Niemann-Pick Type C2 Protein Regulates Free Cholesterol Accumulation and Influences Hepatic Stellate Cell Proliferation and Mitochondrial Respiration Function

 ,
,
Abstract

1. Introduction
2. Results
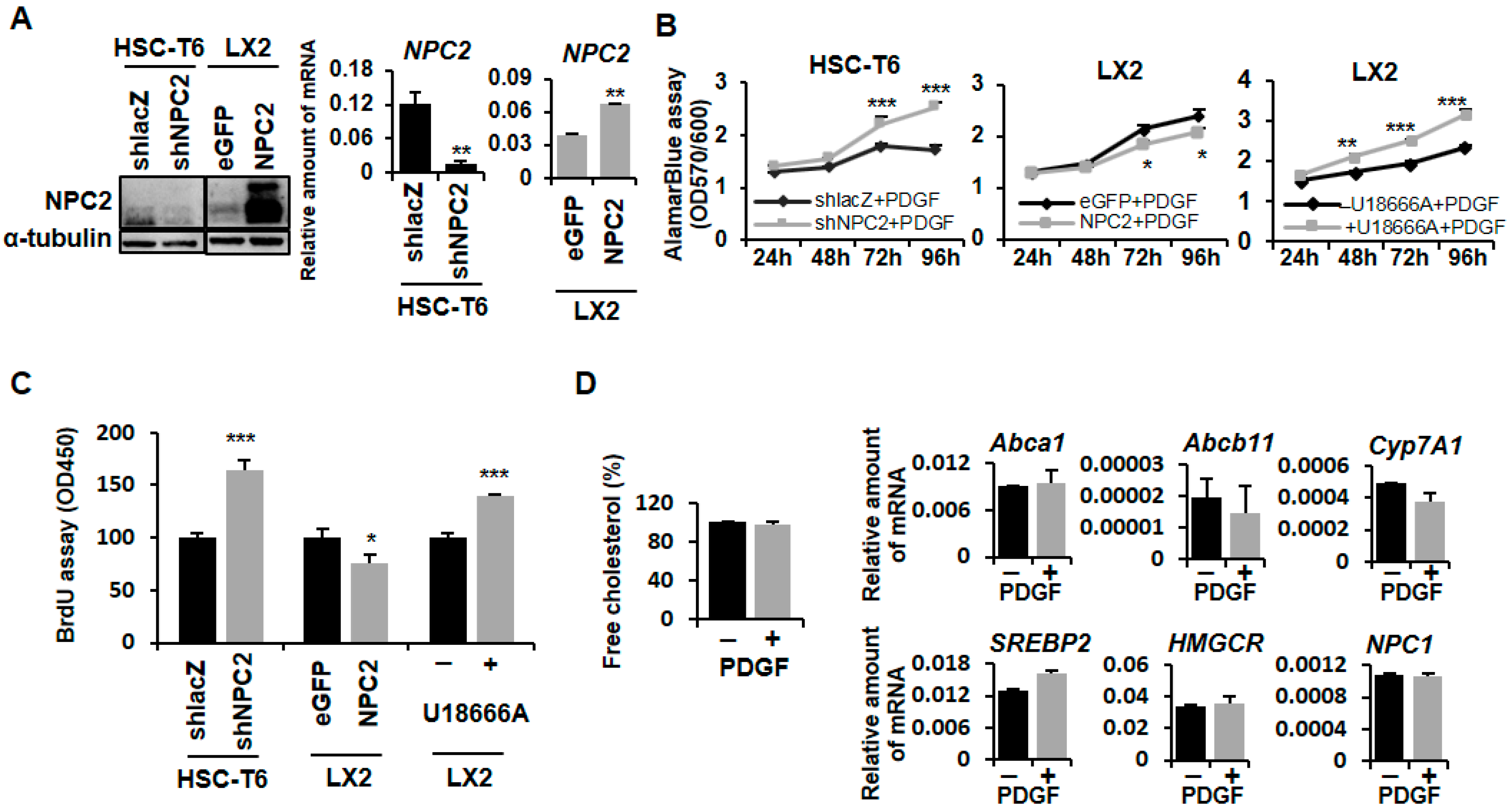
2.1. NPC2 Downregulation and Free Cholesterol Accumulation Enhanced PDGF-BB-Induced Proliferation in HSCs
2.2. NPC2 Downregulation and Free Cholesterol Accumulation Enhanced PDGF-BB-Induced MAPK and AKT Activation in HSCs
2.3. NPC2 Expression and Free Cholesterol Accumulation Did Not Alter HSC Apoptosis
2.4. NPC2 Downregulated and Free Cholesterol Accumulated HSCs Disrupt Mitochondrial Function
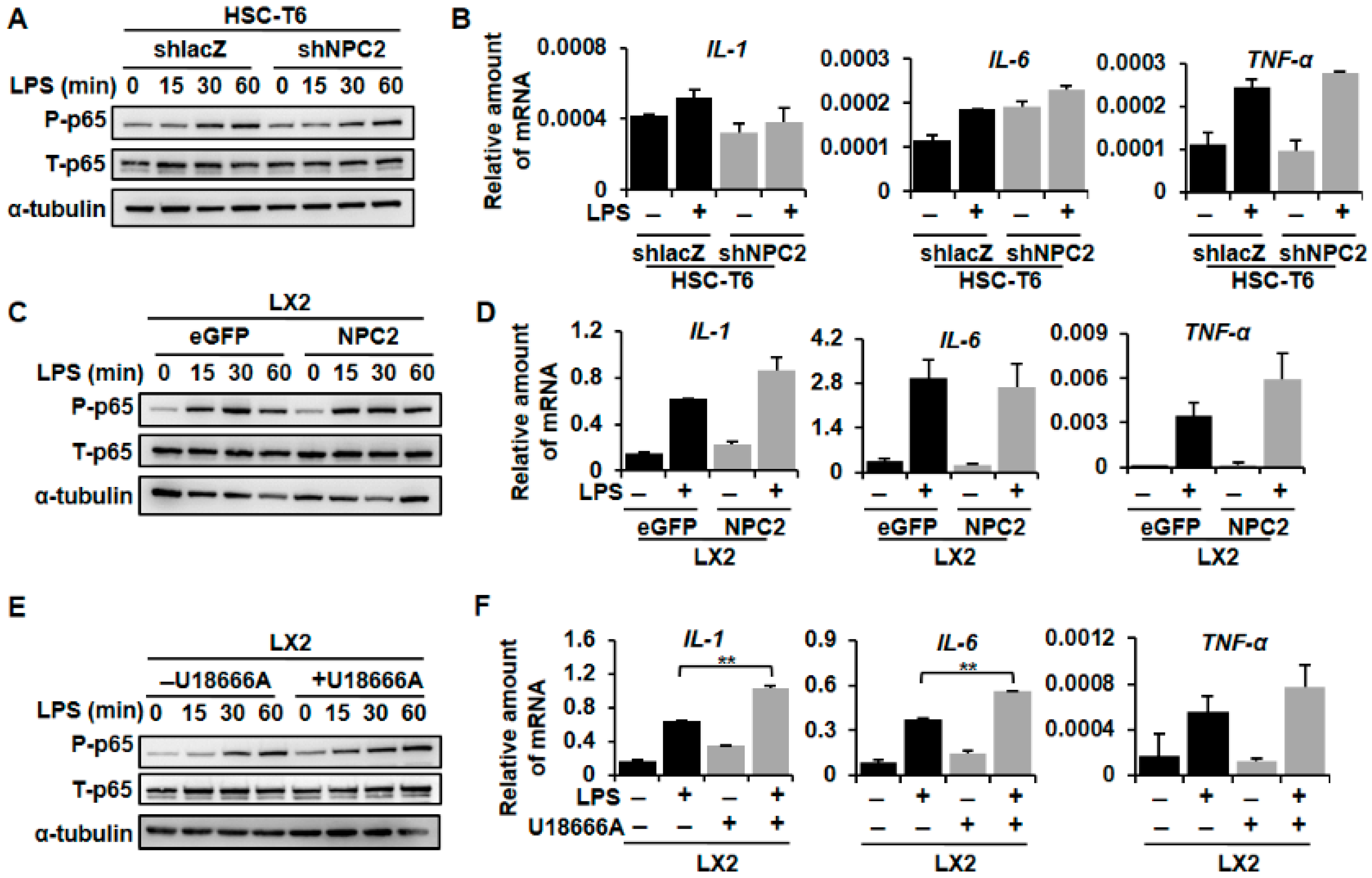
2.5. NPC2 Expression Was Not Involved in LPS-Induced Inflammatory Response, While U18666A-Treated HSCs Enhanced LPS-Induced Inflammation
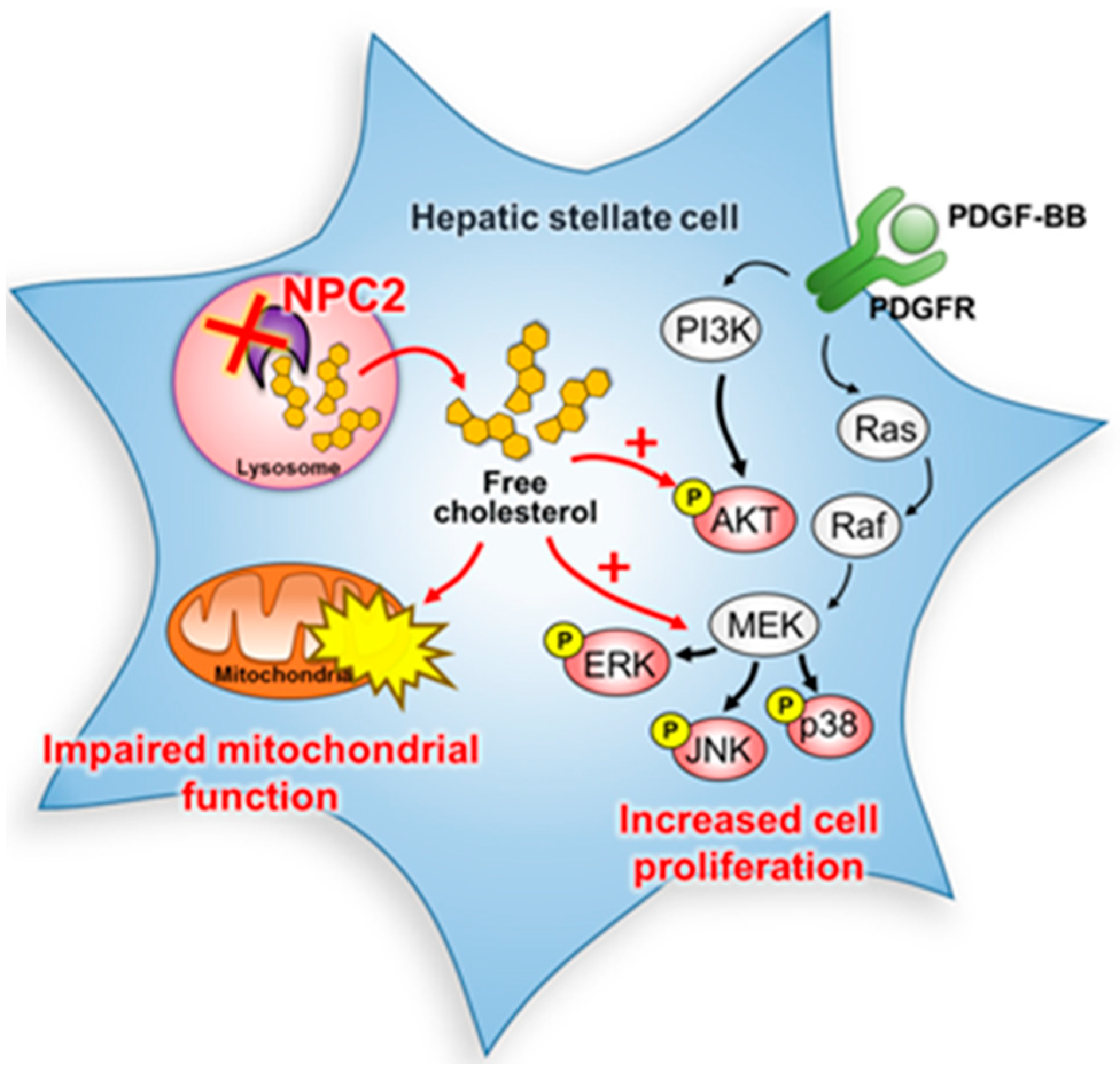
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Plasmids, and Lentiviral Infection
4.2. AlamarBlue® Cell Viability Assay
4.3. BrdU Assay
4.4. PDGF-BB, LPS, and U18666A Treatment
4.5. Western Blot
4.6. Real-Time PCR
4.7. Free Cholesterol Quantification
4.8. Seahorse Assay
4.9. Statistical Analysis
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| HSCs | Hepatic stellate cells |
| NPC2 | Niemann-Pick type C2 |
| PDGF-BB | Platelet-derived growth factor BB |
| MAPK/ERK | Mitogen-activated protein kinases |
| α-SMA | α-Smooth muscle actin |
| LPS | Lipopolysaccharide |
| TLR4 | Toll-like receptor 4 |
| OCR | Oxygen consumption rate |
| Oligo | Oligomycin |
| AA | Rotenone and antimycin A |
References
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Invest. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Geerts, A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin. Liver Dis. 2001, 21, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Carson, J.P.; Ramm, G.A.; Robinson, M.W.; McManus, D.P.; Gobert, G.N. Schistosome-induced fibrotic disease: The role of hepatic stellate cells. Trends Parasitol. 2018, 34, 524–540. [Google Scholar] [CrossRef] [PubMed]
- Moreira, R.K. Hepatic stellate cells and liver fibrosis. Arch. Pathol. Lab. Med. 2007, 131, 1728–1734. [Google Scholar] [PubMed]
- Lee, U.E.; Friedman, S.L. Mechanisms of hepatic fibrogenesis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.Y.; Friedman, S.L. Fibrosis-dependent mechanisms of hepatocarcinogenesis. Hepatology 2012, 56, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Borkham-Kamphorst, E.; Weiskirchen, R. The PDGF system and its antagonists in liver fibrosis. Cytokine Growth Factor Rev. 2016, 28, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, F.; Olinga, P.; Poelstra, K.; Beljaars, L. Targeted therapies in liver fibrosis: Combining the best parts of platelet-derived growth factor BB and interferon gamma. Front. Med. 2015, 2, 72. [Google Scholar] [CrossRef] [PubMed]
- Pinzani, M.; Milani, S.; Herbst, H.; DeFranco, R.; Grappone, C.; Gentilini, A.; Caligiuri, A.; Pellegrini, G.; Ngo, D.V.; Romanelli, R.G.; et al. Expression of platelet-derived growth factor and its receptors in normal human liver and during active hepatic fibrogenesis. Am. J. Pathol. 1996, 148, 785–800. [Google Scholar] [PubMed]
- Shi, H.; Dong, L.; Dang, X.; Liu, Y.; Jiang, J.; Wang, Y.; Lu, X.; Guo, X. Effect of chlorogenic acid on LPS-induced proinflammatory signaling in hepatic stellate cells. Inflamm. Res. 2013, 62, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Schwabe, R.F. Hepatic inflammation and fibrosis: Functional links and key pathways. Hepatology 2015, 61, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Paik, Y.H.; Schwabe, R.F.; Bataller, R.; Russo, M.P.; Jobin, C.; Brenner, D.A. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology 2003, 37, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Seki, E.; Brenner, D.A. Toll-like receptor signaling in the liver. Gastroenterology 2006, 130, 1886–1900. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N.; Morrow, O.B.; Connole, M.L.; Lee, S.P. Association between dietary nutrient composition and the incidence of cirrhosis or liver cancer in the united states population. Hepatology 2009, 50, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Teratani, T.; Tomita, K.; Suzuki, T.; Oshikawa, T.; Yokoyama, H.; Shimamura, K.; Tominaga, S.; Hiroi, S.; Irie, R.; Okada, Y.; et al. A high-cholesterol diet exacerbates liver fibrosis in mice via accumulation of free cholesterol in hepatic stellate cells. Gastroenterology 2012, 142, 152–164 e110. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Teratani, T.; Suzuki, T.; Shimizu, M.; Sato, H.; Narimatsu, K.; Usui, S.; Furuhashi, H.; Kimura, A.; Nishiyama, K.; et al. Acyl-CoA:Cholesterol acyltransferase 1 mediates liver fibrosis by regulating free cholesterol accumulation in hepatic stellate cells. J. Hepatol. 2014, 61, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Ko, D.C.; Binkley, J.; Sidow, A.; Scott, M.P. The integrity of a cholesterol-binding pocket in Niemann-Pick C2 protein is necessary to control lysosome cholesterol levels. Proc. Natl. Acad. Sci. USA 2003, 100, 2518–2525. [Google Scholar] [CrossRef] [PubMed]
- Storch, J.; Xu, Z. Niemann-Pick C2 (NPC2) and intracellular cholesterol trafficking. Biochim. Biophys. Acta 2009, 1791, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Reif, S.; Spirer, Z.; Messer, G.; Baratz, M.; Bembi, B.; Bujanover, Y. Severe failure to thrive and liver dysfunction as the main manifestations of a new variant of Niemann-Pick disease. Clin. Pediatr. 1994, 33, 628–630. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.A.; Portmann, B.; Mowat, A.P.; Sherlock, S.; Lake, B.D. Niemann-Pick disease type C: Diagnosis and outcome in children, with particular reference to liver disease. J. Pediatr. 1993, 123, 242–247. [Google Scholar] [CrossRef]
- Liao, Y.J.; Lin, M.W.; Yen, C.H.; Lin, Y.T.; Wang, C.K.; Huang, S.F.; Chen, K.H.; Yang, C.P.; Chen, T.L.; Hou, M.F.; et al. Characterization of Niemann-Pick type C2 protein expression in multiple cancers using a novel NPC2 monoclonal antibody. PLoS ONE 2013, 8, e77586. [Google Scholar] [CrossRef] [PubMed]
- Twu, Y.C.; Lee, T.S.; Lin, Y.L.; Hsu, S.M.; Wang, Y.H.; Liao, C.Y.; Wang, C.K.; Liang, Y.C.; Liao, Y.J. Niemann-Pick type C2 protein mediates hepatic stellate cells activation by regulating free cholesterol accumulation. Int. J. Mol. Sci. 2016, 17, 1122. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.; Yamasaki, G.; Johnson, R.J.; Friedman, S.L. Induction of β-platelet-derived growth factor receptor in rat hepatic lipocytes during cellular activation in vivo and in culture. J. Clin. Invest. 1994, 94, 1563–1569. [Google Scholar] [CrossRef] [PubMed]
- Copetti-Santos, D.; Moraes, V.; Weiler, D.F.; de Mello, A.S.; Machado Fde, S.; Marinho, J.P.; Siebert, C.; Kolling, J.; Funchal, C.; Wyse, A.T.; et al. U18666a treatment results in cholesterol accumulation, reduced Na+, K+-ATPase activity, and increased oxidative stress in rat cortical astrocytes. Lipids 2015, 50, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Liscum, L.; Faust, J.R. The intracellular transport of low density lipoprotein-derived cholesterol is inhibited in chinese hamster ovary cells cultured with 3-β-[2-(diethylamino)ethoxy]androst-5-en-17-one. J. Biol. Chem. 1989, 264, 11796–11806. [Google Scholar] [PubMed]
- Liscum, L.; Munn, N.J. Intracellular cholesterol transport. Biochim. Biophys. Acta 1999, 1438, 19–37. [Google Scholar] [CrossRef]
- Ko, D.C.; Gordon, M.D.; Jin, J.Y.; Scott, M.P. Dynamic movements of organelles containing Niemann-Pick C1 protein: NPC1 involvement in late endocytic events. Mol. Biol. Cell 2001, 12, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Investig. 2005, 115, 2679–2688. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial cristae: Where beauty meets functionality. Trends Biochem. Sci. 2016, 41, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Mason, E.F.; Rathmell, J.C. Cell metabolism: An essential link between cell growth and apoptosis. Biochim. Biophys. Acta 2011, 1813, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Li, Y.; Wang, H.; Feng, Z.; Li, Y.; Long, J.; Liu, J. Mitochondrial accumulation under oxidative stress is due to defects in autophagy. J. Cell. Biochem. 2013, 114, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Kirchhoff, C.; Osterhoff, C.; Young, L. Molecular cloning and characterization of he1, a major secretory protein of the human epididymis. Biol. Reprod. 1996, 54, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.J.; Fang, C.C.; Yen, C.H.; Hsu, S.M.; Wang, C.K.; Huang, S.F.; Liang, Y.C.; Lin, Y.Y.; Chu, Y.T.; Arthur Chen, Y.M. Niemann-Pick type C2 protein regulates liver cancer progression via modulating ERK1/2 pathway: Clinicopathological correlations and therapeutical implications. Int. J. Cancer 2015, 137, 1341–1351. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Hepatic fibrosis—Overview. Toxicology 2008, 254, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.J.; Parola, M. Liver fibrogenic cells. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326 Pt 1, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Kanaseki, T.; Mizushima, N.; Mizuta, T.; Arakawa-Kobayashi, S.; Thompson, C.B.; Tsujimoto, Y. Role of BCL-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 2004, 6, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Bergamini, E.; Brunk, U.T.; Droge, W.; Ffrench, M.; Terman, A. Autophagy and aging: The importance of maintaining “clean” cells. Autophagy 2005, 1, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Eaton, S.; Bartlett, K.; Pourfarzam, M. Mammalian mitochondrial β-oxidation. Biochem. J. 1996, 320 Pt 2, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Pessayre, D.; Fromenty, B. Nash: A mitochondrial disease. J. Hepatol. 2005, 42, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Perez-Carreras, M.; Del Hoyo, P.; Martin, M.A.; Rubio, J.C.; Martin, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Wos, M.; Szczepanowska, J.; Pikula, S.; Tylki-Szymanska, A.; Zablocki, K.; Bandorowicz-Pikula, J. Mitochondrial dysfunction in fibroblasts derived from patients with Niemann-Pick type C disease. Arch. Biochem. Biophys. 2016, 593, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.; Balboa, E.; Zanlungo, S.; Enrich, C.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Lysosomal and mitochondrial liaisons in Niemann-Pick disease. Front. Physiol. 2017, 8, 982. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zhao, M.; Qiu, X.; Deis, J.A.; Huang, H.; Tang, Q.Q.; Chen, X. Niemann-Pick type C2 deficiency impairs autophagy-lysosomal activity, mitochondrial function, and TLR signaling in adipocytes. J. Lipid Res. 2016, 57, 1644–1658. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Gea, V.; Ghiassi-Nejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Thoen, L.F.; Guimaraes, E.L.; Dolle, L.; Mannaerts, I.; Najimi, M.; Sokal, E.; van Grunsven, L.A. A role for autophagy during hepatic stellate cell activation. J. Hepatol. 2011, 55, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S.P.; Qian, T.; Grissom, S.F.; Lemasters, J.J. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001, 15, 2286–2287. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Teratani, T.; Suzuki, T.; Shimizu, M.; Sato, H.; Narimatsu, K.; Okada, Y.; Kurihara, C.; Irie, R.; Yokoyama, H.; et al. Free cholesterol accumulation in hepatic stellate cells: Mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology 2014, 59, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Sugimoto, Y.; Ohsaki, Y.; Ueno, M.; Kato, S.; Kitamura, Y.; Hosokawa, H.; Davies, J.P.; Ioannou, Y.A.; Vanier, M.T.; et al. Endosomal accumulation of toll-like receptor 4 causes constitutive secretion of cytokines and activation of signal transducers and activators of transcription in Niemann-Pick disease type C (NPC) fibroblasts: A potential basis for glial cell activation in the npc brain. J. Neurosci. 2007, 27, 1879–1891. [Google Scholar] [PubMed]
- Appelqvist, H.; Nilsson, C.; Garner, B.; Brown, A.J.; Kagedal, K.; Ollinger, K. Attenuation of the lysosomal death pathway by lysosomal cholesterol accumulation. Am. J. Pathol. 2011, 178, 629–639. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′→3′) | Reverse Primer (3′→5′) |
|---|---|---|
| GAPDH | TCACCACCATGGAGAAGGC | GCTAAGCAGTTGGTGGTGCA |
| NPC2 | CGGAGCCCCTGCACTTC | GGGCTCACATTCACCTCCTTTA |
| Bax | GATCCAGGATCGAGCAGA | AAGTAGAAGAGGGCAACCAC |
| Bcl-xl | GCTGCATTGTTCCCGTAGAG | GTTGGATGGCCACCTATCTG |
| Abca1 | CCCCTGCTTCCGTTATCCA | GGACCTTGTGCATGTCCTTAATG |
| Abcb11 | CAGAACATGACAAACGGAACAAG | CCTGCGTAGATGCCAGAAAATT |
| Cyp7A1 | ACTCTCTGAAGCCATGATGCAA | ACCCAGACAGCGCTCTTTGA |
| SREBP2 | GCTACCGGTCCTCCATCAAC | ACGCCAGACTTGTGCATCTTG |
| HMGCR | TGTGGTTTGTGAAGCCGTCAT | TCAACCATAGCTTCCGTAGTTGTC |
| NPC1 | AACAAAGTAGACATCGGGTTGGA | ACTGAGCGAGTGATTTGAAATAGTCA |
| h-IL-1 | CTGAGCACCTTCTTTCCCTTCA | TGGACCAGACATCACCAAGCT |
| h-IL-6 | TGGCTGAAAAAGATGGATGCT | TCTGCACAGCTCTGGCTTGT |
| h-TNF-α | TGTAGCCCATGTTGTAGCAAACC | GAGGACCTGGGAGTAGATGAGGTA |
| m-IL-1 | TCCAGGATGAGGACATGAGCAC | GAACGTCACACACCAGCAGGTTA |
| m-IL-6 | GGACTGATGCTGGTGAC | CATTTCTTTGTATCTCTGGAAGTT |
| m-TNF-α | CAAGACTGACAACCAGACG | ACAGAAGCAGAGATTATGACC |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.-H.; Twu, Y.-C.; Wang, C.-K.; Lin, F.-Z.; Lee, C.-Y.; Liao, Y.-J. Niemann-Pick Type C2 Protein Regulates Free Cholesterol Accumulation and Influences Hepatic Stellate Cell Proliferation and Mitochondrial Respiration Function. Int. J. Mol. Sci. 2018, 19, 1678. https://doi.org/10.3390/ijms19061678
Wang Y-H, Twu Y-C, Wang C-K, Lin F-Z, Lee C-Y, Liao Y-J. Niemann-Pick Type C2 Protein Regulates Free Cholesterol Accumulation and Influences Hepatic Stellate Cell Proliferation and Mitochondrial Respiration Function. International Journal of Molecular Sciences. 2018; 19(6):1678. https://doi.org/10.3390/ijms19061678
Chicago/Turabian StyleWang, Yuan-Hsi, Yuh-Ching Twu, Chung-Kwe Wang, Fu-Zhen Lin, Chun-Ya Lee, and Yi-Jen Liao. 2018. "Niemann-Pick Type C2 Protein Regulates Free Cholesterol Accumulation and Influences Hepatic Stellate Cell Proliferation and Mitochondrial Respiration Function" International Journal of Molecular Sciences 19, no. 6: 1678. https://doi.org/10.3390/ijms19061678
APA StyleWang, Y.-H., Twu, Y.-C., Wang, C.-K., Lin, F.-Z., Lee, C.-Y., & Liao, Y.-J. (2018). Niemann-Pick Type C2 Protein Regulates Free Cholesterol Accumulation and Influences Hepatic Stellate Cell Proliferation and Mitochondrial Respiration Function. International Journal of Molecular Sciences, 19(6), 1678. https://doi.org/10.3390/ijms19061678