Ketamine, a Clinically Used Anesthetic, Inhibits Vascular Smooth Muscle Cell Proliferation via PP2A-Activated PI3K/Akt/ERK Inhibition



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
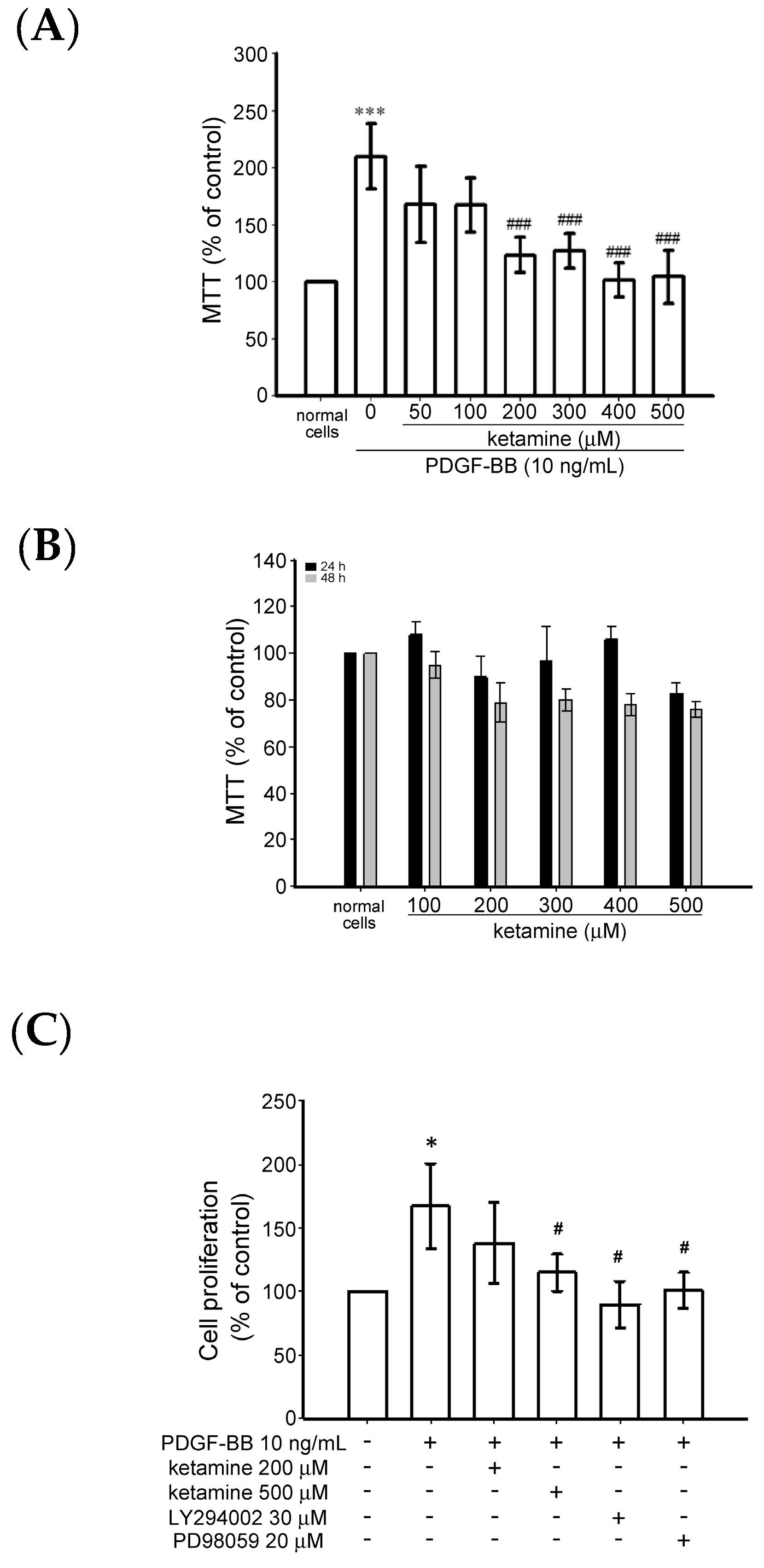
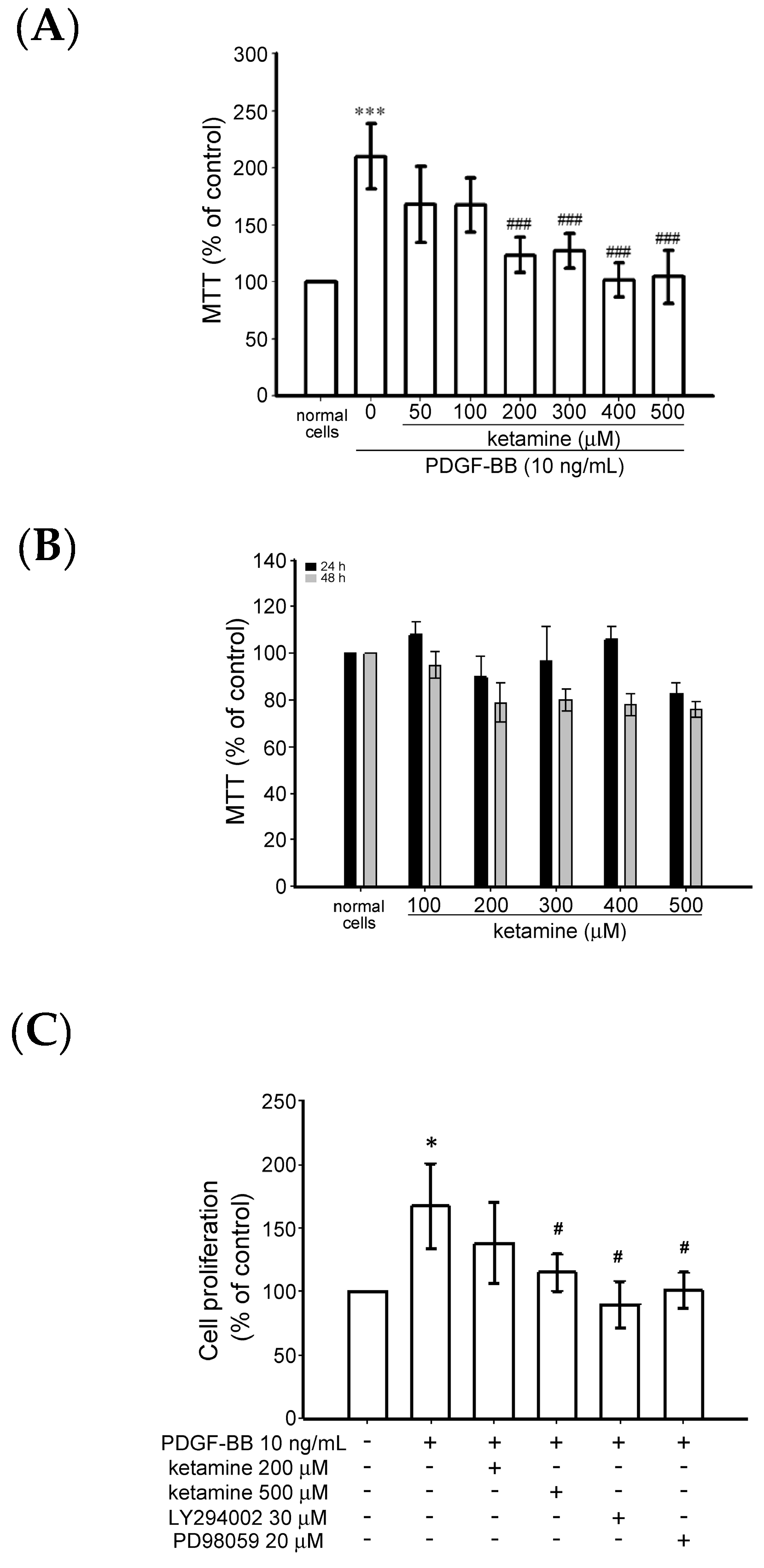
2.1.1. Effects of Ketamine on Platelet-Derived Growth Factor BB (PDGF-BB)-Induced Toxicity and Proliferation of Vascular Smooth Muscle Cells (VSMCs)
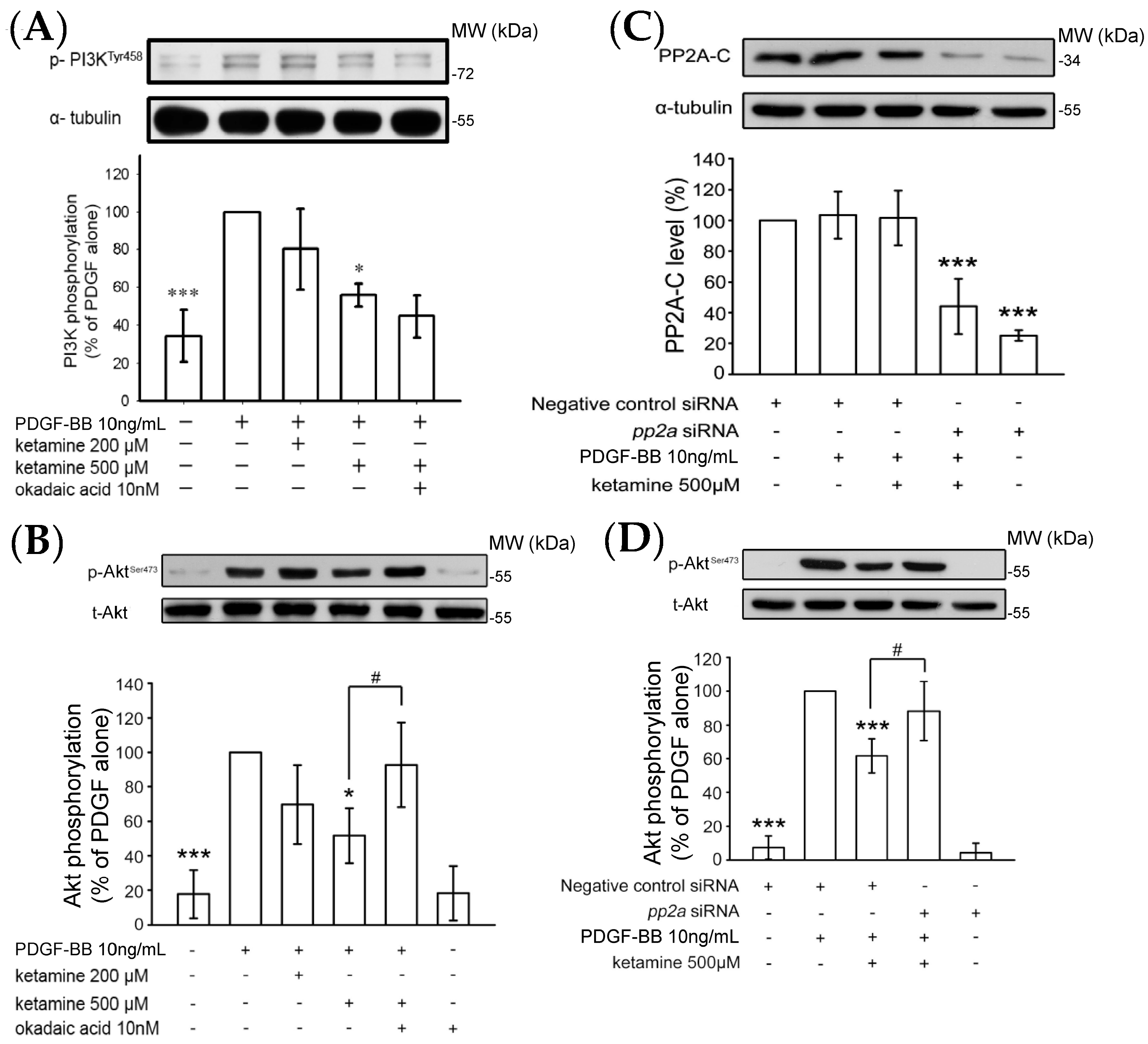
2.1.2. Ketamine Suppressed PDGF-BB-Induced Phosphatidylinositol 3-Kinase (PI3K) and Akt Signaling Pathway
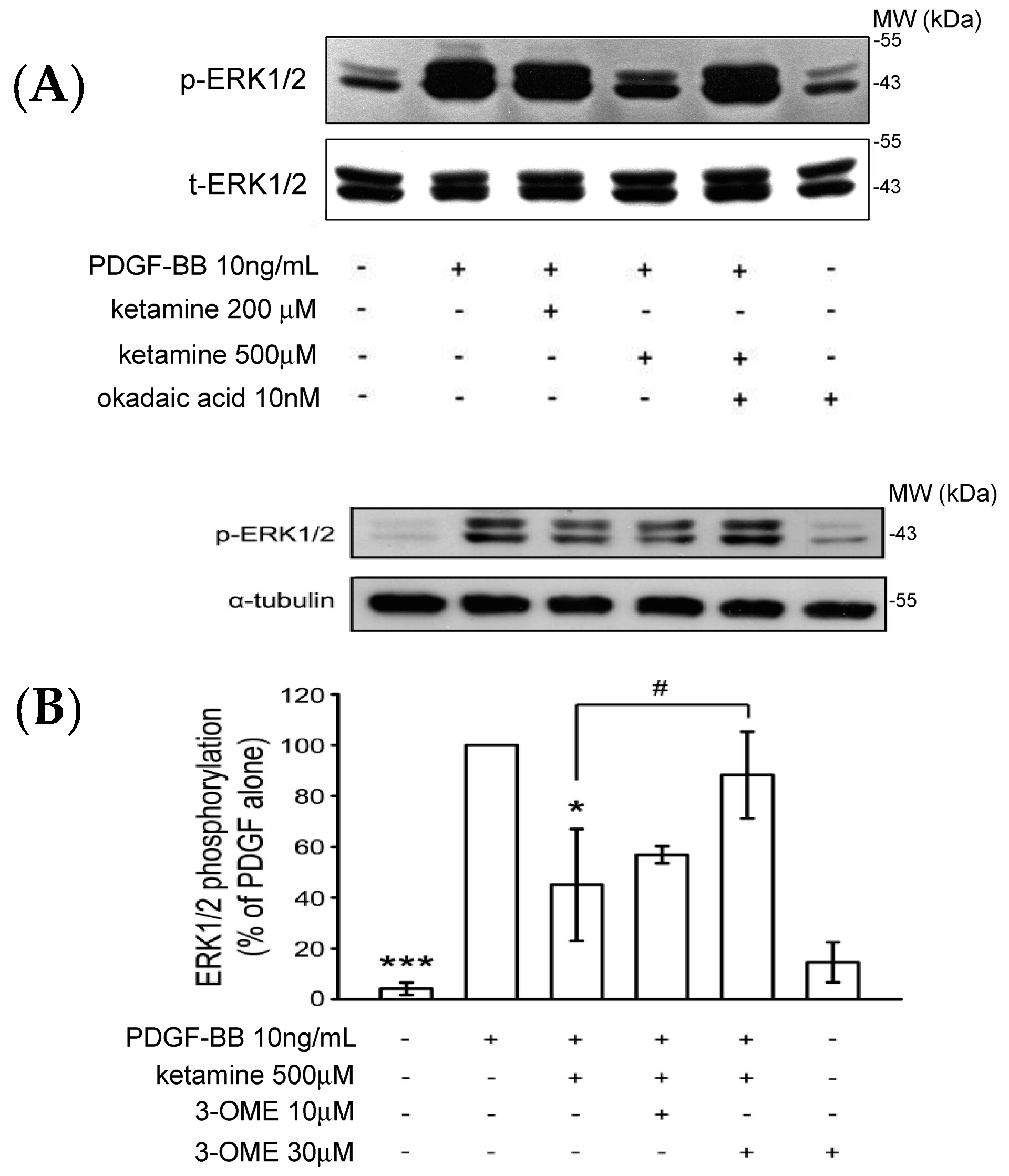
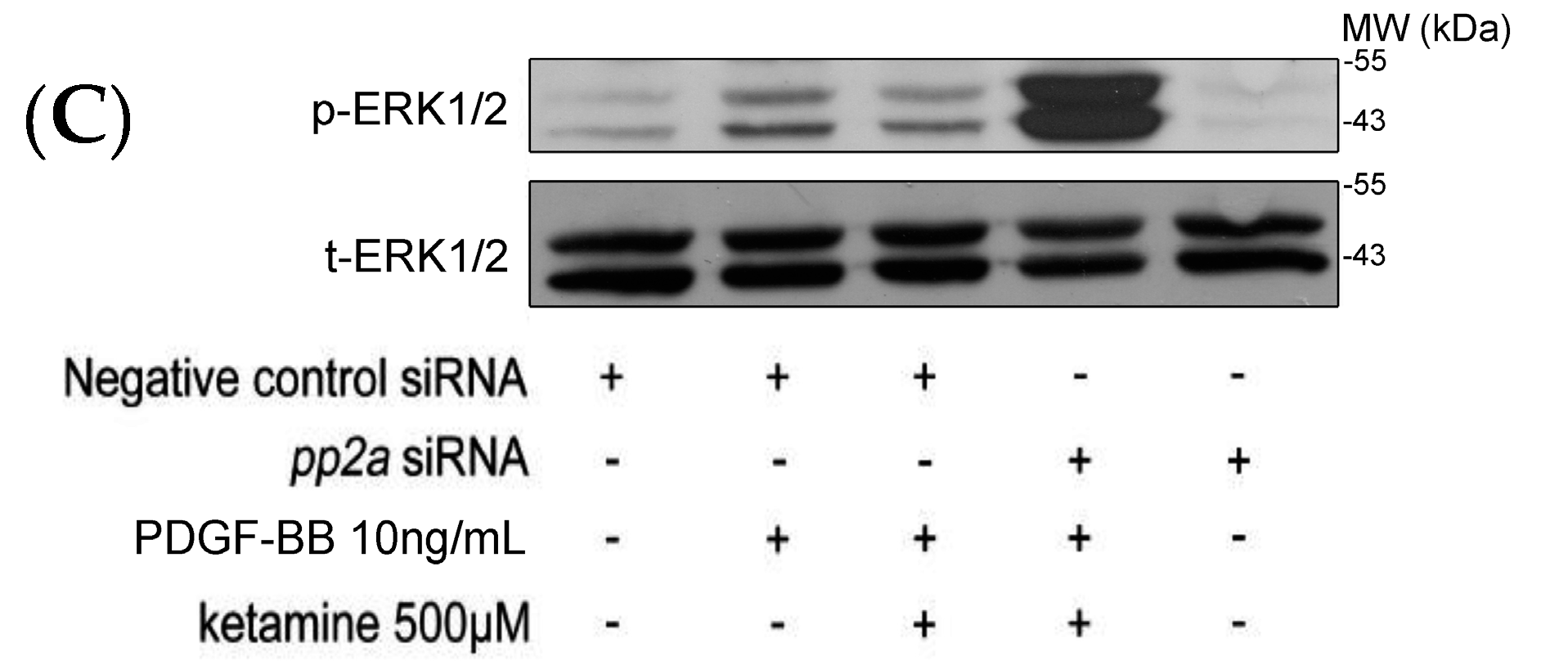
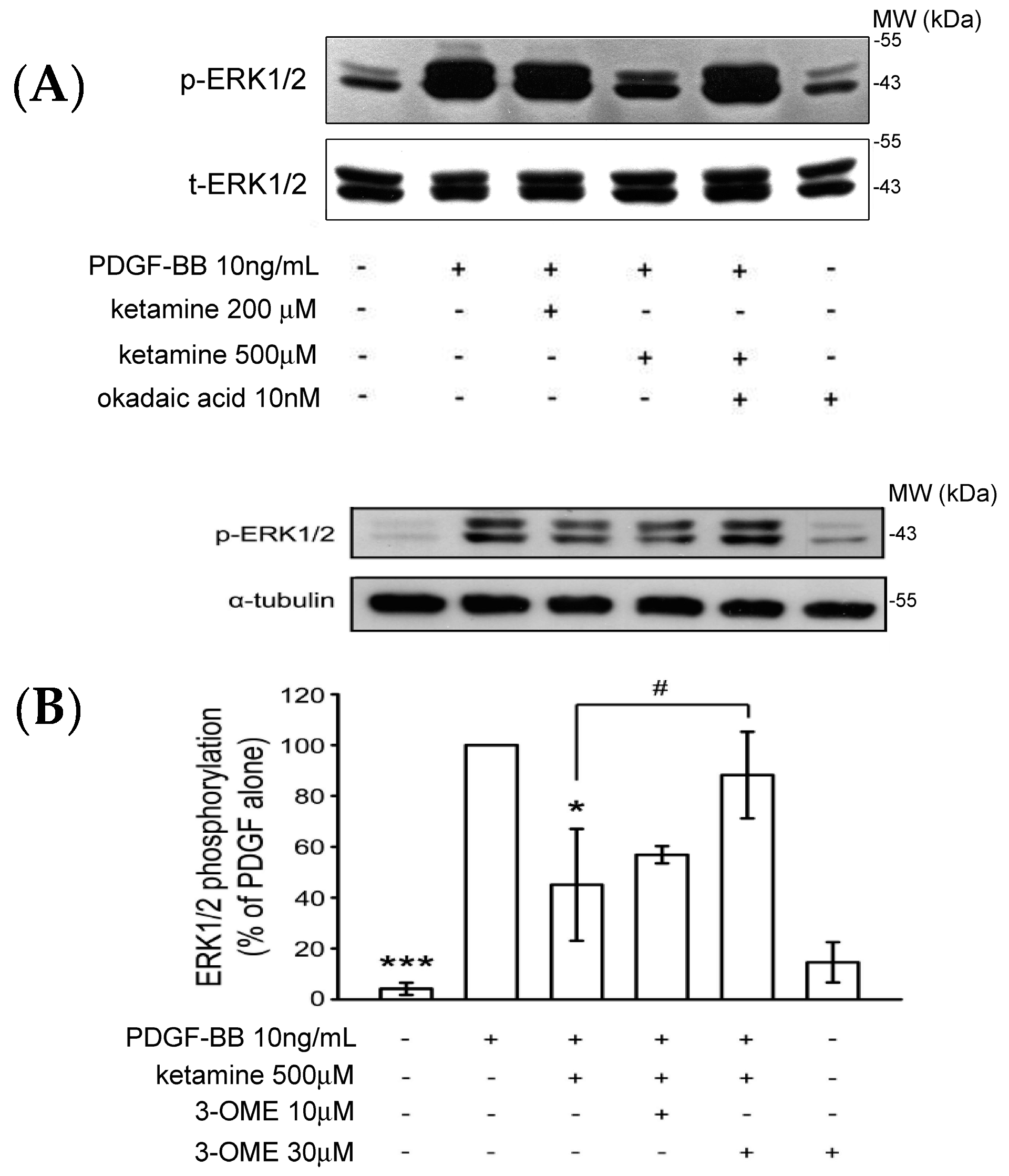
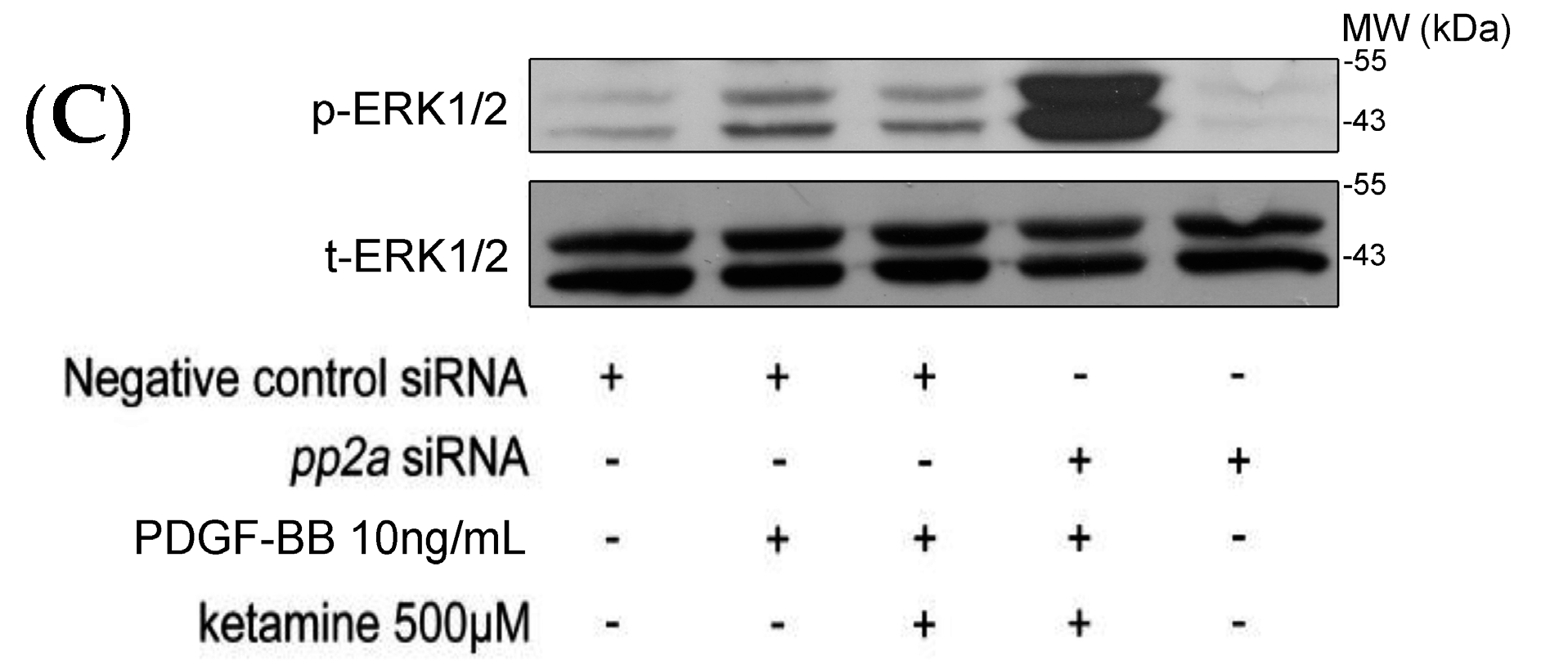
2.1.3. Effects of Ketamine, Okadaic Acid, 3-O-Methyl-Sphingomyeline (3-OME), and pp2a SiRNA on the PDGF-BB-Induced Phosphorylation of ERK1/2 in VSMCs
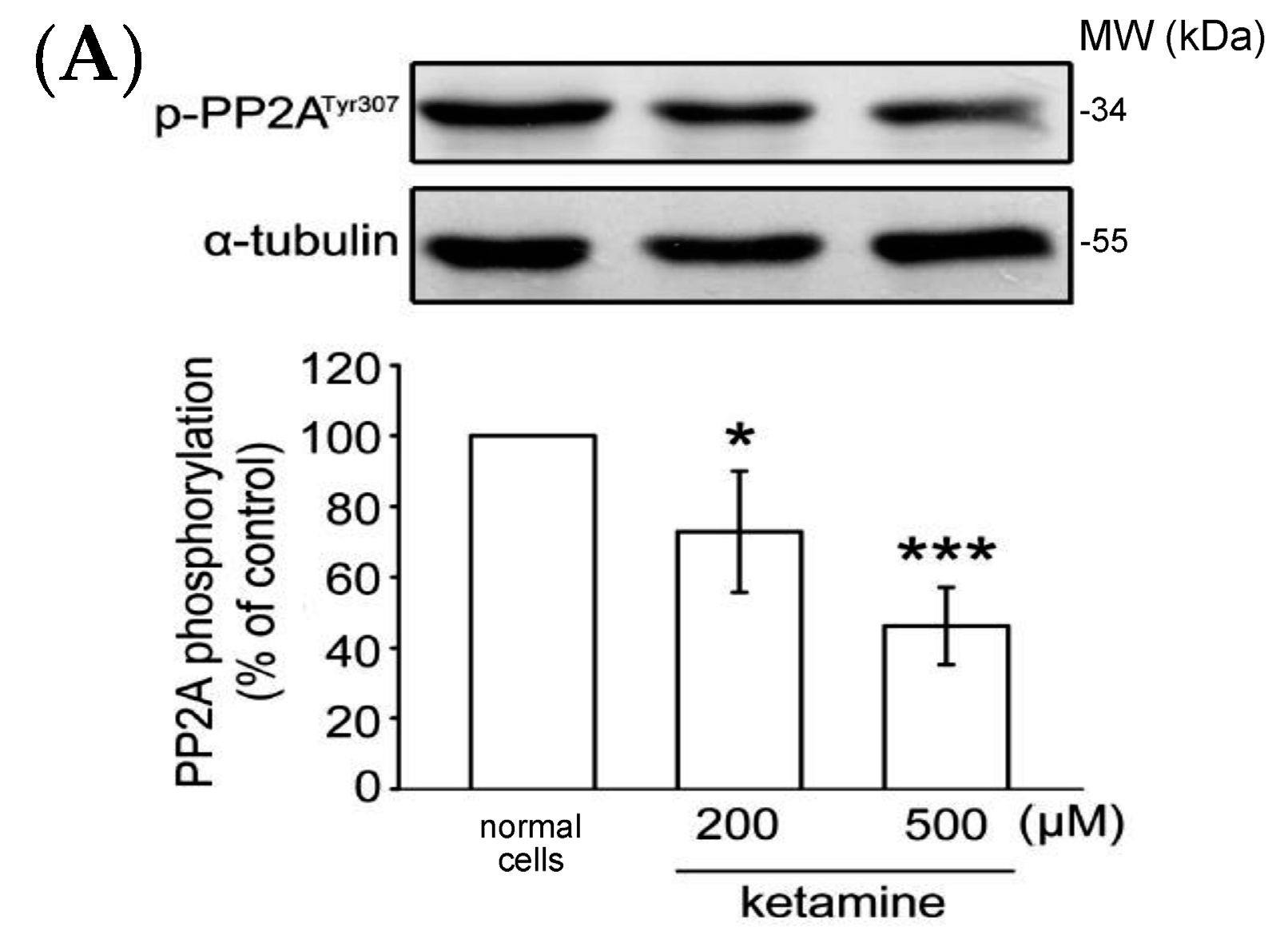
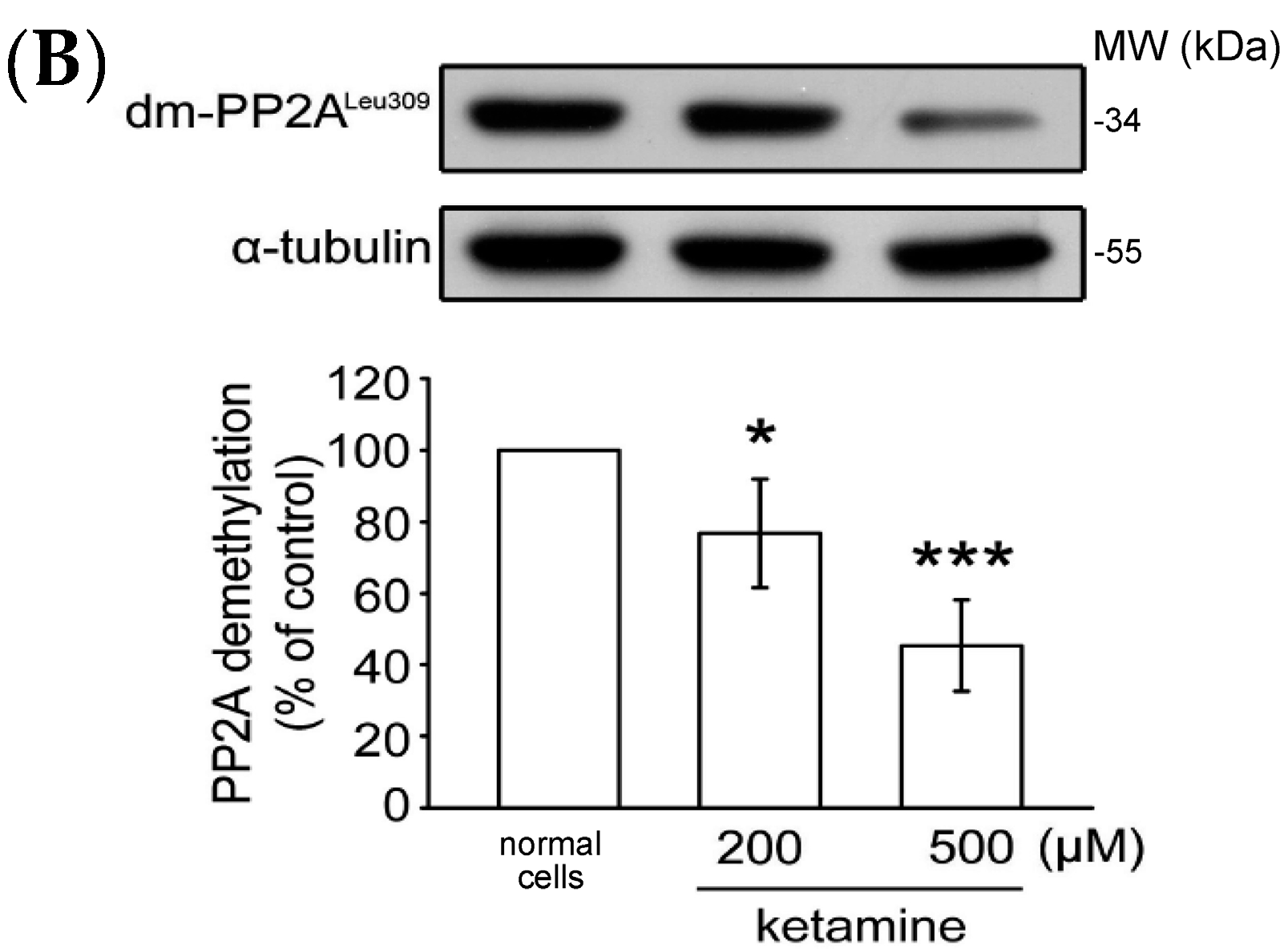
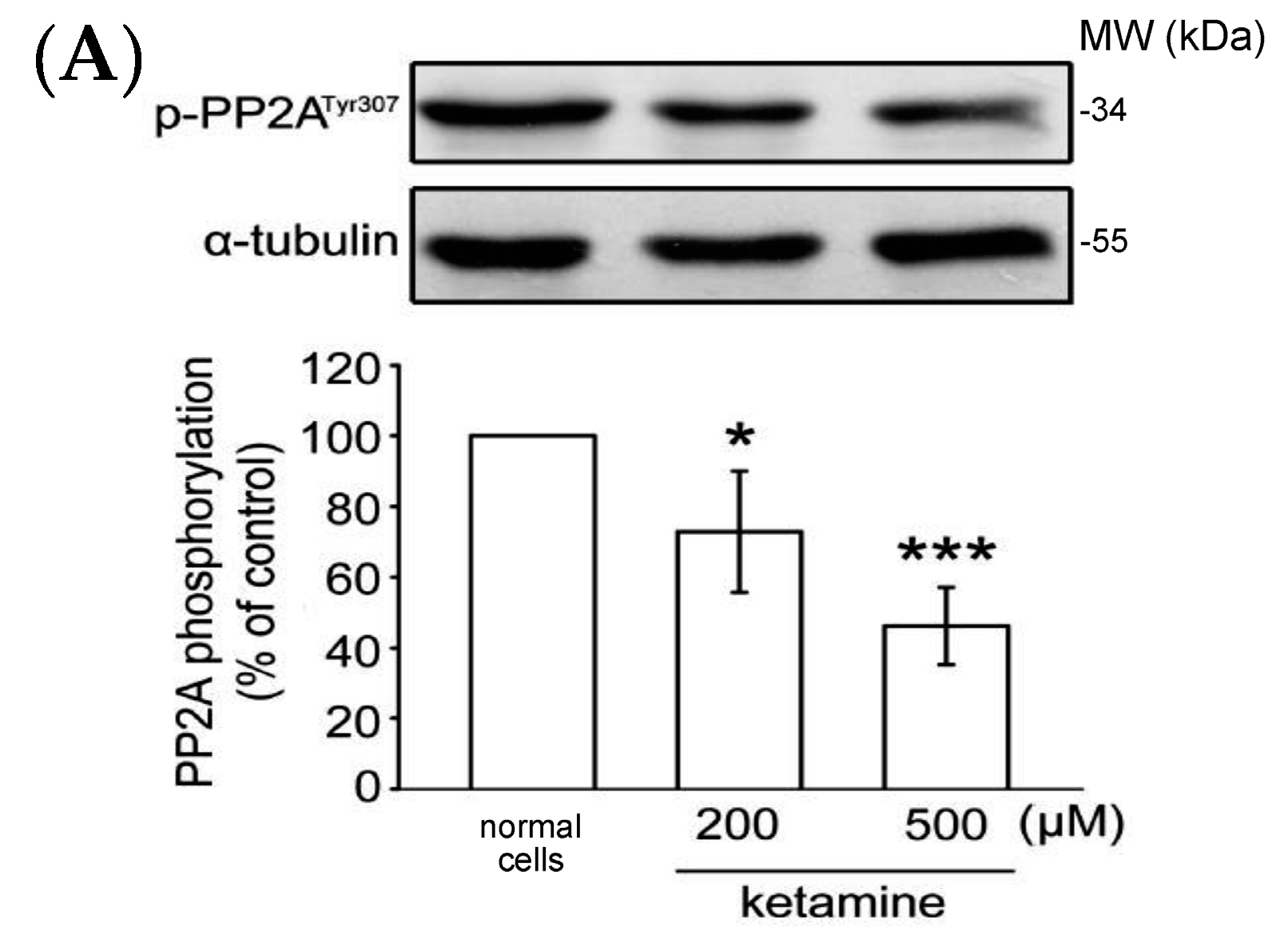
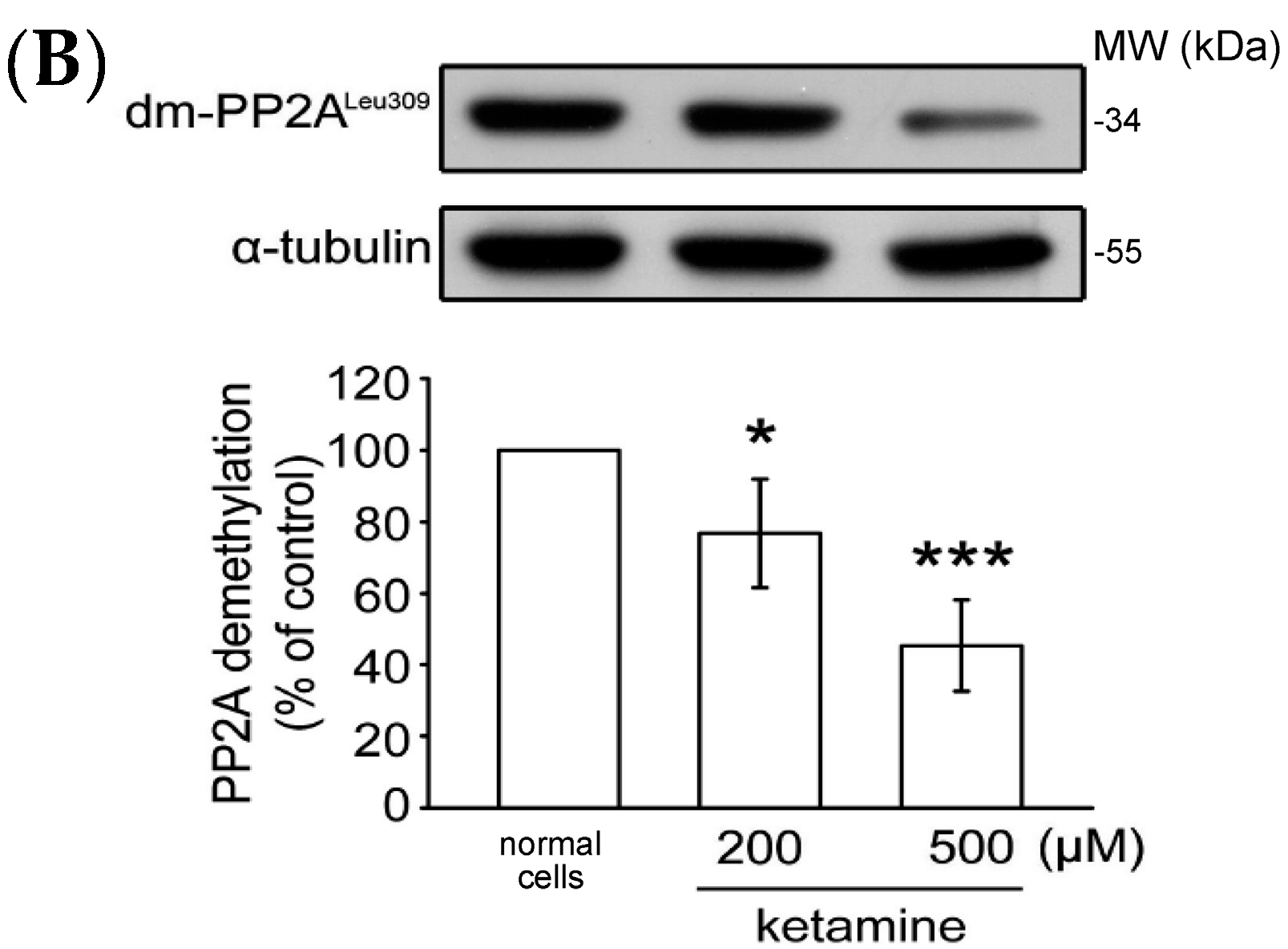
2.1.4. Influence of Ketamine and pp2a-SiRNA on Phosphorylated PP2A (Tyr307) and Methylated PP2A (Leu309) in VSMCs
2.2. Discussion
3. Materials and Methods
3.1. Materials
3.2. Rat VSMC Primary Culture
3.3. Cell Viability Assay
3.4. DNA Synthesis Assay
3.5. Immunoblot Analysis
3.6. Suppression of PP2A Expression
3.7. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lagraauw, H.M.; Kuiper, J.; Bot, I. Acute and chronic psychological stress as risk factors for cardiovascular disease: Insights gained from epidemiological, clinical and experimental studies. Brain Behav. Immun. 2015, 50, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.B.; Mengi, S.A.; Xu, Y.J.; Arneja, A.S.; Dhalla, N.S. Pathogenesis of atherosclerosis: A multifactorial process. Exp. Clin. Cardiol. 2002, 7, 40–53. [Google Scholar] [PubMed]
- Ross, R. The pathogenesis of atherosclerosis: A perspective for the 1990s. Nature 1993, 362, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis is an inflammatory disease. Am. Heart J. 1999, 138, S419–S420. [Google Scholar] [CrossRef]
- Glass, C.K.; Witztum, J.L. Atherosclerosis: The road ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef]
- Zhan, Y.; Kim, S.; Izumi, Y.; Izumiya, Y.; Nakao, T.; Miyazaki, H.; Iwao, H. Role of JNK, p38, and ERK in platelet derived growth factor-induced vascular proliferation, migration, and gene expression. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Janssens, V.; Goris, J. Protein phosphatase 2A: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signaling. Biochem. J. 2001, 353, 417–439. [Google Scholar] [CrossRef] [PubMed]
- Richman, J.G.; Regan, J.W. α2-Adrenergic receptors increase cell migration and decrease F-actin labeling in rat aortic smooth muscle cells. Am. J. Physiol. Cell Physiol. 1998, 274, C654–C662. [Google Scholar]
- Weinbroum, A.A. Non-opioid IV adjuvants in the perioperative period: Pharmacological and clinical aspects of ketamine and gabapentinoids. Pharmacol. Res. 2012, 65, 411–429. [Google Scholar] [CrossRef] [PubMed]
- McGrane, S.; Pandharipande, P.P. Sedation in the intensive care unit. Minerva Anestesiol. 2012, 78, 369–380. [Google Scholar] [PubMed]
- Akata, T. General anesthetics and vascular smooth muscle: Direct actions of general anesthetics on cellular mechanisms regulating vascular tone. Anesthesiology 2007, 106, 365–391. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.M.; West, D.C.; Lodge, D. An N-methylaspartate receptor-mediated synapse in rat cerebral cortex: A site of action of ketamine? Nature 1985, 313, 479–481. [Google Scholar] [CrossRef] [PubMed]
- Zeilhofer, H.U. Synaptic modulation in pain pathways. Rev. Physiol. Biochem. Pharmacol. 2005, 154, 73–100. [Google Scholar] [PubMed]
- Ueki, K.; Fruman, D.A.; Brachmann, S.M.; Tseng, Y.H.; Cantley, L.C.; Kahn, C.R. Molecular balance between the regulatory and catalytic subunits of phosphoinositide 3-kinase regulates cell signaling and survival. Mol. Cell. Biol. 2002, 22, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Iida, M.; Tanabe, K.; Matsushima-Nishiwaki, R.; Kozawa, O.; Iida, H. Adenosine monophosphate-activated protein kinase regulates platelet-derived growth factor-BB-induced vascular smooth muscle cell migration. Arch. Biochem. Biophys. 2013, 530, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Sangodkar, J.; Farrington, C.C.; McClinch, K.; Galsky, M.D.; Kastrinsky, D.B.; Narla, G. All roads lead to PP2A: Exploiting the therapeutic potential of this phosphatase. FEBS J. 2016, 283, 1004–1024. [Google Scholar] [CrossRef] [PubMed]
- Lacolley, P.; Regnault, V.; Nicoletti, A.; Li, Z.; Michel, J.B. The vascular smooth muscle cell in arterial pathology: A cell that can take on multiple roles. Cardiovasc. Res. 2012, 95, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Jung, F.; Haendeler, J.; Goebel, C.; Zeiher, A.M.; Dimmeler, S. Growth factor-induced phosphoinositide 3-OH kinase/Akt phosphorylation in smooth muscle cells: Induction of cell proliferation and inhibition of cell death. Cardiovasc. Res. 2000, 48, 148–157. [Google Scholar] [CrossRef]
- Aroni, F.; Iacovidou, N.; Dontas, I.; Pourzitaki, C.; Xanthos, T. Pharmacological aspects and potential new clinical applications of ketamine: Reevaluation of an old drug. J. Clin. Pharmacol. 2009, 49, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Niesters, M.; Khalili-Mahani, N.; Martini, C.; Aarts, L.; van Gerven, J.; van Buchem, M.A.; Dahan, A.; Rombouts, S. Effect of subanesthetic ketamine on intrinsic functional brain connectivity: A placebo-controlled functional magnetic resonance imaging study in healthy male volunteers. Anesthesiology 2012, 117, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Slikker, W., Jr.; Zou, X.; Hotchkiss, C.E.; Divine, R.L.; Sadovova, N.; Twaddle, N.C.; Doerge, D.R.; Scallet, A.C.; Patterson, T.A.; Hanig, J.P.; et al. Ketamine-induced neuronal cell death in the perinatal rhesus monkey. Toxicol. Sci. 2007, 98, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Hommes, D.W.; Peppelenbosch, M.P.; van Deventer, S.J. Mitogen activated protein (MAP) kinase signal transduction pathways and novel anti-inflammatory targets. Gut 2003, 52, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Higaki, M.; Shimokado, K. Phosphatidylinositol 3-kinase is required for growth factor-induced amino acid uptake by vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2127–2132. [Google Scholar] [CrossRef] [PubMed]
- Parcellier, A.; Tintignac, L.A.; Zhuravleva, E.; Hemmings, B.A. PKB and the mitochondria: AKTing on apoptosis. Cell. Signal. 2008, 20, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Fougerat, A.; Gayral, S.; Gourdy, P.; Schambourg, A.; Ruckle, T.; Schwarz, M.K.; Rommel, C.; Hirsch, E.; Arnal, J.F.; Salles, J.P.; et al. Genetic and pharmacological targeting of phosphoinositide 3-kinase-γ reduces atherosclerosis and favors plaque stability by modulating inflammatory processes. Circulation 2008, 117, 1310–1317. [Google Scholar] [CrossRef] [PubMed]
- Nakashio, A.; Fujita, N.; Tsuruo, T. Topotecan inhibits VEGF- and bFGF-induced vascular endothelial cell migration via down-regulation of the PI3K-Akt signaling pathway. Int. J. Cancer 2002, 98, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.K. Inflammation and plaque vulnerability. Cardiovasc. Drugs Ther. 2009, 23, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, C.; Haesen, D.; Sents, W.; Ivanova, E.; Janssens, V. Structure, regulation, and pharmacological modulation of PP2A phosphatases. Methods Mol. Biol. 2013, 1053, 283–305. [Google Scholar] [PubMed]
- Wandzioch, E.; Pusey, M.; Werda, A.; Bail, S.; Bhaskar, A.; Nestor, M.; Yang, J.J.; Rice, L.M. PME-1 modulates protein phosphatase 2A activity to promote the malignant phenotype of endometrial cancer cells. Cancer Res. 2014, 74, 4295–4305. [Google Scholar] [CrossRef] [PubMed]
- Okamura, H.; Yoshida, K.; Ochiai, K.; Haneji, T. Reduction of protein phosphatase 2A Cα enhances bone formation and osteoblast differentiation through the expression of bone-specific transcription factor Osterix. Bone 2011, 49, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Brautigan, D.L. Flicking the switches: Phosphorylation of serine/threonine protein phosphatases. Semin. Cancer Biol. 1995, 6, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Tolstykh, T.; Lee, J.; Vafai, S.; Stock, J.B. Carboxyl methylation regulates phosphoprotein phosphatase 2A by controlling the association of regulatory B subunits. EMBO J. 2000, 19, 5682–5691. [Google Scholar] [CrossRef] [PubMed]
- Longin, S.; Zwaenepoel, K.; Louis, J.V.; Dilworth, S.; Goris, J.; Janssens, V. Selection of protein phosphatase 2A regulatory subunits is mediated by the C terminus of the catalytic Subunit. J. Biol. Chem. 2007, 282, 26971–26980. [Google Scholar] [CrossRef] [PubMed]
- Damuni, Z.; Xiong, H.; Li, M. Autophosphorylation-activated protein kinase inactivates the protein tyrosine phosphatase activity of protein phosphatase 2A. FEBS Lett. 1994, 352, 311–314. [Google Scholar] [CrossRef]
- Baharians, Z.; Schonthal, A.H. Autoregulation of protein phosphatase type 2A expression. J. Biol. Chem. 1998, 273, 19019–19024. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Serine/threonine phosphatases: Mechanism through structure. Cell 2009, 139, 468–484. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Martin, B.L.; Brautigan, D.L. Regulation of protein serine–threonine phosphatase type-2A by tyrosine phosphorylation. Science 1992, 257, 1261–1264. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Lee, J.J.; Hsieh, C.Y.; Hsiao, G.; Chou, D.S.; Sheu, J.R. Inhibitory effects of ketamine on lipopolysaccharide-induced microglial activation. Mediat. Inflamm. 2009, 2009, 705379. [Google Scholar] [CrossRef] [PubMed]
- Shibakawa, Y.S.; Sasaki, Y.; Goshima, N.; Echigo, Y.; Kamiya, K.; Kurahashi, Y.; Yamada, T. AndohEffects of ketamine and propofol on inflammatory responses of primary glial cell cultures stimulated with lipopolysaccharide. Br. J. Anaesth. 2005, 95, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Domino, E.F.; Elemer, K.; Zsigmond, M.D.; Laurence, E.; Domino, M.D.; Kenneth, E.; Domino, B.S.; Sarla, P.; Kothary, M.B.; Steven, E. Plasma levels of ketamine and two of its metabolites in surgical patients using a gas chromatographic mass fragmentographic assay. Anesth. Analg. 1982, 61, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, G.; Shen, M.Y.; Chang, W.C.; Cheng, Y.W.; Pan, S.L.; Kuo, Y.H.; Chen, T.F.; Sheu, J.R. A novel antioxidant, octyl caffeate, suppression of LPS/IFN-γ-induced inducible nitric oxide synthase gene expression in rat aortic smooth muscle cells. Biochem. Pharmacol. 2003, 65, 383–1392. [Google Scholar] [CrossRef]
- Hsu, M.J.; Lee, S.S.; Lin, W.W.J. Polysaccharide purified from Ganoderma lucidum inhibits spontaneous and Fas-mediated apoptosis in human neutrophils through activation of the phosphatidylinositol 3 kinase/Akt signaling pathway. J. Leukoc. Biol. 2002, 72, 207–216. [Google Scholar] [PubMed]
- Hsieh, C.Y.; Liu, C.L.; Hsu, M.J.; Jayakumar, T.; Chou, D.S.; Wang, Y.H.; Hsiao, G.; Sheu, J.R. Inhibition of vascular smooth muscle cell proliferation by the vitamin E derivative pentamethyl hydroxychromane in an in vitro and in vivo study: Pivotal role of hydroxyl radical mediated PLCγ1 and JAK2 phosphorylation. Free Radic. Biol. Med. 2010, 49, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.Y.; Hsu, M.J.; Hsiao, G.; Wang, Y.H.; Huang, C.W.; Chen, S.W.; Jayakumar, T.; Chiu, P.T.; Chiu, Y.H.; Sheu, J.R. Andrographolide enhances nuclear factor-κB subunit p65 Ser536 dephosphorylation through activation of protein phosphatase 2A in vascular smooth muscle cells. J. Biol. Chem. 2011, 286, 5942–5955. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, Y.; Li, J.-Y.; Jayakumar, T.; Hung, S.-H.; Lee, W.-C.; Manubolu, M.; Sheu, J.-R.; Hsu, M.-J. Ketamine, a Clinically Used Anesthetic, Inhibits Vascular Smooth Muscle Cell Proliferation via PP2A-Activated PI3K/Akt/ERK Inhibition. Int. J. Mol. Sci. 2017, 18, 2545. https://doi.org/10.3390/ijms18122545
Chang Y, Li J-Y, Jayakumar T, Hung S-H, Lee W-C, Manubolu M, Sheu J-R, Hsu M-J. Ketamine, a Clinically Used Anesthetic, Inhibits Vascular Smooth Muscle Cell Proliferation via PP2A-Activated PI3K/Akt/ERK Inhibition. International Journal of Molecular Sciences. 2017; 18(12):2545. https://doi.org/10.3390/ijms18122545
Chicago/Turabian StyleChang, Yi, Jiun-Yi Li, Thanasekaran Jayakumar, Shou-Huang Hung, Wei-Cheng Lee, Manjunath Manubolu, Joen-Rong Sheu, and Ming-Jen Hsu. 2017. "Ketamine, a Clinically Used Anesthetic, Inhibits Vascular Smooth Muscle Cell Proliferation via PP2A-Activated PI3K/Akt/ERK Inhibition" International Journal of Molecular Sciences 18, no. 12: 2545. https://doi.org/10.3390/ijms18122545
APA StyleChang, Y., Li, J.-Y., Jayakumar, T., Hung, S.-H., Lee, W.-C., Manubolu, M., Sheu, J.-R., & Hsu, M.-J. (2017). Ketamine, a Clinically Used Anesthetic, Inhibits Vascular Smooth Muscle Cell Proliferation via PP2A-Activated PI3K/Akt/ERK Inhibition. International Journal of Molecular Sciences, 18(12), 2545. https://doi.org/10.3390/ijms18122545