The Influence of EGFR Inactivation on the Radiation Response in High Grade Glioma

Abstract
1. Introduction
2. Results
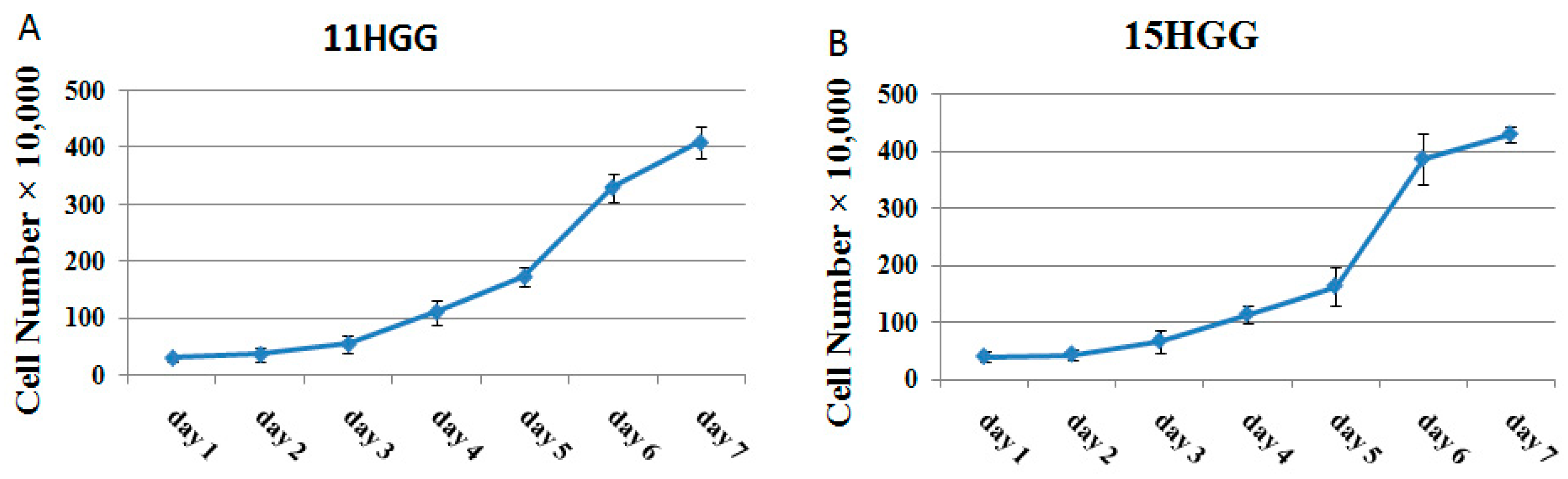
2.1. Growth Pattern of Untreated HGG Cells
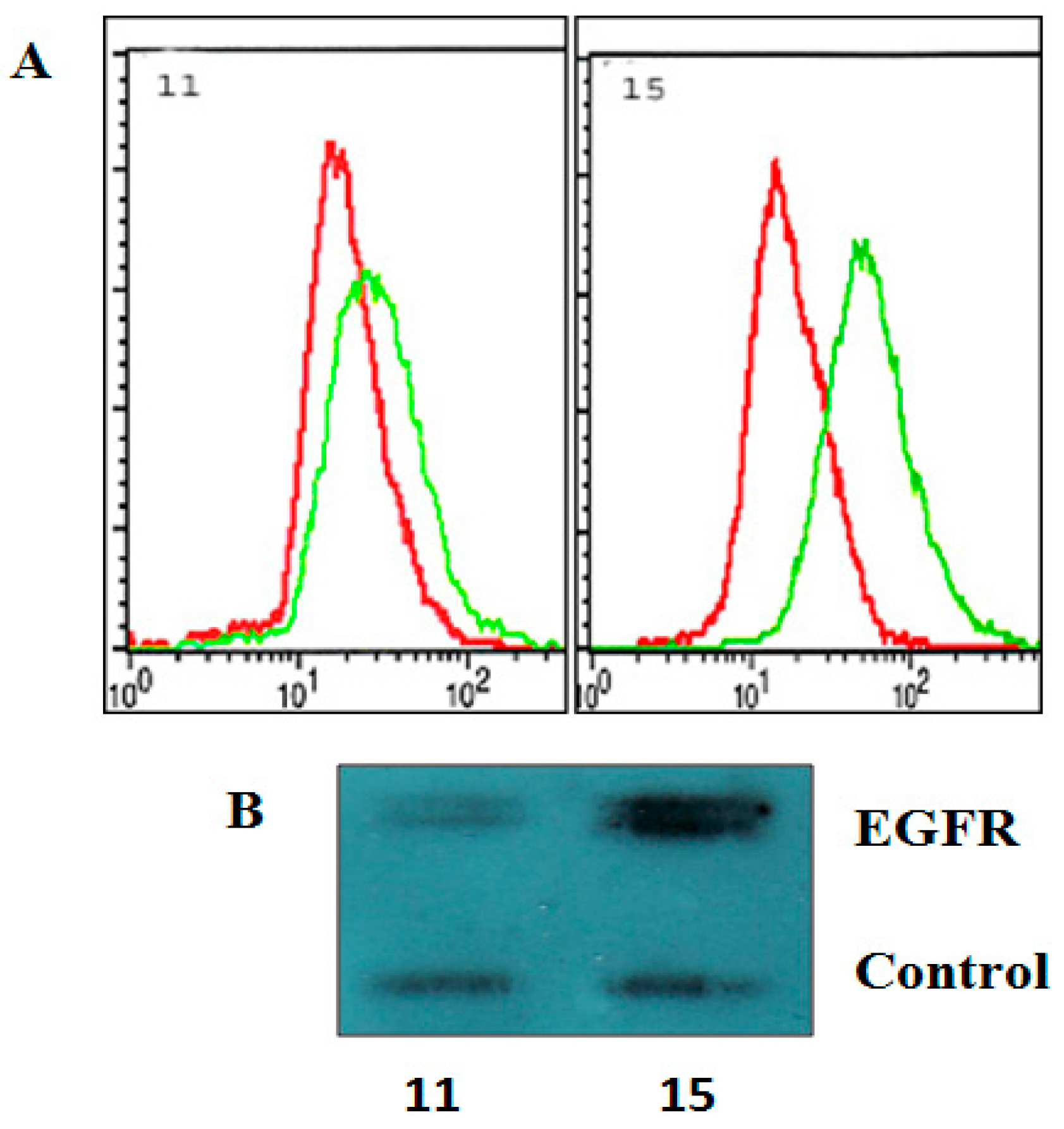
2.2. The Effect of EGFR Inactivation on HGG Cells
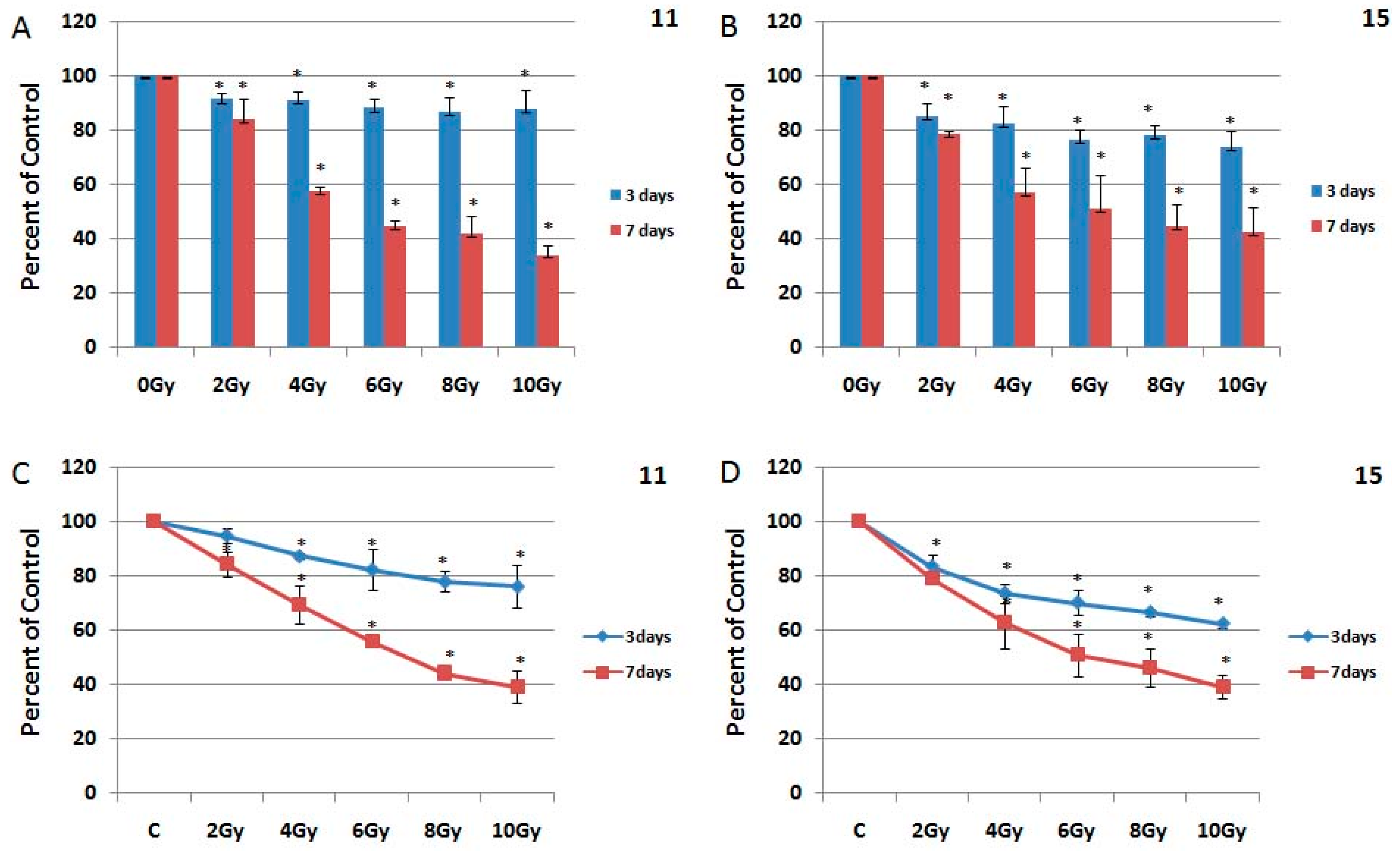
2.3. The Effect of Ionizing Radiation on HGG Cells
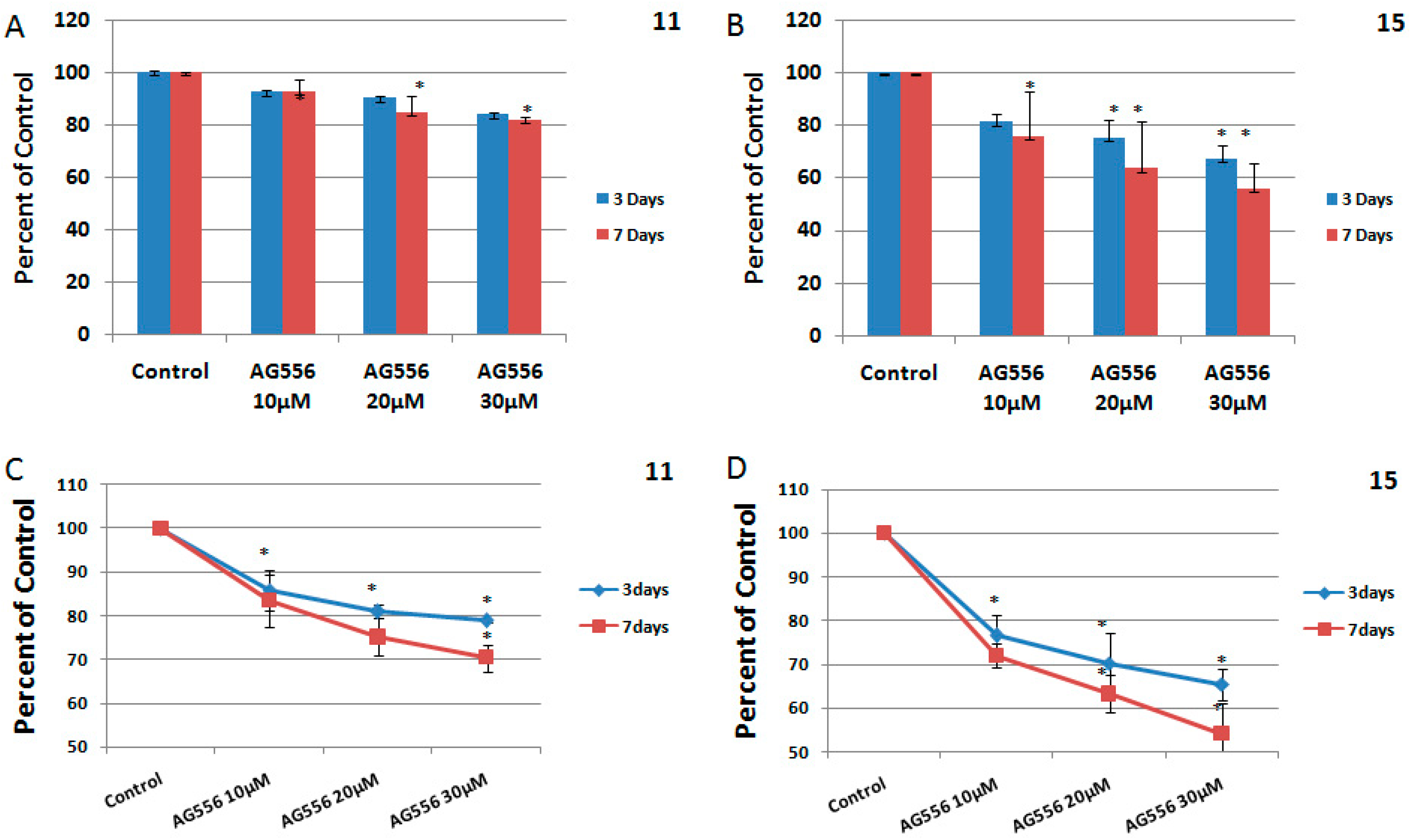
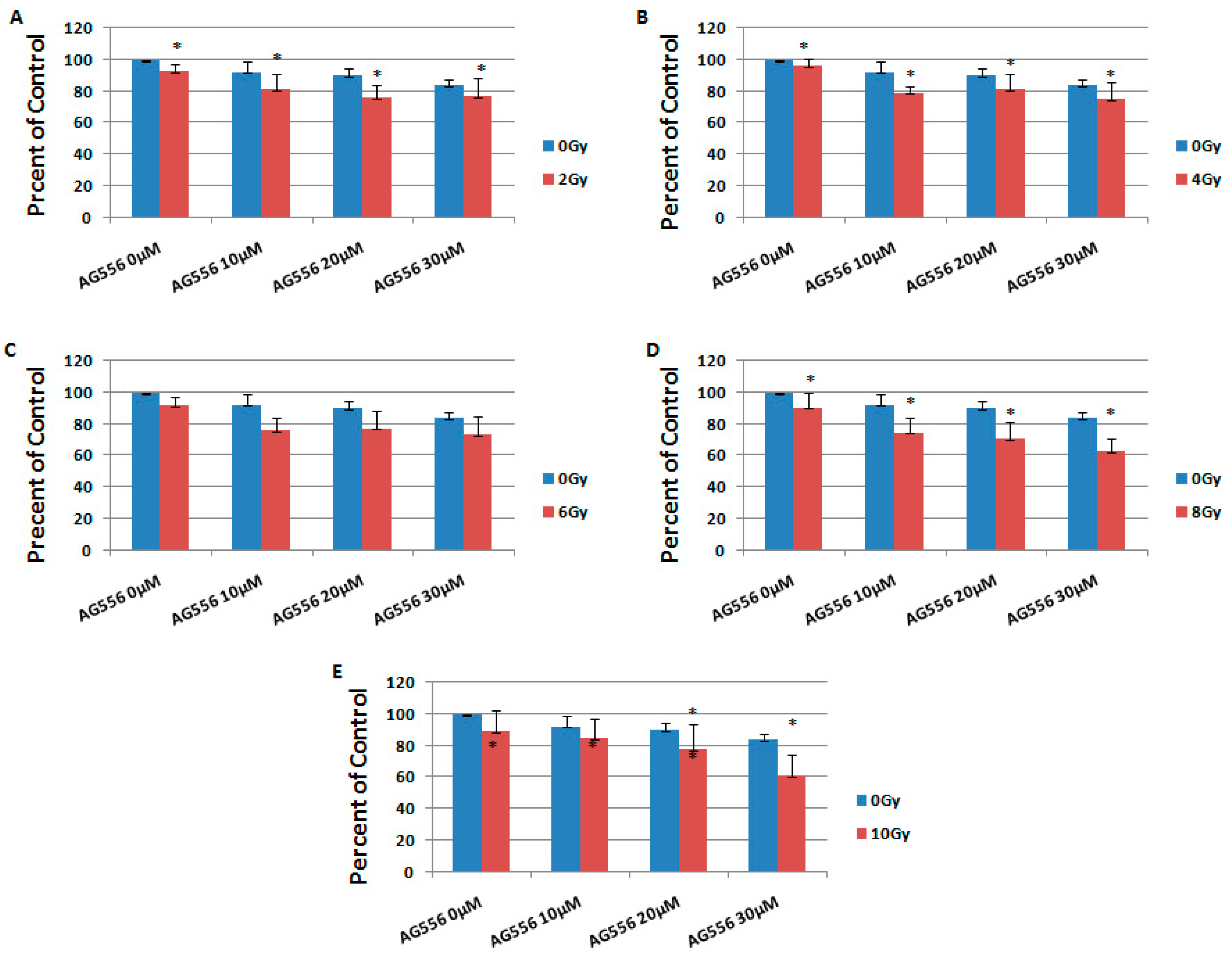
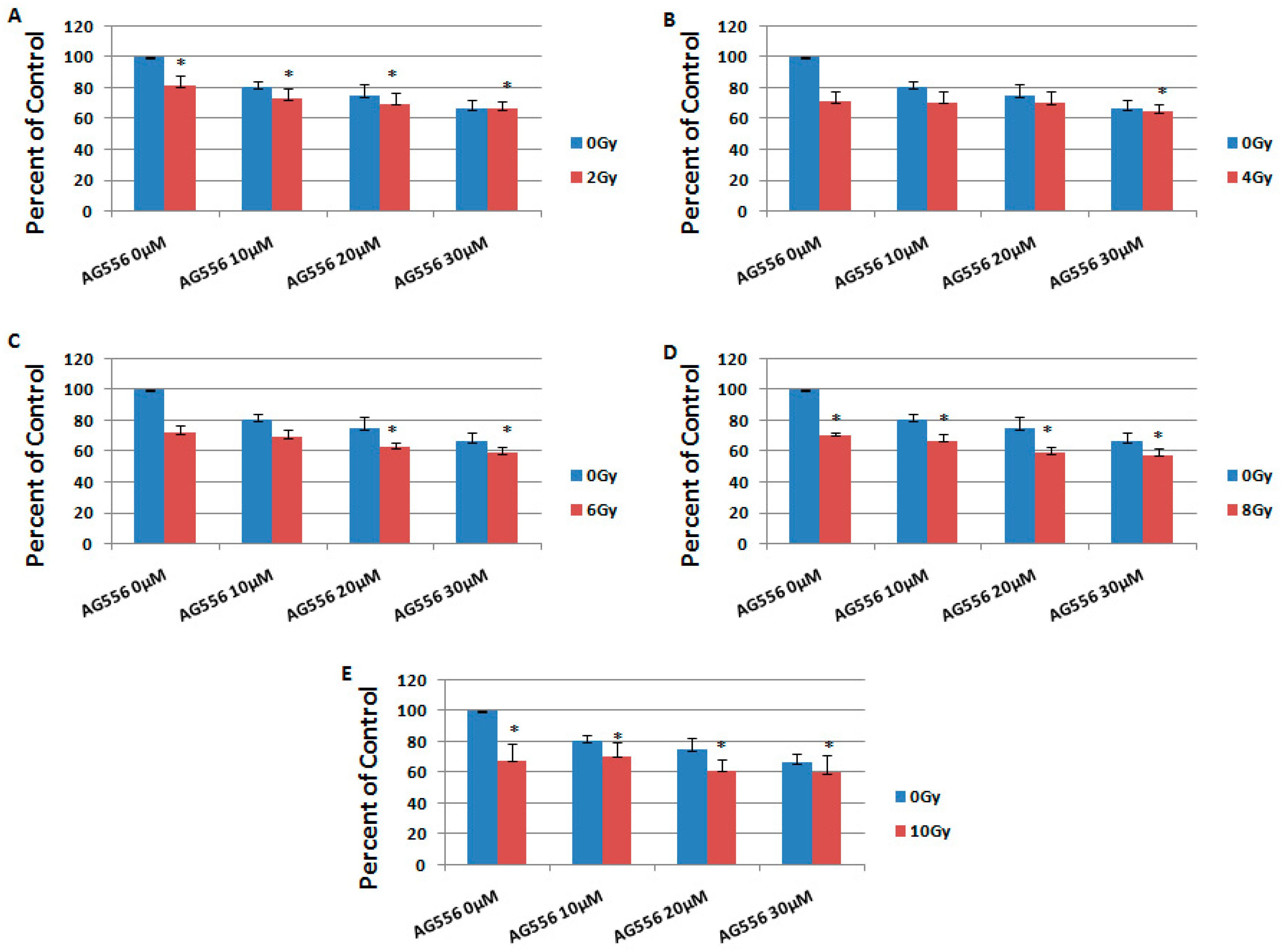
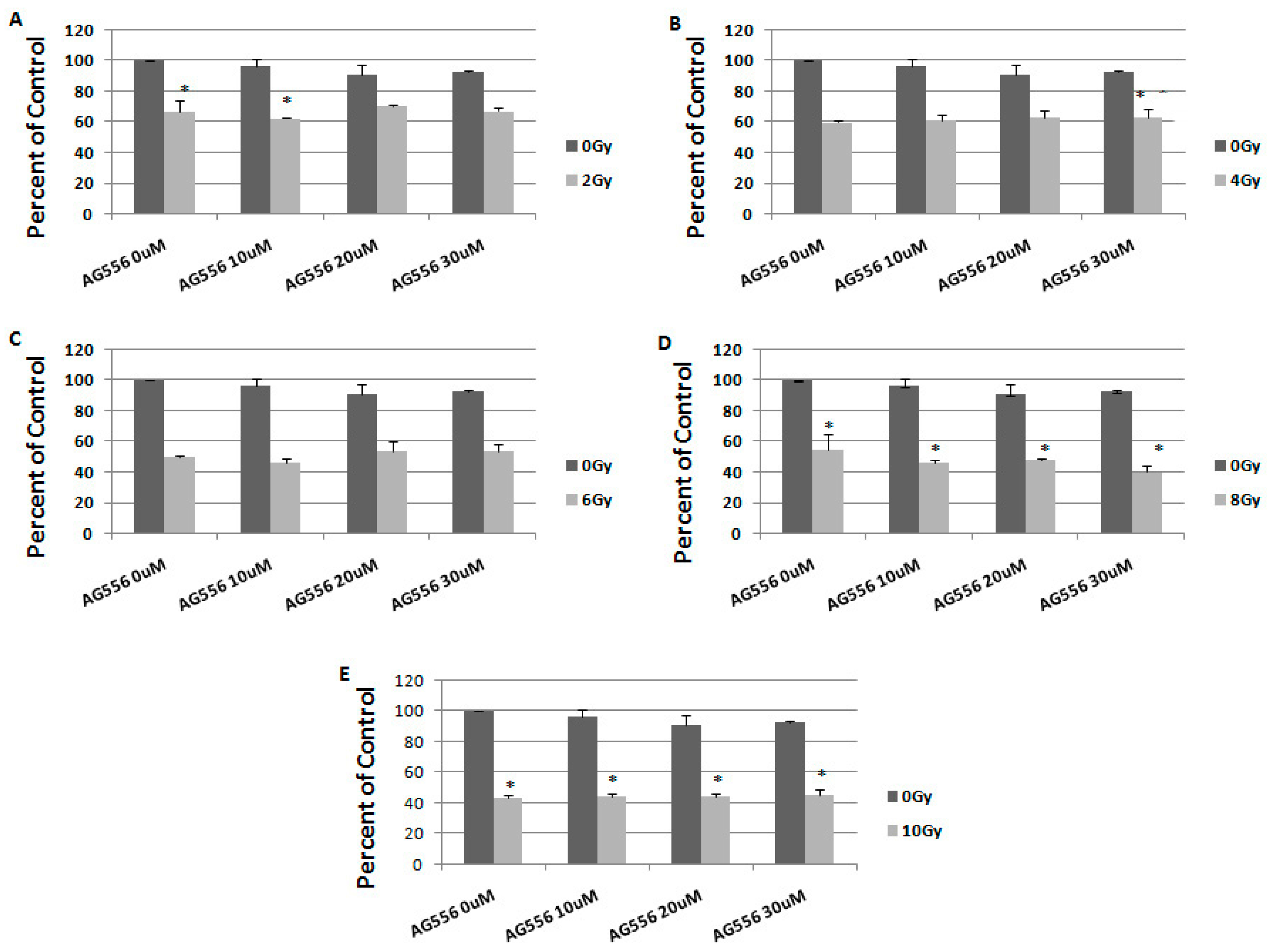
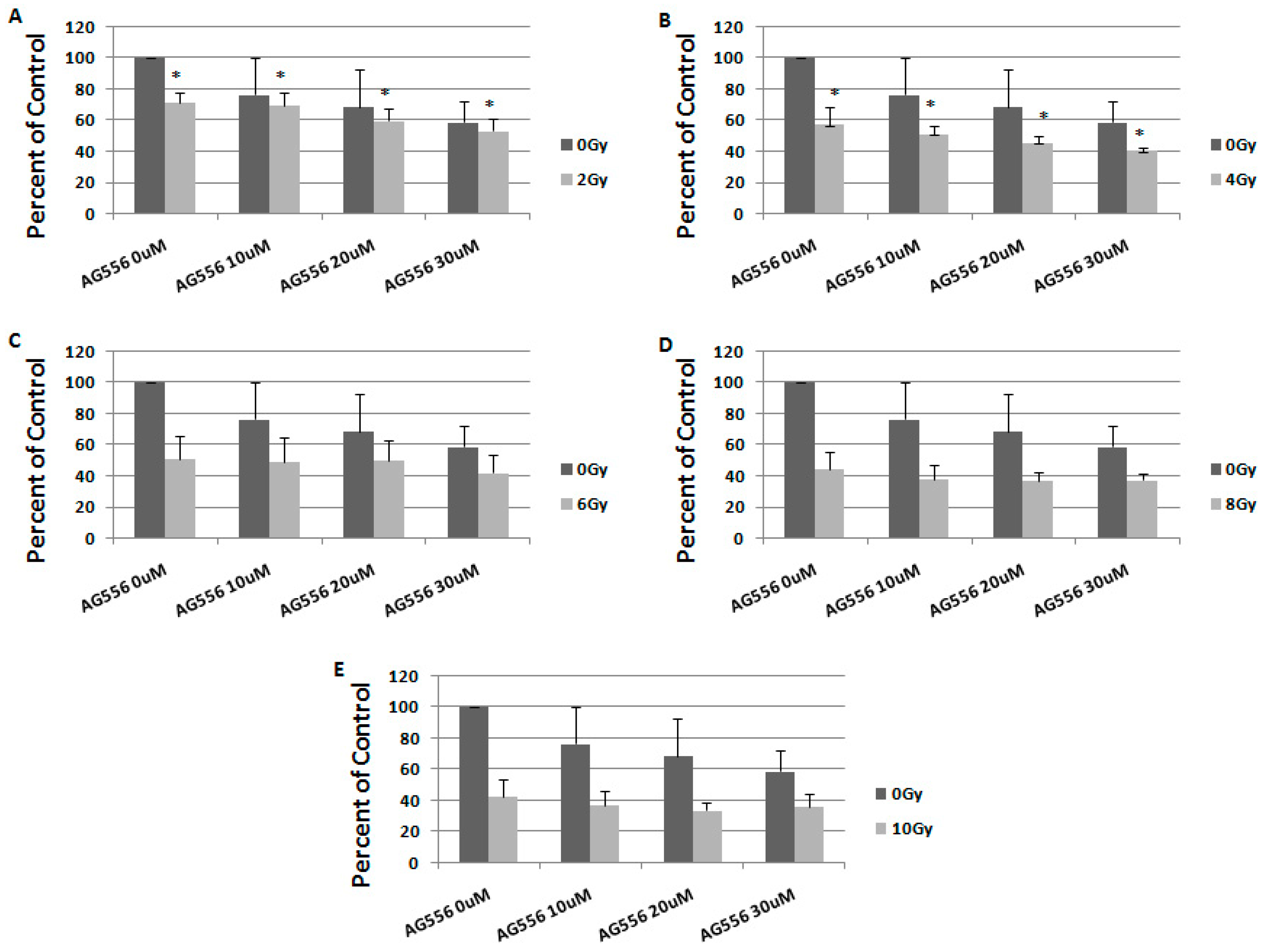
2.4. The Effect of Combined Treatment on HGG Cells Viability
2.5. The Interaction between Combined Treatment with AG556 and Ionizing Radiation in 11 HGG Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Cultures and Treatment
4.3. Growth Kinetic Assay and Calculation of Cell Population Doubling Time
4.4. Irradiation
4.5. MTT Assay
4.6. Trypan Blue Exclusion Test
4.7. Flow Cytometry
4.8. Western Blotting
4.9. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Salmaggi, A.; Duri, S.; Silvani, A.; Gaviani, P.; Milanesi, I.; Casali, C.; Di Meco, F. Loco-regional treatments in first-diagnosis glioblastoma: Literature review on association between stupp protocol and gliadel. Neurol. Sci. 2011, 32 (Suppl. 2), S241–S245. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Schlenska-Lange, A.; Knupfer, H.; Lange, T.J.; Kiess, W.; Knupfer, M. Cell proliferation and migration in glioblastoma multiforme cell lines are influenced by insulin-like growth factor i in vitro. Anticancer Res. 2008, 28, 1055–1060. [Google Scholar] [PubMed]
- Puputti, M.; Tynninen, O.; Sihto, H.; Blom, T.; Maenpaa, H.; Isola, J.; Paetau, A.; Joensuu, H.; Nupponen, N.N. Amplification of kit, pdgfra, vegfr2, and egfr in gliomas. Mol. Cancer Res. MCR 2006, 4, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, T.; Brennan, C.W.; Wang, L.; Squatrito, M.; Sasayama, T.; Nakada, M.; Huse, J.T.; Pedraza, A.; Utsuki, S.; Yasui, Y.; et al. Pdgfra gene rearrangements are frequent genetic events in pdgfra-amplified glioblastomas. Genes Dev. 2010, 24, 2205–2218. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, H.; Puputti, M.; Sihto, H.; Tynninen, O.; Nupponen, N.N. Amplification of genes encoding kit, pdgfralpha and vegfr2 receptor tyrosine kinases is frequent in glioblastoma multiforme. J. Pathol. 2005, 207, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Frederick, L.; Wang, X.Y.; Eley, G.; James, C.D. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000, 60, 1383–1387. [Google Scholar] [PubMed]
- Aldape, K.D.; Ballman, K.; Furth, A.; Buckner, J.C.; Giannini, C.; Burger, P.C.; Scheithauer, B.W.; Jenkins, R.B.; James, C.D. Immunohistochemical detection of egfrviii in high malignancy grade astrocytomas and evaluation of prognostic significance. J. Neuropathol. Exp. Neurol. 2004, 63, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.S.; Nagane, M.; Klingbeil, C.K.; Lin, H.; Nishikawa, R.; Ji, X.D.; Huang, C.M.; Gill, G.N.; Wiley, H.S.; Cavenee, W.K. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J. Biol. Chem. 1997, 272, 2927–2935. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Backlund, L.M.; Nilsson, B.R.; Grander, D.; Ichimura, K.; Goike, H.M.; Collins, V.P. Clinical significance of egfr amplification and the aberrant egfrviii transcript in conventionally treated astrocytic gliomas. J. Mol. Med. 2005, 83, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Shinojima, N.; Tada, K.; Shiraishi, S.; Kamiryo, T.; Kochi, M.; Nakamura, H.; Makino, K.; Saya, H.; Hirano, H.; Kuratsu, J.; et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003, 63, 6962–6970. [Google Scholar] [PubMed]
- Francis, J.M.; Zhang, C.Z.; Maire, C.L.; Jung, J.; Manzo, V.E.; Adalsteinsson, V.A.; Homer, H.; Haidar, S.; Blumenstiel, B.; Pedamallu, C.S.; et al. Egfr variant heterogeneity in glioblastoma resolved through single-nucleus sequencing. Cancer Discov. 2014, 4, 956–971. [Google Scholar] [CrossRef] [PubMed]
- Krex, D.; Klink, B.; Hartmann, C.; von Deimling, A.; Pietsch, T.; Simon, M.; Sabel, M.; Steinbach, J.P.; Heese, O.; Reifenberger, G.; et al. Long-term survival with glioblastoma multiforme. Brain A J. Neurol. 2007, 130, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Halatsch, M.E.; Gehrke, E.E.; Vougioukas, V.I.; Botefur, I.C.; A-Borhani, F.; Efferth, T.; Gebhart, E.; Domhof, S.; Schmidt, U.; Buchfelder, M. Inverse correlation of epidermal growth factor receptor messenger rna induction and suppression of anchorage-independent growth by osi-774, an epidermal growth factor receptor tyrosine kinase inhibitor, in glioblastoma multiforme cell lines. J. Neurosurg. 2004, 100, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Knight, L.A.; Di Nicolantonio, F.; Whitehouse, P.; Mercer, S.; Sharma, S.; Glaysher, S.; Johnson, P.; Cree, I.A. The in vitro effect of gefitinib (‘iressa’) alone and in combination with cytotoxic chemotherapy on human solid tumours. BMC cancer 2004, 4, 83. [Google Scholar] [CrossRef] [PubMed]
- Neyns, B.; Sadones, J.; Joosens, E.; Bouttens, F.; Verbeke, L.; Baurain, J.F.; D’Hondt, L.; Strauven, T.; Chaskis, C.; In’t Veld, P.; et al. Stratified phase ii trial of cetuximab in patients with recurrent high-grade glioma. Ann. Oncol. 2009, 20, 1596–1603. [Google Scholar] [CrossRef] [PubMed]
- Combs, S.E.; Heeger, S.; Haselmann, R.; Edler, L.; Debus, J.; Schulz-Ertner, D. Treatment of primary glioblastoma multiforme with cetuximab, radiotherapy and temozolomide (gert)—Phase i/ii trial: Study protocol. BMC Cancer 2006, 6, 133. [Google Scholar] [CrossRef] [PubMed]
- Bode, U.; Massimino, M.; Bach, F.; Zimmermann, M.; Khuhlaeva, E.; Westphal, M.; Fleischhack, G. Nimotuzumab treatment of malignant gliomas. Expert Opin. Biol. Ther. 2012, 12, 1649–1659. [Google Scholar] [CrossRef] [PubMed]
- Van den Bent, M.J.; Brandes, A.A.; Rampling, R.; Kouwenhoven, M.C.; Kros, J.M.; Carpentier, A.F.; Clement, P.M.; Frenay, M.; Campone, M.; Baurain, J.F.; et al. Randomized phase ii trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: Eortc brain tumor group study 26034. J. Clin. Oncol. 2009, 27, 1268–1274. [Google Scholar] [CrossRef] [PubMed]
- Raizer, J.J.; Abrey, L.E.; Lassman, A.B.; Chang, S.M.; Lamborn, K.R.; Kuhn, J.G.; Yung, W.K.; Gilbert, M.R.; Aldape, K.A.; Wen, P.Y.; et al. A phase ii trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro Oncol. 2010, 12, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Thiessen, B.; Stewart, C.; Tsao, M.; Kamel-Reid, S.; Schaiquevich, P.; Mason, W.; Easaw, J.; Belanger, K.; Forsyth, P.; McIntosh, L.; et al. A phase i/ii trial of gw572016 (lapatinib) in recurrent glioblastoma multiforme: Clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother. Pharmacol. 2010, 65, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Kreisl, T.N.; McNeill, K.A.; Sul, J.; Iwamoto, F.M.; Shih, J.; Fine, H.A. A phase i/ii trial of vandetanib for patients with recurrent malignant glioma. Neuro Oncol. 2012, 14, 1519–1526. [Google Scholar] [CrossRef] [PubMed]
- Swartz, A.M.; Li, Q.J.; Sampson, J.H. Rindopepimut: A promising immunotherapeutic for the treatment of glioblastoma multiforme. Immunotherapy 2014, 6, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, egfrviii-expressing glioblastoma (act iv): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Dumont, F.J.; Bischoff, P. Disrupting the mtor signaling network as a potential strategy for the enhancement of cancer radiotherapy. Curr. Cancer Drug Targets 2012, 12, 899–924. [Google Scholar] [CrossRef] [PubMed]
- Nyati, M.K.; Morgan, M.A.; Feng, F.Y.; Lawrence, T.S. Integration of egfr inhibitors with radiochemotherapy. Nat. Rev. Cancer 2006, 6, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Balaban, N.; Moni, J.; Shannon, M.; Dang, L.; Murphy, E.; Goldkorn, T. The effect of ionizing radiation on signal transduction: Antibodies to egf receptor sensitize a431 cells to radiation. Biochim. Biophys. Acta 1996, 1314, 147–156. [Google Scholar] [CrossRef]
- Das, A.K.; Chen, B.P.; Story, M.D.; Sato, M.; Minna, J.D.; Chen, D.J.; Nirodi, C.S. Somatic mutations in the tyrosine kinase domain of epidermal growth factor receptor (egfr) abrogate egfr-mediated radioprotection in non-small cell lung carcinoma. Cancer Res. 2007, 67, 5267–5274. [Google Scholar] [CrossRef] [PubMed]
- Das, A.K.; Sato, M.; Story, M.D.; Peyton, M.; Graves, R.; Redpath, S.; Girard, L.; Gazdar, A.F.; Shay, J.W.; Minna, J.D.; et al. Non-small-cell lung cancers with kinase domain mutations in the epidermal growth factor receptor are sensitive to ionizing radiation. Cancer Res. 2006, 66, 9601–9608. [Google Scholar] [CrossRef] [PubMed]
- Hagerstrand, D.; Lindh, M.B.; Pena, C.; Garcia-Echeverria, C.; Nister, M.; Hofmann, F.; Ostman, A. Pi3k/pten/akt pathway status affects the sensitivity of high-grade glioma cell cultures to the insulin-like growth factor-1 receptor inhibitor nvp-aew541. Neuro Oncol. 2010, 12, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Hagerstrand, D.; He, X.; Bradic Lindh, M.; Hoefs, S.; Hesselager, G.; Ostman, A.; Nister, M. Identification of a sox2-dependent subset of tumor- and sphere-forming glioblastoma cells with a distinct tyrosine kinase inhibitor sensitivity profile. Neuro Oncol. 2011, 13, 1178–1191. [Google Scholar] [CrossRef] [PubMed]
- Dittmann, K.; Mayer, C.; Fehrenbacher, B.; Schaller, M.; Raju, U.; Milas, L.; Chen, D.J.; Kehlbach, R.; Rodemann, H.P. Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of DNA-dependent protein kinase. J. Biol. Chem. 2005, 280, 31182–31189. [Google Scholar] [CrossRef] [PubMed]
- Kiyozuka, M.; Akimoto, T.; Fukutome, M.; Motegi, A.; Mitsuhashi, N. Radiation-induced dimer formation of egfr: Implications for the radiosensitizing effect of cetuximab. Anticancer Res. 2013, 33, 4337–4346. [Google Scholar] [PubMed]
- Carapancea, M.; Alexandru, O.; Fetea, A.S.; Dragutescu, L.; Castro, J.; Georgescu, A.; Popa-Wagner, A.; Backlund, M.L.; Lewensohn, R.; Dricu, A. Growth factor receptors signaling in glioblastoma cells: Therapeutic implications. J. Neuro Oncol. 2009, 92, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Carapancea, M.; Cosaceanu, D.; Budiu, R.; Kwiecinska, A.; Tataranu, L.; Ciubotaru, V.; Alexandru, O.; Banita, M.; Pisoschi, C.; Backlund, M.L.; et al. Dual targeting of igf-1r and pdgfr inhibits proliferation in high-grade gliomas cells and induces radiosensitivity in jnk-1 expressing cells. J. Neuro Oncol. 2007, 85, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Valerie, K.; Yacoub, A.; Hagan, M.P.; Curiel, D.T.; Fisher, P.B.; Grant, S.; Dent, P. Radiation-induced cell signaling: Inside-out and outside-in. Mol. Cancer Ther. 2007, 6, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Hirao, T.; Urata, Y.; Kageyama, K.; Ikezaki, M.; Kawakatsu, M.; Matsuse, M.; Matsuo, T.; Akishita, M.; Nagata, I.; Kondo, T. Dehydroepiandrosterone augments sensitivity to gamma-ray irradiation in human h4 neuroglioma cells through down-regulation of akt signaling. Free Radic. Res. 2008, 42, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Li, H.F.; Kim, J.S.; Waldman, T. Radiation-induced akt activation modulates radioresistance in human glioblastoma cells. Radiat. Oncol. 2009, 4, 43. [Google Scholar] [CrossRef] [PubMed]
- Cosaceanu, D.; Budiu, R.A.; Carapancea, M.; Castro, J.; Lewensohn, R.; Dricu, A. Ionizing radiation activates igf-1r triggering a cytoprotective signaling by interfering with ku-DNA binding and by modulating ku86 expression via a p38 kinase-dependent mechanism. Oncogene 2007, 26, 2423–2434. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Wen, P.Y.; Mellinghoff, I.K. Targeted molecular therapies against epidermal growth factor receptor: Past experiences and challenges. Neuro Oncol. 2014, 16 (Suppl 8), viii7-13. [Google Scholar] [CrossRef] [PubMed]
- Del Vecchio, C.A.; Giacomini, C.P.; Vogel, H.; Jensen, K.C.; Florio, T.; Merlo, A.; Pollack, J.R.; Wong, A.J. Egfrviii gene rearrangement is an early event in glioblastoma tumorigenesis and expression defines a hierarchy modulated by epigenetic mechanisms. Oncogene 2013, 32, 2670–2681. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in pdgfra, idh1, egfr, and nf1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Frattini, V.; Trifonov, V.; Chan, J.M.; Castano, A.; Lia, M.; Abate, F.; Keir, S.T.; Ji, A.X.; Zoppoli, P.; Niola, F.; et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat. Genet. 2013, 45, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Azuaje, F.; Tiemann, K.; Niclou, S.P. Therapeutic control and resistance of the egfr-driven signaling network in glioblastoma. Cell Commun. Signal. CCS 2015, 13, 23. [Google Scholar] [CrossRef] [PubMed]
- Barker, F.G., 2nd; Simmons, M.L.; Chang, S.M.; Prados, M.D.; Larson, D.A.; Sneed, P.K.; Wara, W.M.; Berger, M.S.; Chen, P.; Israel, M.A.; et al. Egfr overexpression and radiation response in glioblastoma multiforme. Int. J. Radiat. Oncol. Biol. Phys. 2001, 51, 410–418. [Google Scholar] [CrossRef]
- Derer, S.; Bauer, P.; Lohse, S.; Scheel, A.H.; Berger, S.; Kellner, C.; Peipp, M.; Valerius, T. Impact of epidermal growth factor receptor (egfr) cell surface expression levels on effector mechanisms of egfr antibodies. J. Immunol. 2012, 189, 5230–5239. [Google Scholar] [CrossRef] [PubMed]
- Damiano, V.; Melisi, D.; Bianco, C.; Raben, D.; Caputo, R.; Fontanini, G.; Bianco, R.; Ryan, A.; Bianco, A.R.; De Placido, S.; et al. Cooperative antitumor effect of multitargeted kinase inhibitor zd6474 and ionizing radiation in glioblastoma. Clin. Cancer Res. 2005, 11, 5639–5644. [Google Scholar] [CrossRef] [PubMed]
- Popescu, A.M.; Alexandru, O.; Brindusa, C.; Purcaru, S.O.; Tache, D.E.; Tataranu, L.G.; Taisescu, C.; Dricu, A. Targeting the vegf and pdgf signaling pathway in glioblastoma treatment. Int. J. Clin. Exp. Pathol. 2015, 8, 7825–7837. [Google Scholar] [PubMed]
- Danciulescu, O.T.; Folcuti, R.; Dricu, A. Temozolomide and targeted therapy against epidermal growth factor receptor in glioma. Int. J. Clin. Exp. Med. 2016, 9, 15249–15261. [Google Scholar]
- Jasny, B.R.; Pfeffer, L.M.; Tamm, I. Effects of beta interferon on human fibroblasts at different population doubling levels. Proliferation, cell volume, thymidine uptake, and DNA synthesis. J. Exp. Med. 1984, 159, 1741–1749. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rad (Gy) | AG556 (µM) | Predicted Survival | Observed Survival | Effect |
|---|---|---|---|---|
| 2 | 10 | 0.9 | 0.8 | SYN |
| 20 | 0.8 | 0.8 | ADD | |
| 30 | 0.8 | 0.8 | ADD | |
| 4 | 10 | 0.9 | 0.8 | SYN |
| 20 | 0.9 | 0.8 | SYN | |
| 30 | 0.8 | 0.8 | ADD | |
| 6 | 10 | 0.9 | 0.8 | SYN |
| 20 | 0.8 | 0.8 | ADD | |
| 30 | 0.8 | 0.7 | SYN | |
| 8 | 10 | 0.8 | 0.8 | ADD |
| 20 | 0.8 | 0.7 | SYN | |
| 30 | 0.8 | 0.6 | SYN | |
| 10 | 10 | 0.8 | 0.9 | SUB |
| 20 | 0.8 | 0.8 | ADD | |
| 30 | 0.8 | 0.6 | SYN |
| Rad (Gy) | AG556 (µM) | Predicted Survival | Observed Survival | Effect |
|---|---|---|---|---|
| 2 | 10 | 0.7 | 0.7 | ADD |
| 20 | 0.6 | 0.7 | SUB | |
| 30 | 0.6 | 0.6 | ADD | |
| 4 | 10 | 0.6 | 0.7 | SUB |
| 20 | 0.5 | 0.7 | SUB | |
| 30 | 0.5 | 0.7 | SUB | |
| 6 | 10 | 0.5 | 0.7 | SUB |
| 20 | 0.5 | 0.6 | SUB | |
| 30 | 0.5 | 0.6 | SUB | |
| 8 | 10 | 0.5 | 0.7 | SUB |
| 20 | 0.5 | 0.6 | SUB | |
| 30 | 0.5 | 0.6 | SUB | |
| 10 | 10 | 0.6 | 0.7 | SUB |
| 20 | 0.5 | 0.6 | SUB | |
| 30 | 0.5 | 0.6 | SUB |
| Rad (Gy) | AG556 (µM) | Predicted Survival | Observed Survival | Effect |
|---|---|---|---|---|
| 2 | 10 | 0.6 | 0.6 | ADD |
| 20 | 0.6 | 0.7 | SUB | |
| 30 | 0.6 | 0.7 | SUB | |
| 4 | 10 | 0.6 | 0.6 | ADD |
| 20 | 0.6 | 0.6 | ADD | |
| 30 | 0.6 | 0.6 | ADD | |
| 6 | 10 | 0.5 | 0.5 | ADD |
| 20 | 0.5 | 0.5 | ADD | |
| 30 | 0.5 | 0.5 | ADD | |
| 8 | 10 | 0.5 | 0.5 | ADD |
| 20 | 0.5 | 0.5 | ADD | |
| 30 | 0.5 | 0.40 | SYN | |
| 10 | 10 | 0.4 | 0.5 | SUB |
| 20 | 0.4 | 0.5 | SUB | |
| 30 | 0.4 | 0.5 | SUB |
| Rad (Gy) | AG556 (µM) | Predicted Survival | Observed Survival | Effect |
|---|---|---|---|---|
| 2 | 10 | 0.6 | 0.7 | SUB |
| 20 | 0.5 | 0.6 | SUB | |
| 30 | 0.5 | 0.5 | ADD | |
| 4 | 10 | 0.4 | 0.5 | SUB |
| 20 | 0.4 | 0.5 | SUB | |
| 30 | 0.3 | 0.4 | SUB | |
| 6 | 10 | 0.4 | 0.5 | SUB |
| 20 | 0.4 | 0.5 | SUB | |
| 30 | 0.3 | 0.4 | SUB | |
| 8 | 10 | 0.4 | 0.4 | ADD |
| 20 | 0.3 | 0.4 | SUB | |
| 30 | 0.3 | 0.4 | SUB | |
| 10 | 10 | 0.3 | 0.4 | SUB |
| 20 | 0.3 | 0.4 | SUB | |
| 30 | 0.3 | 0.4 | SUB |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alexandru, O.; Purcaru, S.O.; Tataranu, L.G.; Lucan, L.; Castro, J.; Folcuţi, C.; Artene, S.-A.; Tuţă, C.; Dricu, A. The Influence of EGFR Inactivation on the Radiation Response in High Grade Glioma. Int. J. Mol. Sci. 2018, 19, 229. https://doi.org/10.3390/ijms19010229
Alexandru O, Purcaru SO, Tataranu LG, Lucan L, Castro J, Folcuţi C, Artene S-A, Tuţă C, Dricu A. The Influence of EGFR Inactivation on the Radiation Response in High Grade Glioma. International Journal of Molecular Sciences. 2018; 19(1):229. https://doi.org/10.3390/ijms19010229
Chicago/Turabian StyleAlexandru, Oana, Stefana Oana Purcaru, Ligia Gabriela Tataranu, Laura Lucan, Juan Castro, Catalin Folcuţi, Stefan-Alexandru Artene, Cristian Tuţă, and Anica Dricu. 2018. "The Influence of EGFR Inactivation on the Radiation Response in High Grade Glioma" International Journal of Molecular Sciences 19, no. 1: 229. https://doi.org/10.3390/ijms19010229
APA StyleAlexandru, O., Purcaru, S. O., Tataranu, L. G., Lucan, L., Castro, J., Folcuţi, C., Artene, S.-A., Tuţă, C., & Dricu, A. (2018). The Influence of EGFR Inactivation on the Radiation Response in High Grade Glioma. International Journal of Molecular Sciences, 19(1), 229. https://doi.org/10.3390/ijms19010229