
Induction of Syndecan-4 by Organic–Inorganic Hybrid Molecules with a 1,10-Phenanthroline Structure in Cultured Vascular Endothelial Cells


 ,
, 
Abstract
:
1. Introduction
2. Results
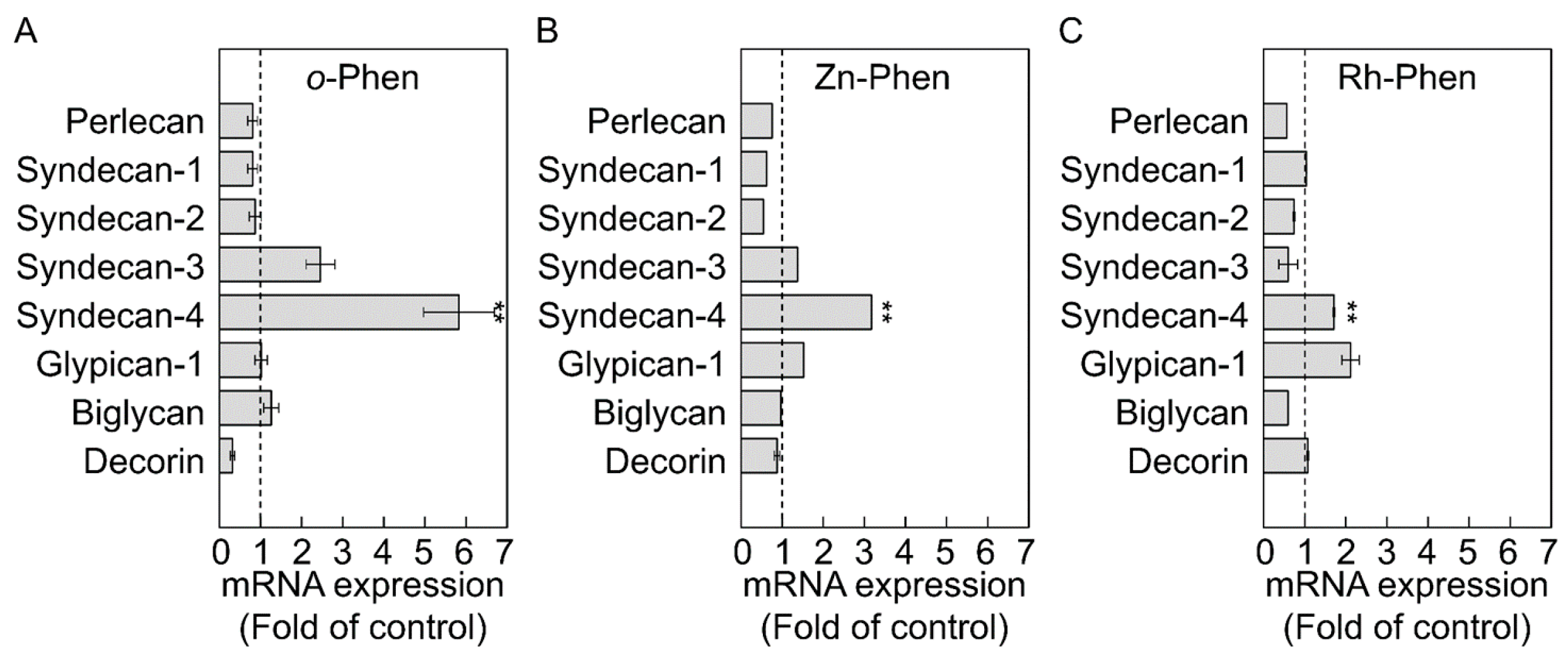
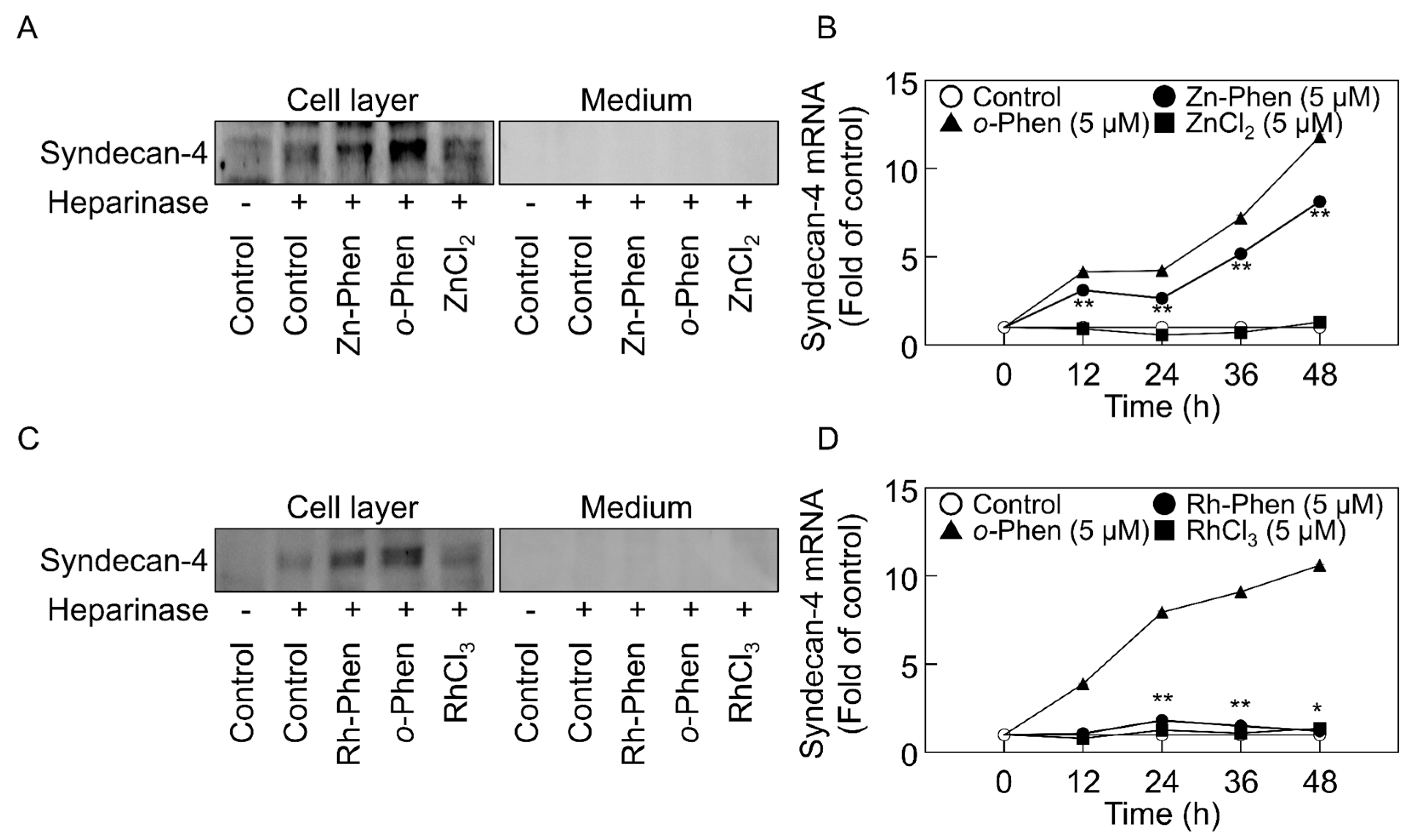
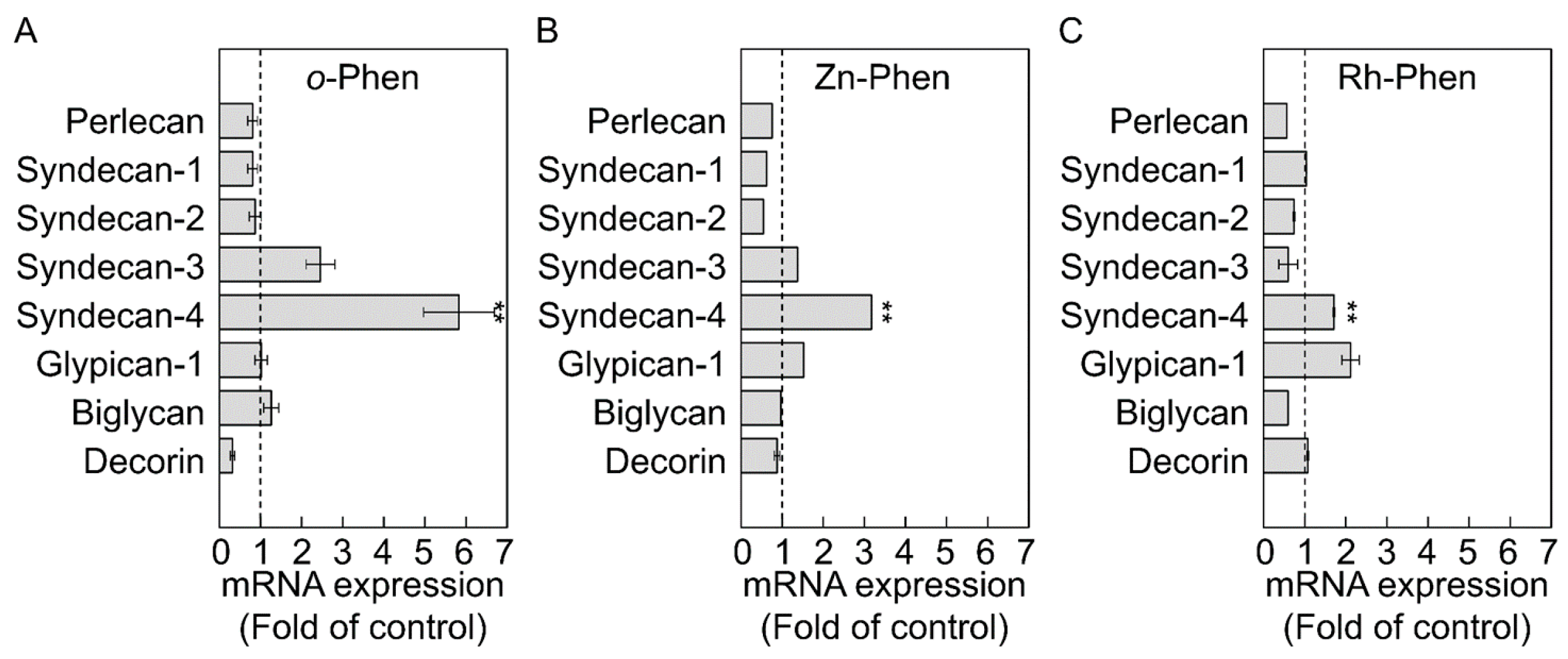
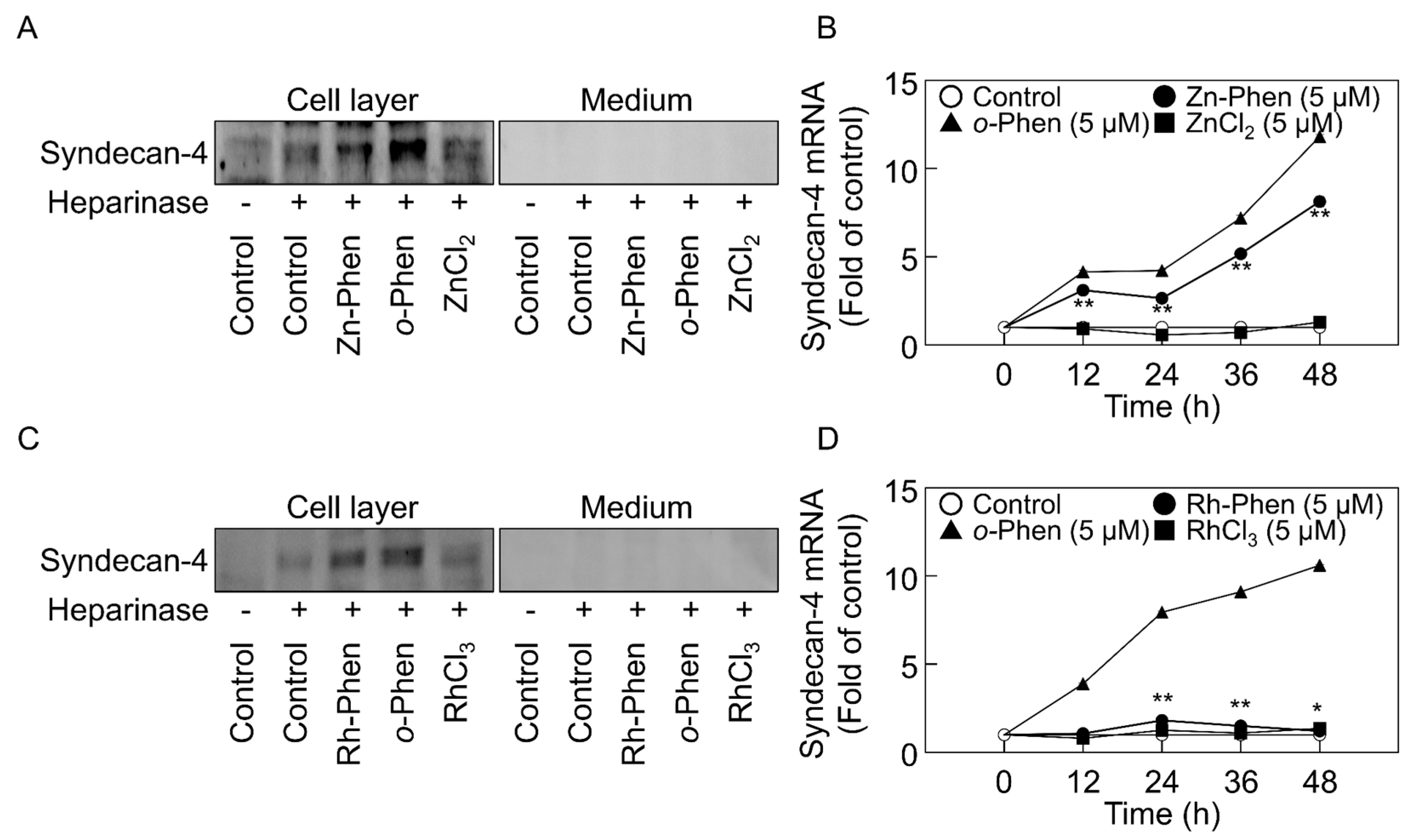
2.1. o-Phen, Zn-Phen, and Rh-Phen Induce Syndecan-4 Expression in Vascular Endothelial Cells
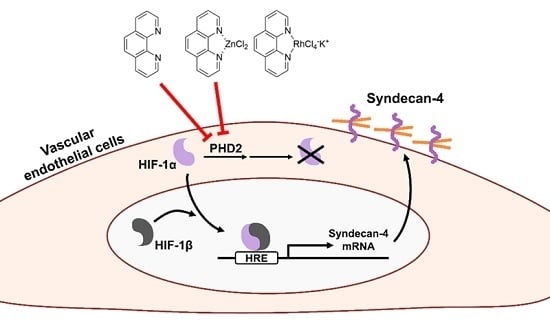
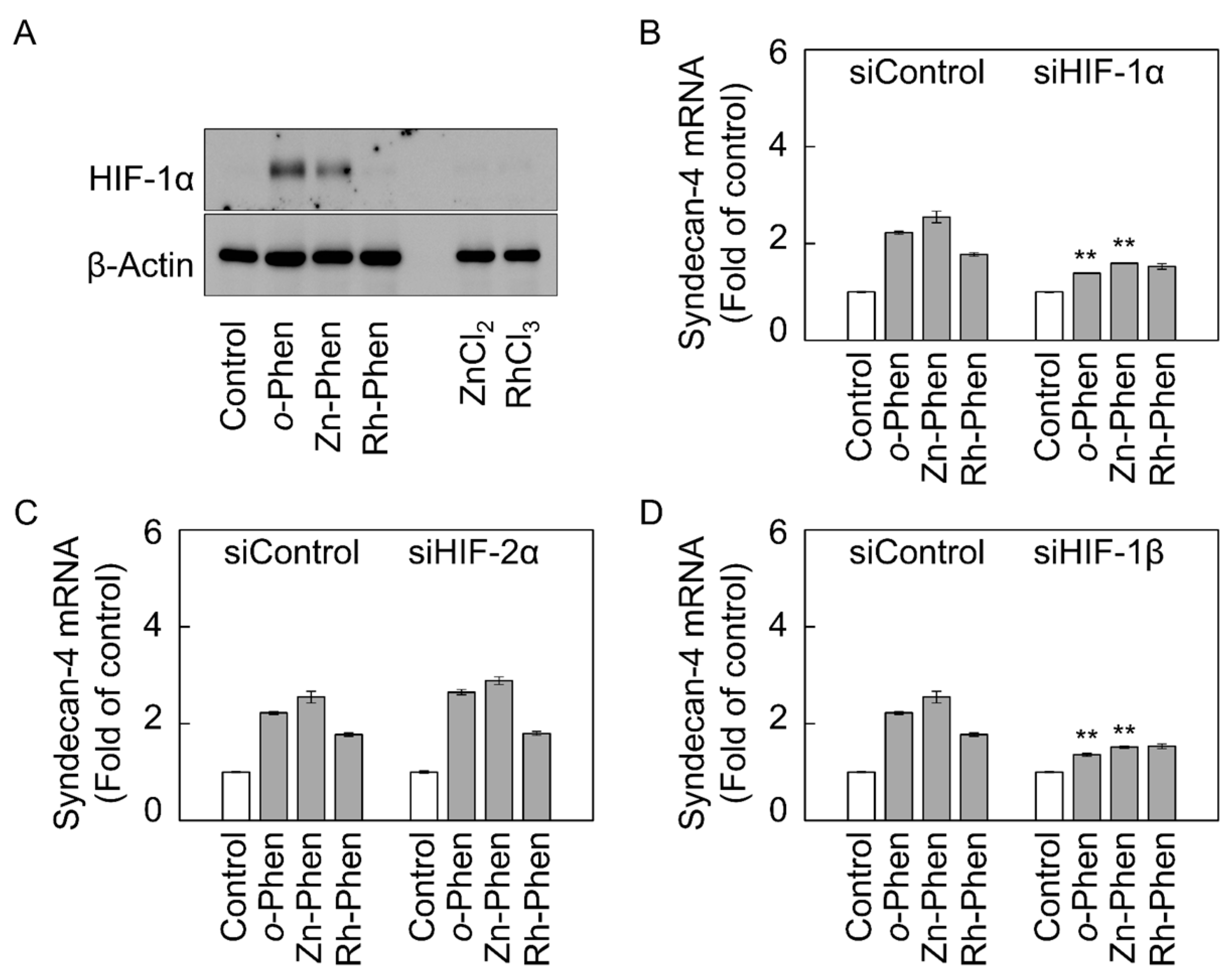
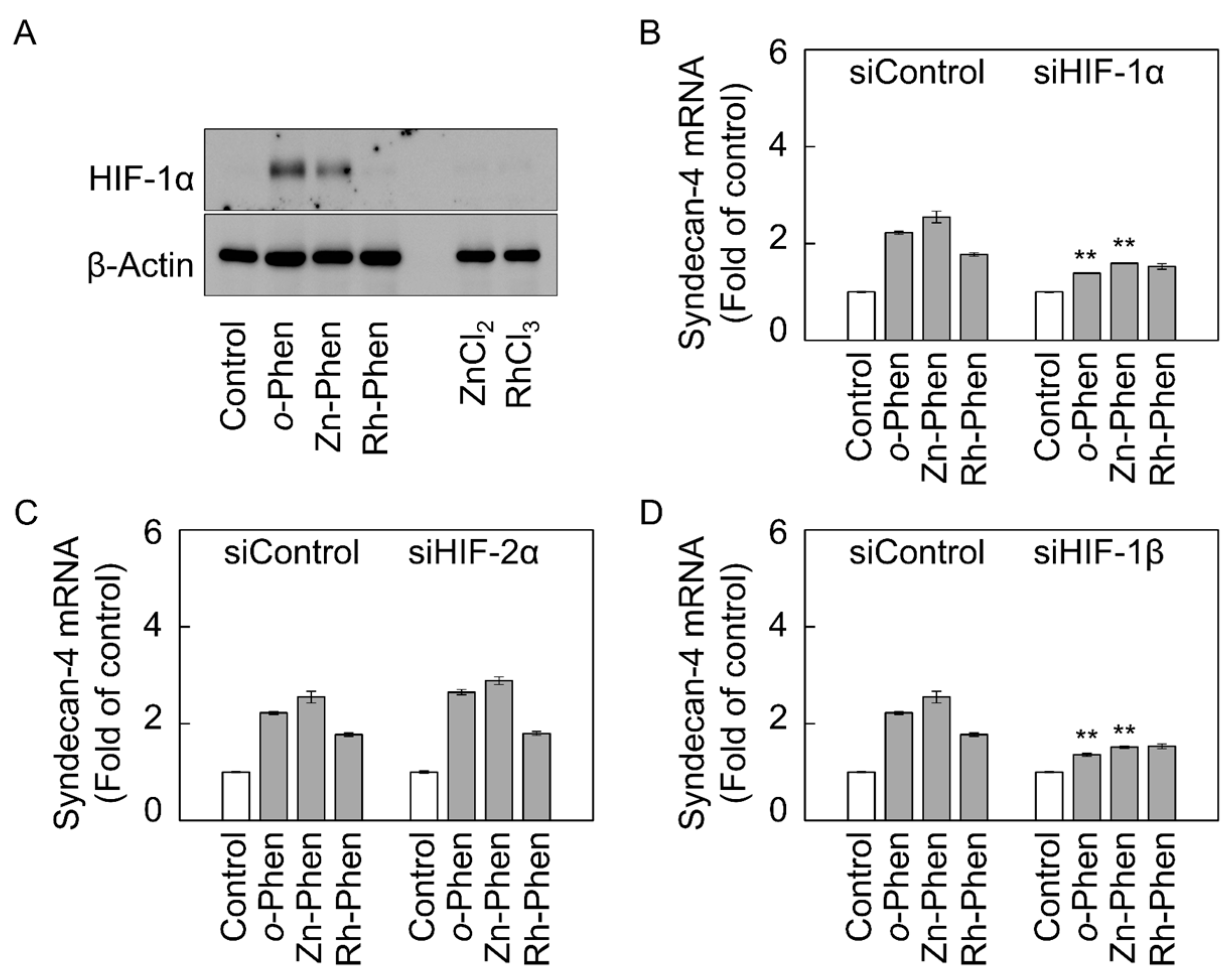
2.2. The Hypoxia-Inducible Factor (HIF)-1α/β Pathway Specifically Mediates Upregulation of Syndecan-4 Expression by o-Phen and Zn-Phen
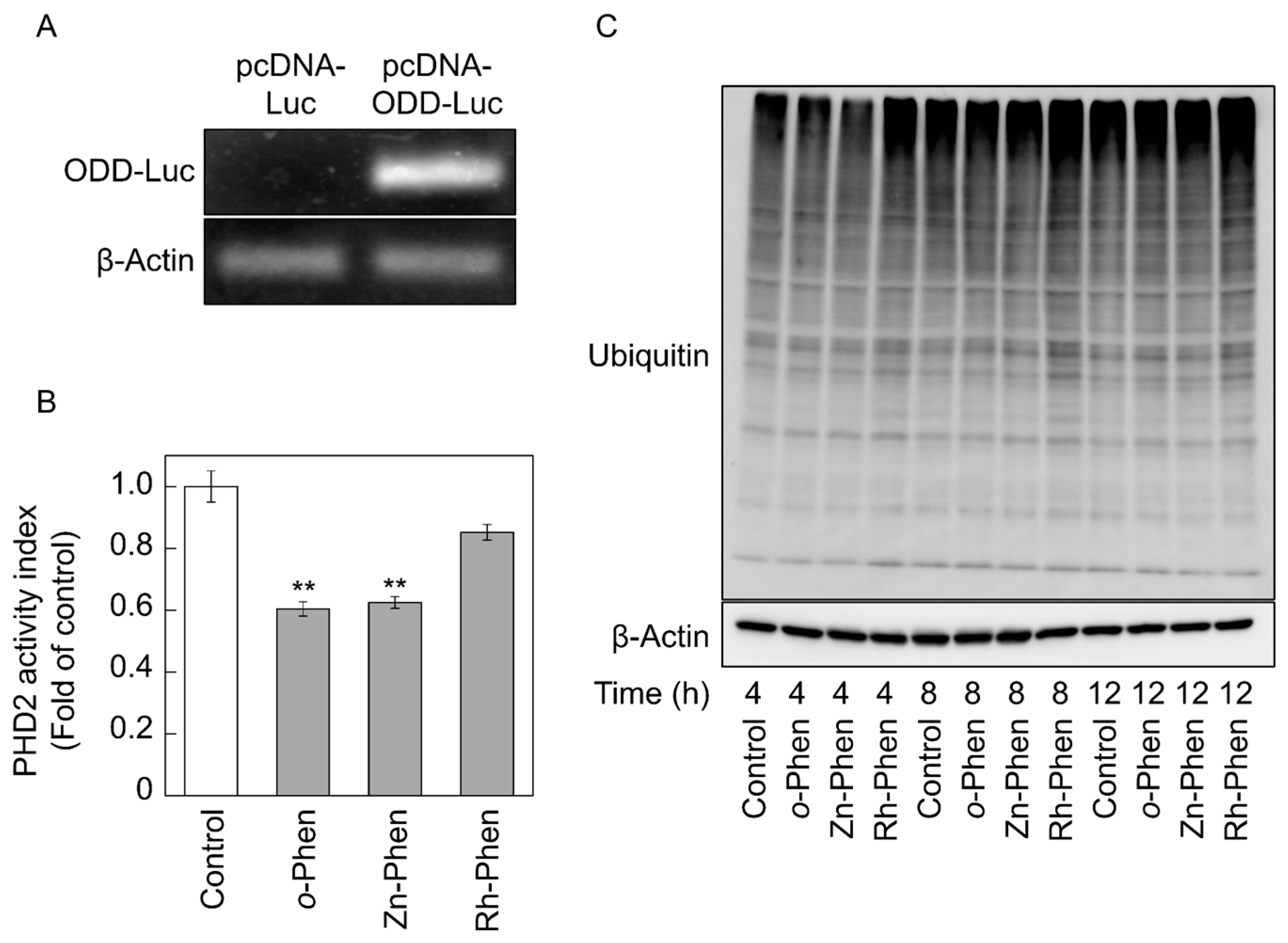
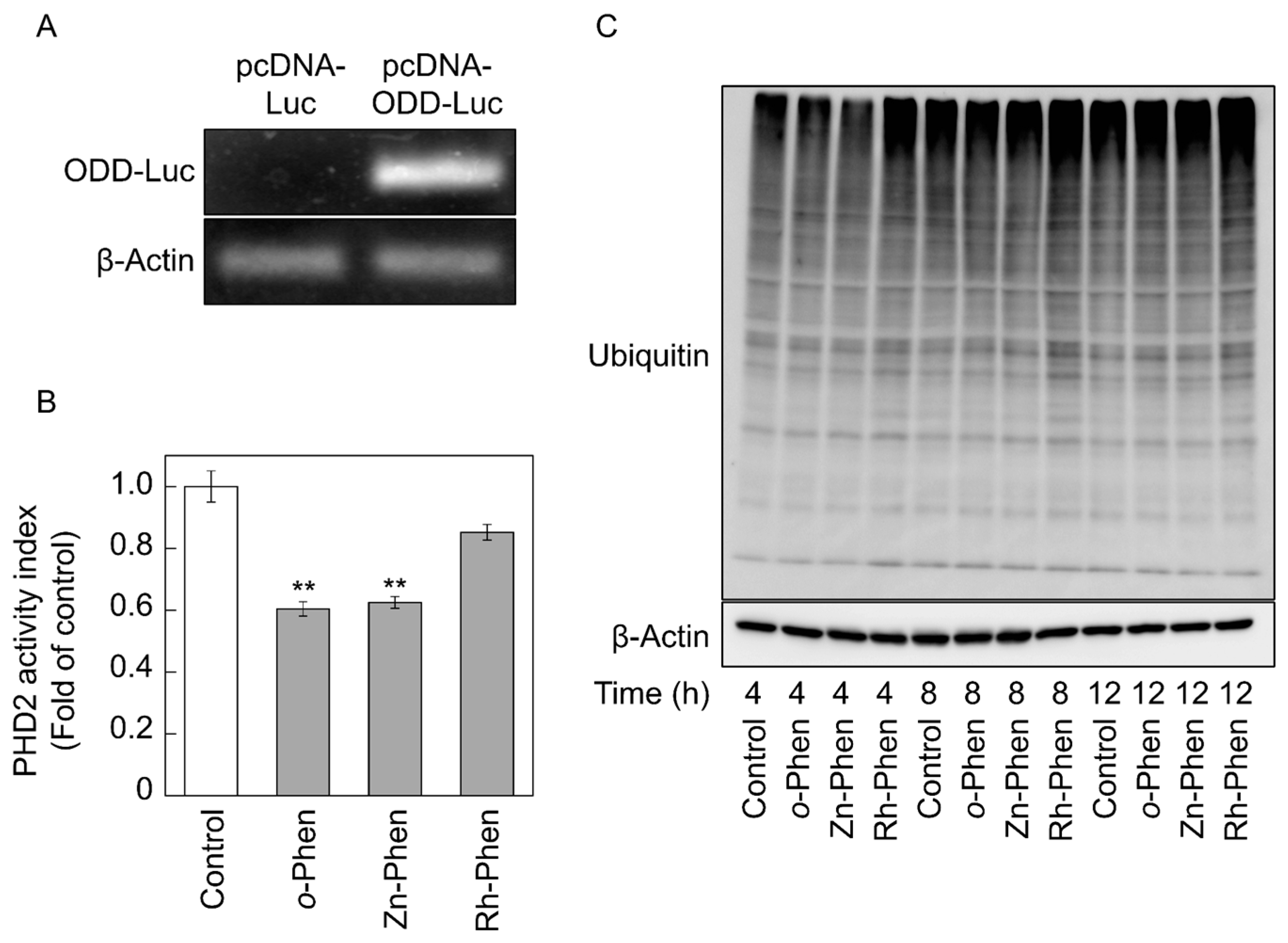
2.3. o-Phen and Zn-Phen Inhibit Prolyl Hydroxylase Domain-Containing Protein 2 (PHD2) Activity
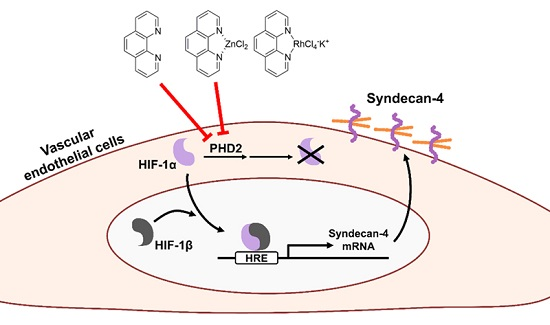
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Treatments
4.3. siRNA Transfection
4.4. Real-Time RT-PCR
4.5. Proteoglycan Core-Protein Extraction and Western Blot Analysis
4.6. Plasmid Construction
4.7. Luciferase Assay
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AhR | Aryl hydrocarbon receptor |
| B2M | β2-Microgloblin |
| DMEM | Dulbecco’s modified Eagle medium |
| PBS | Phosphate-buffered saline |
| FBS | Fetal bovine serum |
| HIF | Hypoxia-inducible factor |
| FGF-2 | Fibroblast growth factor-2 |
| HRE | Hypoxia-response element |
| ODD | Oxygen-dependent degradation domain |
| o-Phen | 1,10-Phenanthroline |
| PHD2 | Prolyl hydroxylase domain-containing protein 2 |
| Rh-Phen | Potassium tetrachloro(1,10-phenanthroline)rhodate(III) |
| siRNA | Small interfering RNA |
| Zn-Phen | Dichloro(1,10-phenanthroline)zinc |
References
- Jaffe, E.A.; Hoyer, L.W.; Nachman, R.L. Synthesis of von Willebrand factor by cultured human endothelial cells. Proc. Natl. Acad. Sci. USA 1974, 71, 1906–1909. [Google Scholar] [CrossRef] [PubMed]
- Maynard, J.R.; Dreyer, B.E.; Stemerman, M.B.; Pitlick, F.A. Tissue-factor coagulant activity of cultured human endothelial and smooth muscle cells and fibroblasts. Blood 1977, 50, 387–396. [Google Scholar] [PubMed]
- Revtyak, G.E.; Johnson, A.R.; Campbell, W.B. Prostaglandin synthesis in bovine coronary endothelial cells: Comparison with other commonly studied endothelial cells. Thromb. Res. 1987, 48, 671–683. [Google Scholar] [CrossRef]
- Levin, E.G.; Loskutoff, D.J. Cultured bovine endothelial cells produce both urokinase and tissue-type plasminogen activators. J. Cell Biol. 1982, 94, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E. Structure and biology of proteoglycans. Annu. Rev. Cell Biol. 1988, 4, 229–255. [Google Scholar] [CrossRef] [PubMed]
- Saku, T.; Furthmayr, H. Characterization of the major heparan sulfate proteoglycan secreted by bovine aortic endothelial cells in culture. Homology to the large molecular weight molecule of basement membranes. J. Biol. Chem. 1989, 264, 3514–3523. [Google Scholar] [PubMed]
- Kojima, T.; Shworak, N.W.; Rosenberg, R.D. Molecular cloning and expression of two distinct cDNA-encoding heparan sulfate proteoglycan core proteins from a rat endothelial cell line. J. Biol. Chem. 1992, 267, 4870–4877. [Google Scholar] [PubMed]
- Mertens, G.; Cassiman, J.J.; Van den Berghe, H.; Vermylen, J.; David, G. Cell surface heparan sulfate proteoglycans from human vascular endothelial cells. Core protein characterization and antithrombin III binding properties. J. Biol. Chem. 1992, 267, 20435–20443. [Google Scholar] [PubMed]
- Schönherr, E.; O’Connell, B.C.; Schittny, J.; Robenek, H.; Fastermann, D.; Fisher, L.W.; Plenz, G.; Vischer, P.; Young, M.F.; Kresse, H. Paracrine or virus-mediated induction of decorin expression by endothelial cells contributes to tube formation and prevention of apoptosis in collagen lattices. Eur. J. Cell Biol. 1999, 78, 44–55. [Google Scholar] [CrossRef]
- Yamamoto, C.; Deng, X.; Fujiwara, Y.; Kaji, T. Proteoglycans predominantly synthesized by human brain microvascular endothelial cells in culture are perlecan and biglycan. J. Health Sci. 2005, 51, 576–583. [Google Scholar] [CrossRef]
- Tollefsen, D.M.; Pestka, C.A.; Monafo, W.J. Activation of heparin cofactor II by dermatan sulfate. J. Biol. Chem. 1983, 258, 6713–6716. [Google Scholar] [CrossRef] [PubMed]
- Kaji, T.; Yamamoto, C.; Oh-i, M.; Fujiwara, Y.; Yamazaki, Y.; Morita, T.; Plaas, A.H.; Wight, T.N. The vascular endothelial growth factor VEGF165 induces perlecan synthesis via VEGF receptor-2 in cultured human brain microvascular endothelial cells. Biochim. Biophys. Acta 2006, 1760, 1465–1474. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, M.G.; Tsoi, C.K.; Jarvelainen, H.T.; Wight, T.N. Selective expression and processing of biglycan during migration of bovine aortic endothelial cells. The role of endogenous basic fibroblast growth factor. J. Biol. Chem. 1997, 272, 318–325. [Google Scholar] [PubMed]
- Kaji, T.; Yamamoto, C.; Oh-i, M.; Nishida, T.; Takigawa, M. Differential regulation of biglycan and decorin synthesis by connective tissue growth factor in cultured vascular endothelial cells. Biochem. Biophys. Res. Commun. 2004, 322, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Kaji, T.; Yamada, A.; Miyajima, S.; Yamamoto, C.; Fujiwara, Y.; Wight, T.N.; Kinsella, M.G. Cell density-dependent regulation of proteoglycan synthesis by transforming growth factor-β1 in cultured bovine aortic endothelial cells. J. Biol. Chem. 2000, 275, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Yoshida, E.; Shinkai, Y.; Yamamoto, C.; Fujiwara, Y.; Kumagai, Y.; Kaji, T. Biglycan intensifies ALK5-Smad2/3 signaling by TGF-β1 and downregulates syndecan-4 in cultured vascular endothelial cells. J. Cell. Biochem. 2016. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Matsuzaki, H.; Nakamura, T.; Yoshida, E.; Ohkubo, T.; Maruyama, H.; Yamamoto, C.; Saito, S.; Kaji, T. Cytotoxicity of zinc, copper and rhodium complexes with 1,10-phenanthroline or 2,9-dimethyl-1,10-phenanthroline in cultured vascular endothelial cells. Fundam. Toxicol. Sci. 2016, 3, 109–113. [Google Scholar] [CrossRef]
- Kohri, K.; Yoshida, E.; Yasuike, S.; Fujie, T.; Yamamoto, C.; Kaji, T. The cytotoxicity of organobismuth compounds with certain molecular structures can be diminished by replacing the bismuth atom with an antimony atom in the molecules. J. Toxicol. Sci. 2015, 40, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Fujie, T.; Matsumura, M.; Yoshida, E.; Yamamoto, C.; Fujiwara, Y.; Yasuike, S.; Kaji, T. Comparative cytotoxicity of triphenylstibane and fluorine-substituted triarylpnictogens in cultured vascular endothelial cells. Fundam. Toxicol. Sci. 2015, 2, 61–66. [Google Scholar] [CrossRef]
- Fujie, T.; Murakami, M.; Yoshida, E.; Yasuike, S.; Kimura, T.; Fujiwara, Y.; Yamamoto, C.; Kaji, T. Transcriptional induction of metallothionein by tris(pentafluorophenyl)stibane in cultured bovine aortic endothelial cells. Int. J. Mol. Sci. 2016, 17, 1381. [Google Scholar] [CrossRef] [PubMed]
- Fujie, T.; Segawa, Y.; Yoshida, E.; Kimura, T.; Fujiwara, Y.; Yamamoto, C.; Satoh, M.; Naka, H.; Kaji, T. Induction of metallothionein isoforms by copper diethyldithiocarbamate in cultured vascular endothelial cells. J. Toxicol. Sci. 2016, 41, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.A.; Song, H.K.; Lee, S.H.; Chung, B.H.; Lim, H.M.; Lee, M.K. Differential in vitro and cellular effects of iron chelators for hypoxia inducible factor hydroxylases. J. Cell. Biochem. 2013, 114, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFα targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Whiteford, J.R.; Behrends, V.; Kirby, H.; Kusche-Gullberg, M.; Muramatsu, T.; Couchman, J.R. Syndecans promote integrin-mediated adhesion of mesenchymal cells in two distinct pathways. Exp. Cell Res. 2007, 313, 3902–3913. [Google Scholar] [CrossRef] [PubMed]
- Vuong, T.T.; Reine, T.M.; Sudworth, A.; Jenssen, T.G.; Kolset, S.O. Syndecan-4 is a major syndecan in primary human endothelial cells in vitro, modulated by inflammatory stimuli and involved in wound healing. J. Histochem. Cytochem. 2015, 63, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.; Son, H.; Choi, Y.; Lee, J.H.; Choi, S.; Lim, Y.; Han, I.O.; Oh, E.S. Syndecan-4 promotes the retention of phosphatidylinositol 4,5-bisphosphate in the plasma membrane. FEBS Lett. 2009, 583, 2395–2400. [Google Scholar] [CrossRef] [PubMed]
- Cizmeci-Smith, G.; Langan, E.; Youkey, J.; Showalter, L.J.; Carey, D.J. Syndecan-4 is a primary-response gene induced by basic fibroblast growth factor and arterial injury in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Song, S.J.; Cool, S.M.; Nurcombe, V. Regulated expression of syndecan-4 in rat calvaria osteoblasts induced by fibroblast growth factor-2. J. Cell. Biochem. 2007, 100, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Velleman, S.G.; Li, X.; Coy, C.S.; McFarland, D.C. The effect of fibroblast growth factor 2 on the in vitro expression of syndecan-4 and glypican-1 in turkey satellite cells. Poult. Sci. 2008, 87, 1834–1840. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, E.; Suzuki, A.; Murata, M.; Ando, Y.; Kato, I.; Takagi, Y.; Takagi, A.; Murate, T.; Saito, H.; Kojima, T. Molecular mechanisms of syndecan-4 upregulation by TNF-α in the endothelium-like EAhy926 cells. J. Biochem. 2013, 154, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pasparakis, M.; Kollias, G.; Simons, M. Myocyte-dependent regulation of endothelial cell syndecan-4 expression. Role of TNF-α. J. Biol. Chem. 1999, 274, 14786–14790. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Hirose, Y.; Tran, C.M.; Chiba, K.; Miyamoto, T.; Toyama, Y.; Shapiro, I.M.; Risbud, M.V. HIF-1-PHD2 axis controls expression of syndecan 4 in nucleus pulposus cells. FASEB J. 2014, 28, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, S.; Yamada, M.; Inoue, S.; Matsubara, M. Simple and rapid spectrophotometric determination of iron after preconcentration as its 1,10-phenanthroline complex on the natural polymer “chitin”. Talanta 1989, 36, 606–608. [Google Scholar] [CrossRef]
- Wenger, R.H. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J. 2002, 16, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Mole, D.R.; Blancher, C.; Copley, R.R.; Pollard, P.J.; Gleadle, J.M.; Ragoussis, J.; Ratcliffe, P.J. Genome-wide association of hypoxia-inducible factor (HIF)-1α and HIF-2α DNA binding with expression profiling of hypoxia-inducible transcripts. J. Biol. Chem. 2009, 284, 16767–16775. [Google Scholar] [CrossRef] [PubMed]
- Schodel, J.; Oikonomopoulos, S.; Ragoussis, J.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 2011, 117, e207–e217. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.E.; Bagnall, J.; Mason, D.; Levy, R.; Fernig, D.G.; See, V. Differential sub-nuclear distribution of hypoxia-inducible factors (HIF)-1 and -2 α impacts on their stability and mobility. Open Biol. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Shworak, N.W.; Shirakawa, M.; Colliec-Jouault, S.; Liu, J.; Mulligan, R.C.; Birinyi, L.K.; Rosenberg, R.D. Pathway-specific regulation of the synthesis of anticoagulantly active heparan sulfate. J. Biol. Chem. 1994, 269, 24941–24952. [Google Scholar] [PubMed]
- Karlinsky, J.B.; Rounds, S.; Farber, H.W. Effects of hypoxia on heparan sulfate in bovine aortic and pulmonary artery endothelial cells. Circ. Res. 1992, 71, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Kyhse-Andersen, J. Electroblotting of multiple gels: A simple apparatus without buffer tank for rapid transfer of proteins from polycrylamide to nitrocellulose. J. Biochem. Biophys. Methods 1984, 10, 203–209. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′–3′) | Reverse Primer (5’–3’) |
|---|---|---|
| Perlecan | ATGGCAGCGATGAAGCGGAC | TTGTGGACACGCAGCGGAAC |
| Syndecan-1 | CAGTCAGGAGACAGCATCAG | CCGACAGACATTCCATACC |
| Syndecan-2 | CCAGATGAAGAGGACACAAACG | CCAATAACTCCGCCAGCAA |
| Syndecan-3 | CAAGCAGGCGAGCGTC | GGTGGCAGAGATGAAGTGG |
| Syndecan-4 | TTGCCGTCTTCCTCGTGC | AGGCGTAGAACTCATTGGTGG |
| Glypican-1 | GAAGGTCGGCAGGAAGAG | CCAGGAGCAGCAGAGGA |
| Biglycan | GCTGCCACTGCCATCTGAG | CGAGGACCAAGGCGTAG |
| Decorin | CTGCGGTTGACAATGGC | CTCACTCCTGAATAAGAAGCC |
| HIF-1α | GCTTGCTCATCAGTTGCCAC | GCATCCAGAAGTTTCCTCACAC |
| HIF-2α | CAGTGGCAAGGTGGCTGTGTC | GGTCCCGAAATCCAGAGAAATGA |
| HIF-1β | TAAGGAGCGGTTTGCCAGGTC | TTCTGTTATGTAGGCTGTCATCTTGTTC |
| AhR | GTGTCAGTTATCTCAGAGCCAAG | AAAGCCATTTAGTGCCTGTAGTA |
| B2M | CCATCCAGCGTCCTCCAAAGA | TTCAATCTGGGGTGGATGGAA |
| β-Actin | CCTCCCTGGAGAAGAGCTACGA | GGAATTGAAGGTAGTTTCGTGAATG |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hara, T.; Kojima, T.; Matsuzaki, H.; Nakamura, T.; Yoshida, E.; Fujiwara, Y.; Yamamoto, C.; Saito, S.; Kaji, T. Induction of Syndecan-4 by Organic–Inorganic Hybrid Molecules with a 1,10-Phenanthroline Structure in Cultured Vascular Endothelial Cells. Int. J. Mol. Sci. 2017, 18, 352. https://doi.org/10.3390/ijms18020352
Hara T, Kojima T, Matsuzaki H, Nakamura T, Yoshida E, Fujiwara Y, Yamamoto C, Saito S, Kaji T. Induction of Syndecan-4 by Organic–Inorganic Hybrid Molecules with a 1,10-Phenanthroline Structure in Cultured Vascular Endothelial Cells. International Journal of Molecular Sciences. 2017; 18(2):352. https://doi.org/10.3390/ijms18020352
Chicago/Turabian StyleHara, Takato, Takayuki Kojima, Hiroka Matsuzaki, Takehiro Nakamura, Eiko Yoshida, Yasuyuki Fujiwara, Chika Yamamoto, Shinichi Saito, and Toshiyuki Kaji. 2017. "Induction of Syndecan-4 by Organic–Inorganic Hybrid Molecules with a 1,10-Phenanthroline Structure in Cultured Vascular Endothelial Cells" International Journal of Molecular Sciences 18, no. 2: 352. https://doi.org/10.3390/ijms18020352
APA StyleHara, T., Kojima, T., Matsuzaki, H., Nakamura, T., Yoshida, E., Fujiwara, Y., Yamamoto, C., Saito, S., & Kaji, T. (2017). Induction of Syndecan-4 by Organic–Inorganic Hybrid Molecules with a 1,10-Phenanthroline Structure in Cultured Vascular Endothelial Cells. International Journal of Molecular Sciences, 18(2), 352. https://doi.org/10.3390/ijms18020352