73,74] | ||||||||||||
| fHCM | MYH7 | p.A200V | NT | 0.53 | 0.48 * | 1.12 * | F | 19 | 1 | [72,75] | ||||||||||||||||||||
| fHCM | MYH7 | NT | 0.39 * | 0.79 * | 0.65 * | 1 | M/F | 19–61 | 11 | [57] | ||||||||||||||||||||
| fHCM | TPM1 | p.I284V | NT | 0.40 * | 0.70 * | 0.62 * | M | 65 | 1 | [57] | ||||||||||||||||||||
| HCM | TPM1 | p.M281T | NT | 0.07 | 0.55 | 0.48 * | 0.67 * | 0.95 | 0.39 * | 0.72 | −0.2 * | 0.06 * | 1.08 * | M | 65 | 1 | [67] | |||||||||||||
| HCM | TNNT2 | p.K280N | NT | 0.96 | 1.07 | 0.70 | 0.80 * | 0.76 * | 0.45 * | 0.34 | 0.30 * | 0.15 * | 0.12 | M | 26 | 1 | [67] | |||||||||||||
| HCM | TNNT2 | p.K280N | NT | 0.56 | 0.78 * | 0.73 * | M | 26 | 2 | [57] | ||||||||||||||||||||
| HCM | TNNI3 | p.R145W | NT | 0.55 * | 0.58 * | 0.84 | M | 46, 66 | 2 | [57] | ||||||||||||||||||||
| HCM | TNNI3 | p.R145W | NT | 0.09 | 0.49 | 0.29 | 0.88 * | 1.18 * | 0.38 * | 0.03 * | 0.34 * | 0.08 * | 0.99 * | M | 46, 66 | 2 | [67] | |||||||||||||
| HCM | MYH7(2) MYBPC3(1) | NT | 0.25 * | 0.14 * | 0.91 | 0.70 | 0.85 | 0.61 * | 1 | 1.1 * | 0.49 * | 1.26 | M/F | 23–61 | 6 | [65,70] | ||||||||||||||
| HCM | none | 0.18 | 0.4 | 0.81 * | 0.86 * | 0.93 | 0.49 * | 1.2 | 1.06 * | 1.2 | 0.73 | M/F | 46–75 | 3–7 | [67] | |||||||||||||||
| HCM | none | 1 | 0.71 * | 0.92 * | 0.78 * | 1 | M/F | 35–75 | 3–14 | [57] | ||||||||||||||||||||
| HCM | none | 0.60 * | 0.54 * | 1 | 0.75 * | 0.8 * | 0.81 * | 1 | 0.49 * | 0.94 | 0.97 | M/F | 46–75 | 11 | [68] | |||||||||||||||
| HCM | none | 1.0 | 0.84 | 1.01 | 2.1 | 1.59 | M/F | 35–72 | 9 | [71] | ||||||||||||||||||||
| HCM | none | 0.61 | 1.09 | 0.54 | M | 59 | 1 | [65] | ||||||||||||||||||||||
| HCM | none | 0.35 | 1.05 | 0.45 | F | 61 | 1 | [65] | ||||||||||||||||||||||
| HCM | none | 0.42 | 1.12 | 0.49 | M | 58 | 1 | [65] | ||||||||||||||||||||||
| Mutation, Gene Expression and Mutated (mut) Allele Expression | Phosphorylation Level (Cardiomyopathy/Healthy) | Titin Isoforms and Passive Stiffness (Cardiomyopathy/Healthy | Maximal Force and Myofibril Density (Cardiomyopathy/Healthy) | Contractile Kinetics Parameters (Cardiomyopathy/Healthy) | Ca2-Sensitivity of Force (Cardiomyopathy/Healthy) | Length Dependent Activation Changes upon Stretch (Cardiomyopathy/Healthy) | Effect PKA on EC50 (in µM) | Patient Information (Sex, Age and Number of Patients) | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Expression | Myocyte | Myofibril | Activation | Relaxation | +PKA | +PKA | |||||||||||||||||||||||
| Diagnosis | Gene with Mutation | Mutation and Type: Truncated (T) or Not (NT) | Gene | Mut Protein | TnI | MyBP-C | LC2 | N2AB/ N2B | Passive Stiffness | Fmax | Myofibril Density | Fmax | kACT | kTR | tLIN | kLIN | kREL | EC50 | nH | EC50 | ΔEC50 | ΔF | ΔEC50 | ΔEC50 | Sex M/F | Age | N | Refs | |
| IDCM | TTN | p.(R23464Tfs *41) | T | 1.0 | 0 | 1.05 | 0.63 * | 1.02 | 1.05 | 1.06 | 0.97 | 1.06 | 0.91 | 1.00 | M | 22 | 1 | [76] | |||||||||||
| fDCM | TTN | p.(R23464Tfs*41) | T | 1.0 | 0 | 1.11 | 0.66 * | 0.96 | 1.12 | 0.95 | 0.98 | 0.18 | 1.13 | 1.13 | M | 37 | 1 | [76] | |||||||||||
| fDCM | TTN | p.(Y18923 *) | T | 0.96 | 0 | 1.25 | 0.62 * | 1.04 | 1.15 | 1.16 * | 0.98 | 1.06 | 1.17 | 1.03 | F | 22 | 1 | [76] | |||||||||||
| PPCM | TTN | p.(K15664Vfs*13) | T | 1.85 | 0.4 | F | 1 | [77] | |||||||||||||||||||||
| DCM | LMNA | p.(R331Q) | NT | 0.5 | 1.98 * | 1 | 0.64 * | 0.63 | 0.86 * | 1 | 0.78 | M/F | 45.3 | 3 | [78,79] | ||||||||||||||
| fDCM | RMB20 | p.E913K | NT | 0.13 | 13.89 | 0.8 | 0.85 | 0.67 * | 0.58 | 1.13 | 0.36 | 0.81 | 1.77 | M | 19 | 1 | [80] | ||||||||||||
| fDCM | TNNC1 | p.G159D | NT | 1.13 | 0.49 | 0.13 | 0.89 | 0.59 * | 1 | 1.20 | 0.97 | 0.97 | 0.71 * | 0.35 | 1.17 | 0.60 * | 0.59 * | 1.22 | M | 3 | 1 | [76,81] | |||||||
| fDCM | TNNI3 | p.K36Q | NT | 1.0 | 0.58 * | 1.10 | 1.17 | 1.05 | 0.70 * | 1.39 | 1.30 * | 1.34 | M | 15 | 1 | [76] | |||||||||||||
| IDCM | TNNI3 | p.(R98 *) | T | 0.39 | 0 | 1.08 | 1.56 | 1 | 0.95 | 0.94 * | 0.88 * | 0.66 | M | 46 | 1 | [78,82] | |||||||||||||
| IDCM | TNNT2 | p.(K217del) | NT | 0.51 | 1.08 | 1.64 | 1.3 * | 1.11 | 1.07 | 0.97 | M | 19 | 1 | [78] | |||||||||||||||
| fDCM | MYH7 | p.E1426K | NT | 1.08 | 0.65 * | 0.97 | 1.13 | 1.19 * | 0.70 * | 0.76 | 1.35 * | 0.96 | M | 43 | 1 | [76] | |||||||||||||
| IDCM | not stated | 0.54 | 1.98 | 1 | 0.98 | 0.94 | 1 | 0.73 | M/F | 54.6 | 5 | [78] | |||||||||||||||||
| DCM | not stated | 1.24 * | 0.27 * | 51–56 | 3–4 | [83] | |||||||||||||||||||||||
| DCM | not stated | 0.5 | 0.51 | 1 | 0.96 | 3.37 | M | 45,59 | 2 | [84] | |||||||||||||||||||
| IDCM | not stated | 0.35 * | 0.58 * | 1.62 * | 1 | 0.94 | 0.69 * | 1.12 | 0.76 | 1.09 | 8 | [85] | |||||||||||||||||
| IDCM | not stated | 0.4 * | 0.4 * | 1.1 | 1.18 | 0.88 | 0.67 | 1.33 | 0.8 | 1.26 | M/F | 52–62 | 7 | [86] | |||||||||||||||
| IDCM | not stated | 1.78 | 1.2 | 1.34 | 0.68 * | 0.81 | 0.87 | 0.59 | 0.86 | 1.33 | 3 | [80] | |||||||||||||||||
| IDCM | not stated | 1.14 | F | 41–57 | 6 | [87] | |||||||||||||||||||||||
| PPCM | not stated | 0.24 * | 0.61 * | 1.58 | 1.39 * | 0.84 | 0.65 * | 1.01 | 0.48 * | 0.96 | F | 6 | [85] | ||||||||||||||||
| ICM | not stated | 0.95 | 0.93 | 0.71 * | 0.83 * | 3 | [88] | ||||||||||||||||||||||
| ICM | not stated | 0.5 | 0.72 * | 0.98 | 2.13 | M/F | 41–65 | 7 | [84] | ||||||||||||||||||||
| ICM | not stated | 0.5 | 0.5 * | 0.83 | 1 | 1.04 | 0.70 * | 1.11 | 0.96 | 1.19 | 4 | [85] | |||||||||||||||||
| ICM (7) DCM (2) | not stated | 0.5 | 0.57 * | 1.06 | 1.0 | 0.66 * | 0.99 | 0.98 | 2.55 * | M/F | 41–65 | 10 | [84] | ||||||||||||||||
| HF | not stated | 0.33 * | 0.65 * | 1 | 0.53 * | 0.53 * | 0.96 | 0.63 * | 0.87 | 0.98 | M/F | 42–57 | 4 | [89] | |||||||||||||||
| IRCM | not stated | 0.84 | 1.20 * | 0.05 * | 0.52 * | F | 66 | 1 | [90] | ||||||||||||||||||||
| IRCM | not stated | 1.16 | 1.92 * | 0.07 * | 0.33 * | M | 21 | 1 | [90] | ||||||||||||||||||||
| IDCM (9) NCC (2) | not stated | 0.27 * | 0.4–2.8 | 0.7 * | 0.64 * | 0.67 * | 1 | <18 | 11 | [58] | |||||||||||||||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vikhorev, P.G.; Vikhoreva, N.N. Cardiomyopathies and Related Changes in Contractility of Human Heart Muscle. Int. J. Mol. Sci. 2018, 19, 2234. https://doi.org/10.3390/ijms19082234
Vikhorev PG, Vikhoreva NN. Cardiomyopathies and Related Changes in Contractility of Human Heart Muscle. International Journal of Molecular Sciences. 2018; 19(8):2234. https://doi.org/10.3390/ijms19082234
Chicago/Turabian StyleVikhorev, Petr G., and Natalia N. Vikhoreva. 2018. "Cardiomyopathies and Related Changes in Contractility of Human Heart Muscle" International Journal of Molecular Sciences 19, no. 8: 2234. https://doi.org/10.3390/ijms19082234
APA StyleVikhorev, P. G., & Vikhoreva, N. N. (2018). Cardiomyopathies and Related Changes in Contractility of Human Heart Muscle. International Journal of Molecular Sciences, 19(8), 2234. https://doi.org/10.3390/ijms19082234