
In Silico Structure and Sequence Analysis of Bacterial Porins and Specific Diffusion Channels for Hydrophilic Molecules: Conservation, Multimericity and Multifunctionality


Abstract
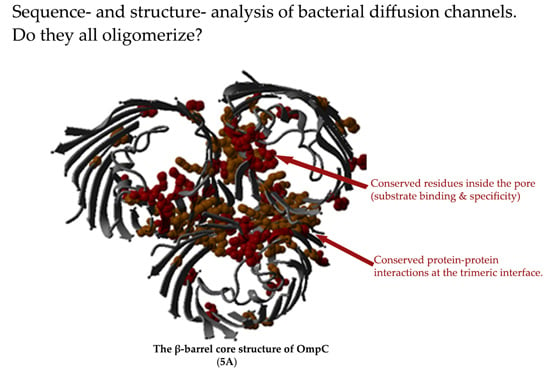
:
1. Introduction
- Non-specific trimeric porins (NTPs): NTPs, also known as general diffusion porins (GDPs), have 16-stranded β-barrels that allow the diffusion of hydrophilic substances smaller than 600 kDa [21].
- Specific monomeric diffusion channels (SMDCs): The oligogalacturonate-specific KdgM channel, outer membrane porin B (OprB) and outer membrane carboxylate channel (Occ) proteins belong to the SMDC group. KdgM is an acidic, sugar-specific channel with a 12-stranded β-barrel [24]. OprB is a carbohydrate-specific channel with a 16-stranded β-barrel [23]. Finally, Occ porins are water-soluble, specific channel for small substrates with a carboxyl group with 18-stranded β-barrels [28,29].
2. Results
2.1. Data Collection
2.2. Non-Specific Porin and Specific Diffusion Channel Secondary Structure Composition
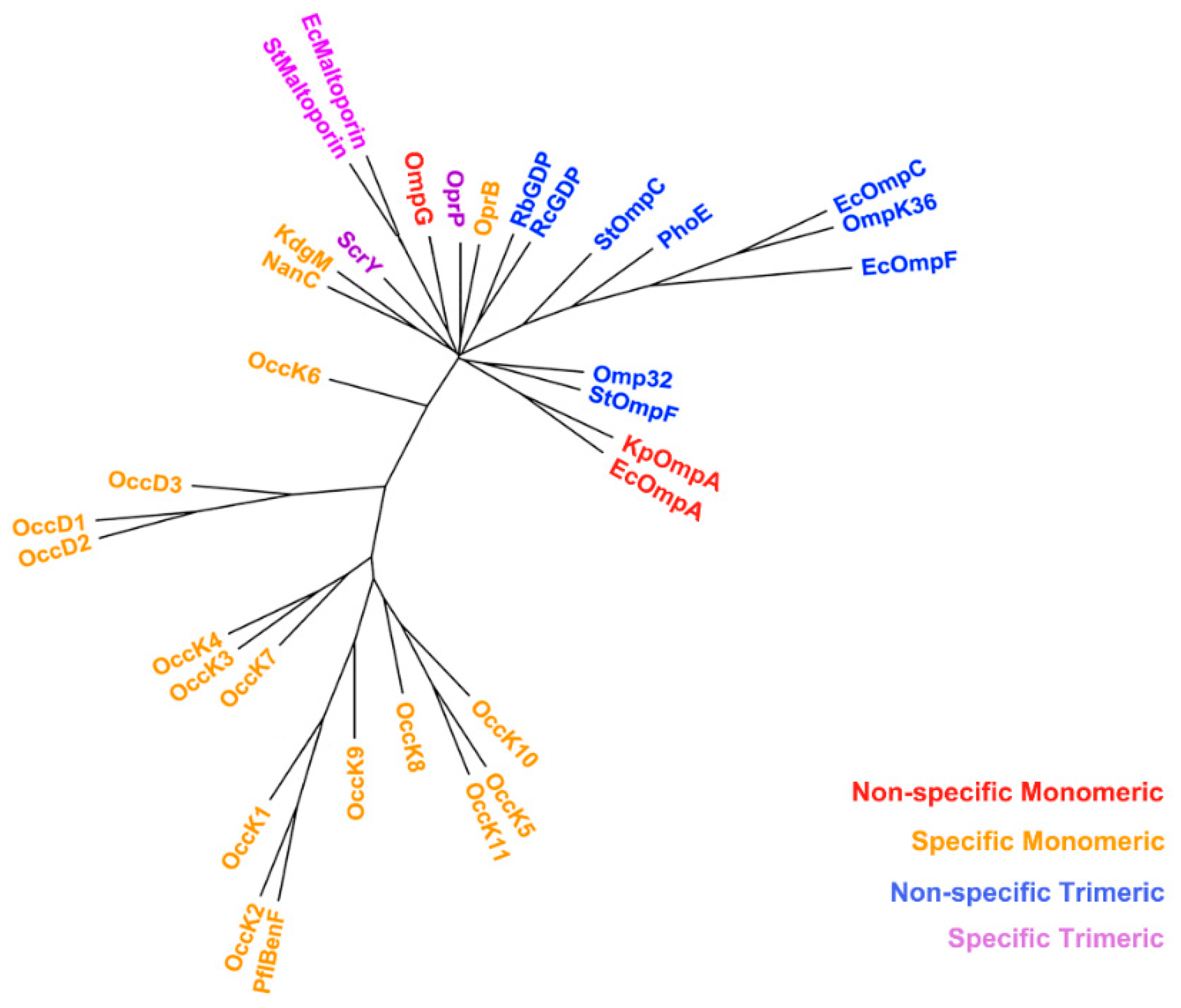
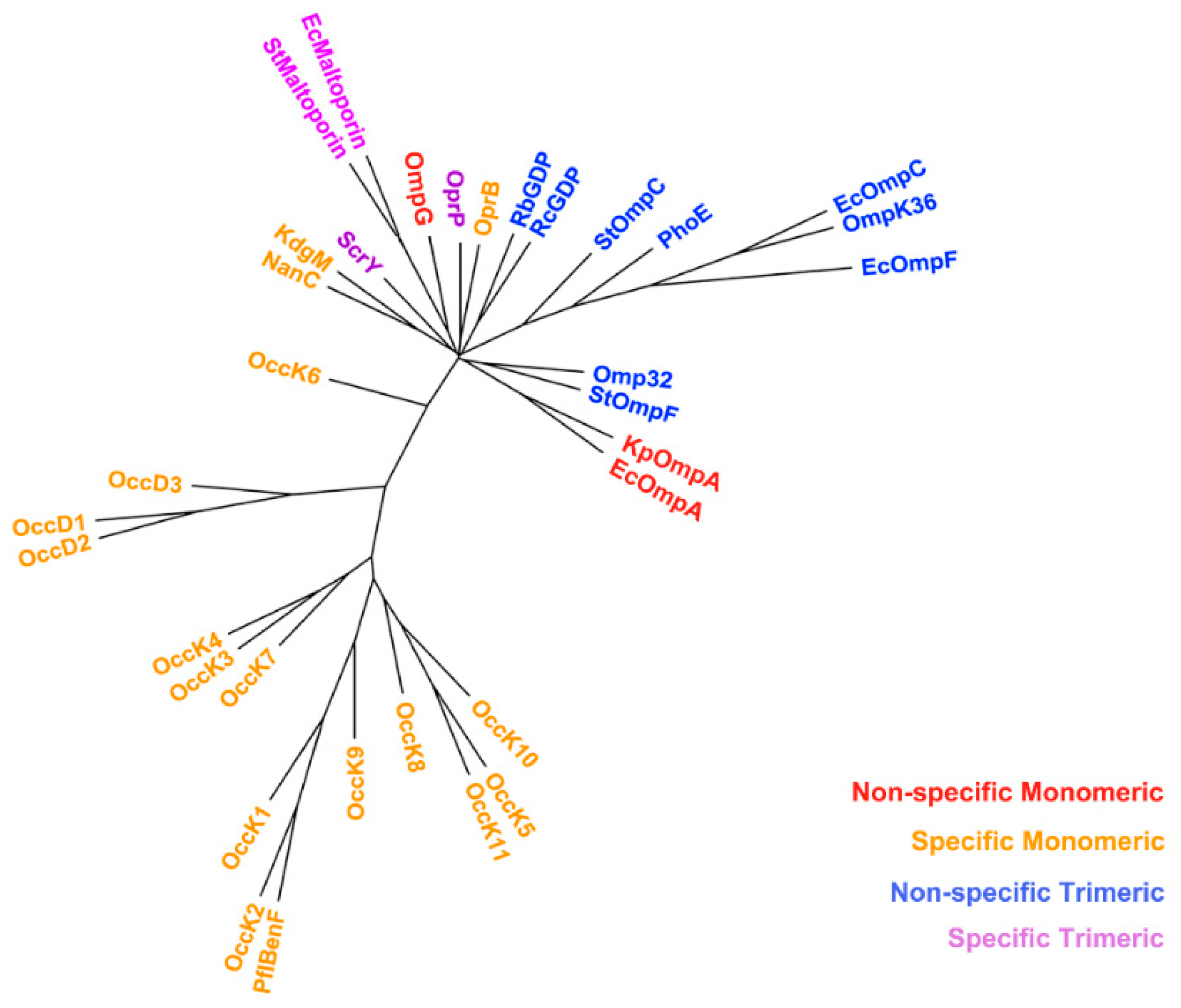
2.3. Non-Specific Porin and Specific Diffusion Channel Phylogenetic Tree
2.4. Non-Specific Porin and Specific Diffusion Channel Classification System
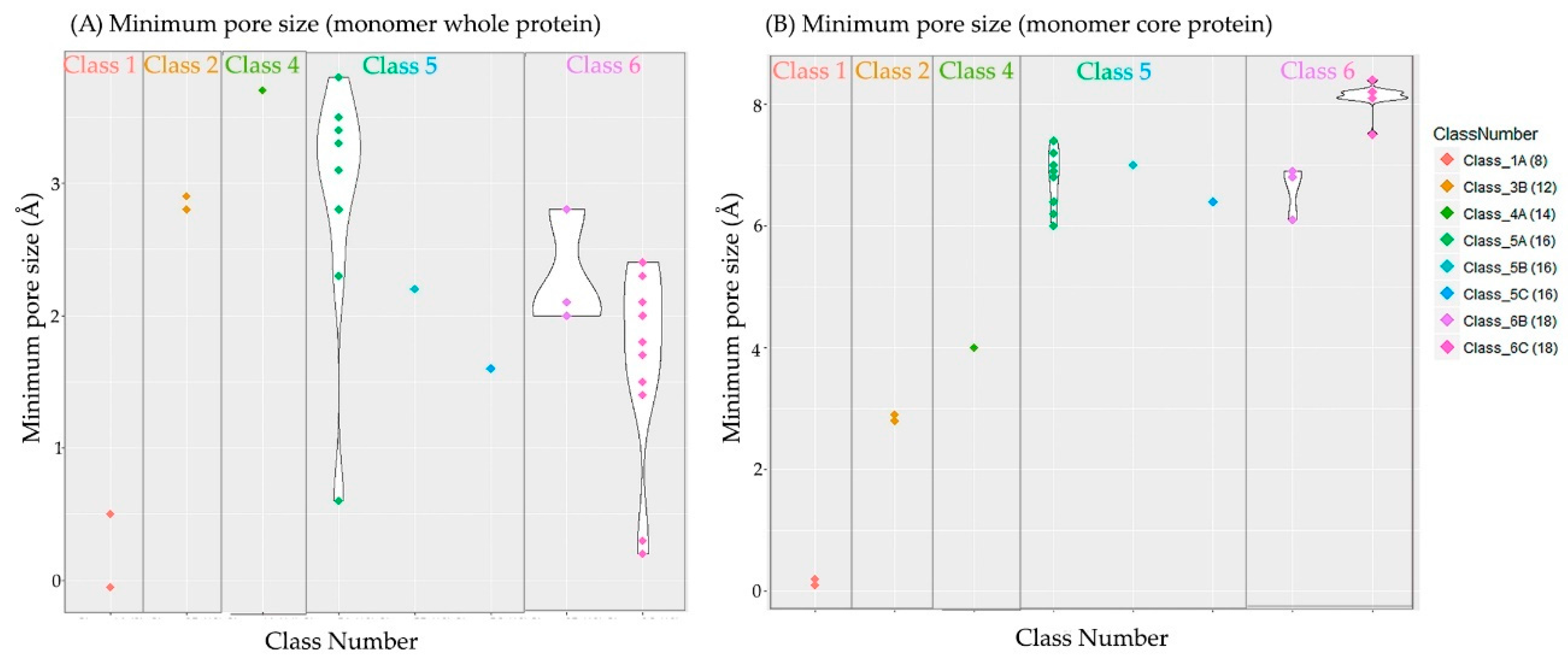
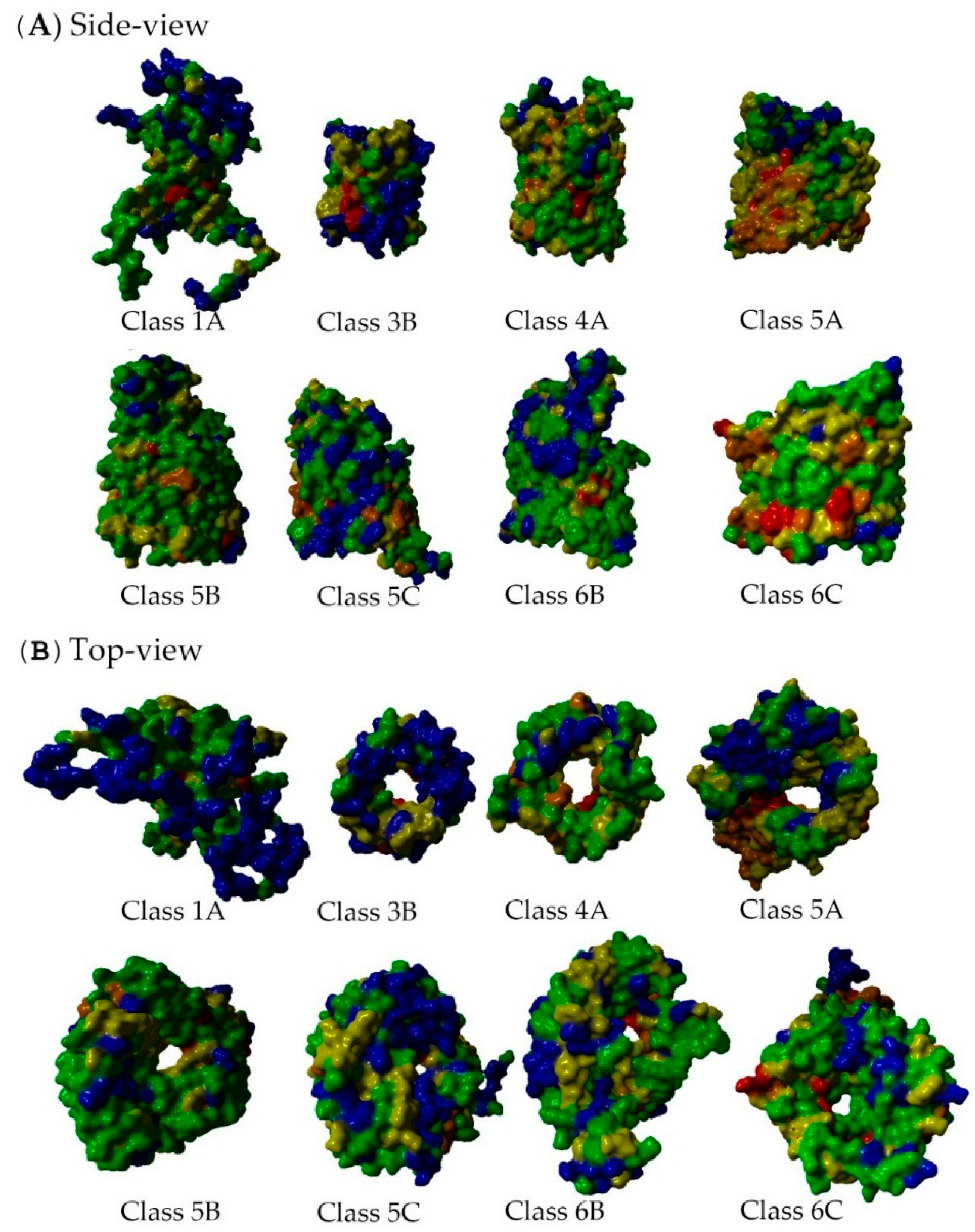
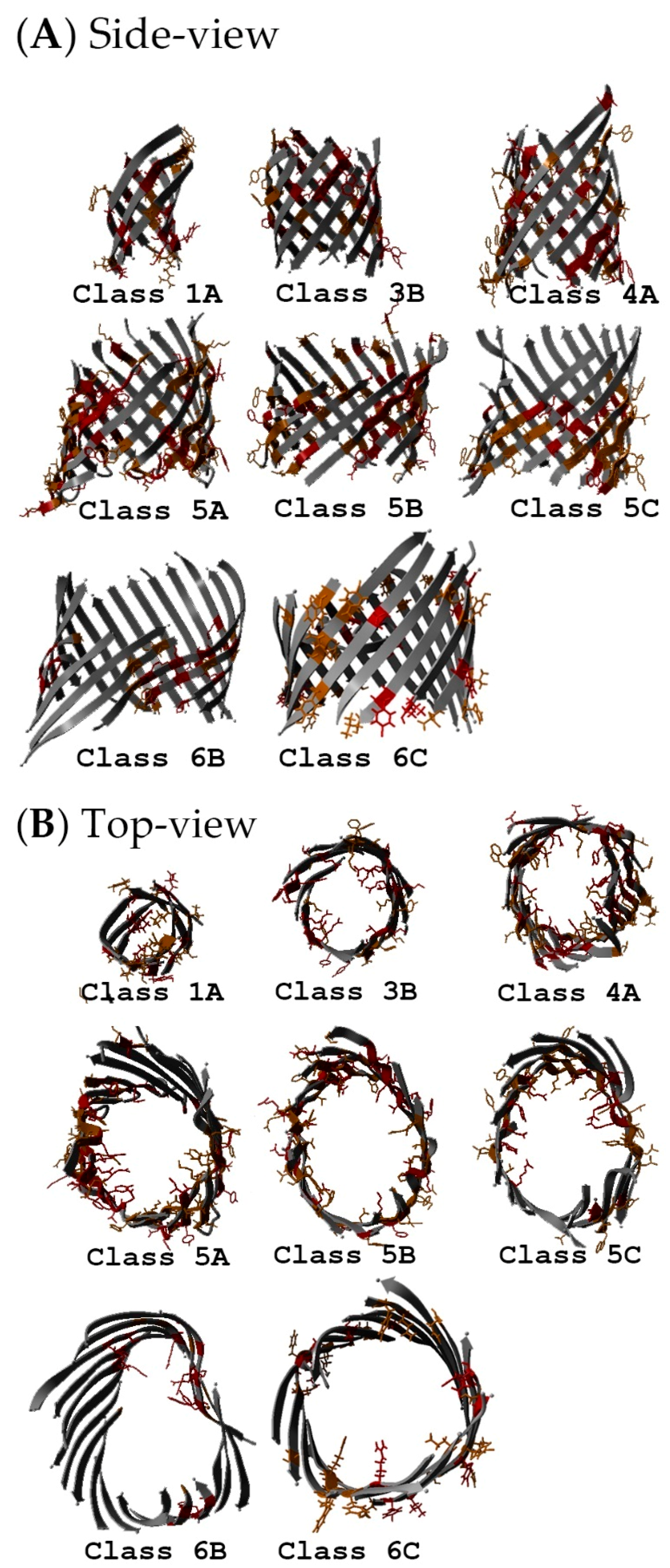
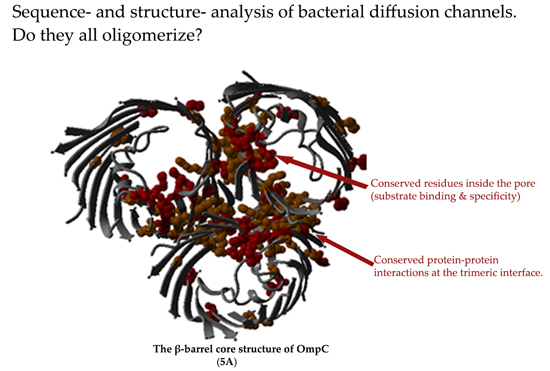
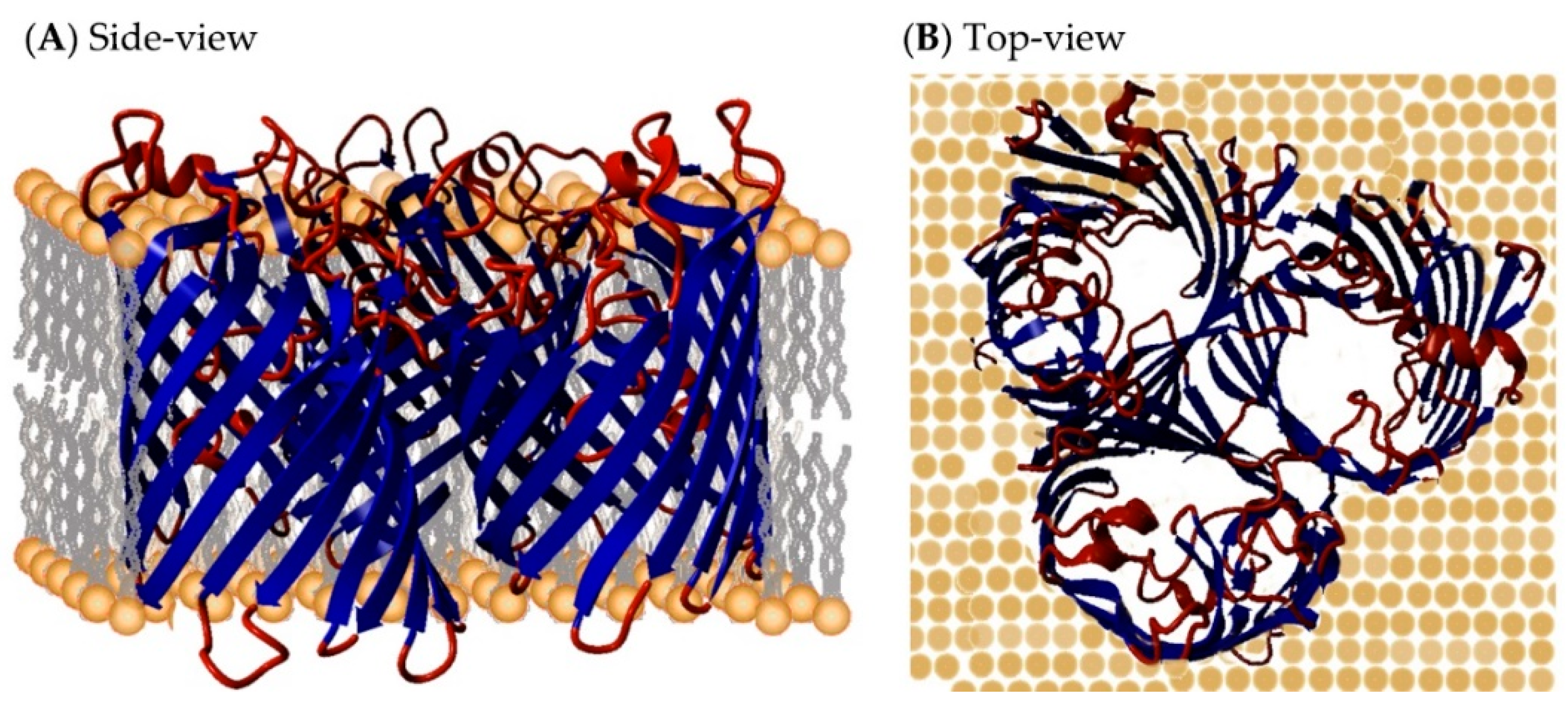
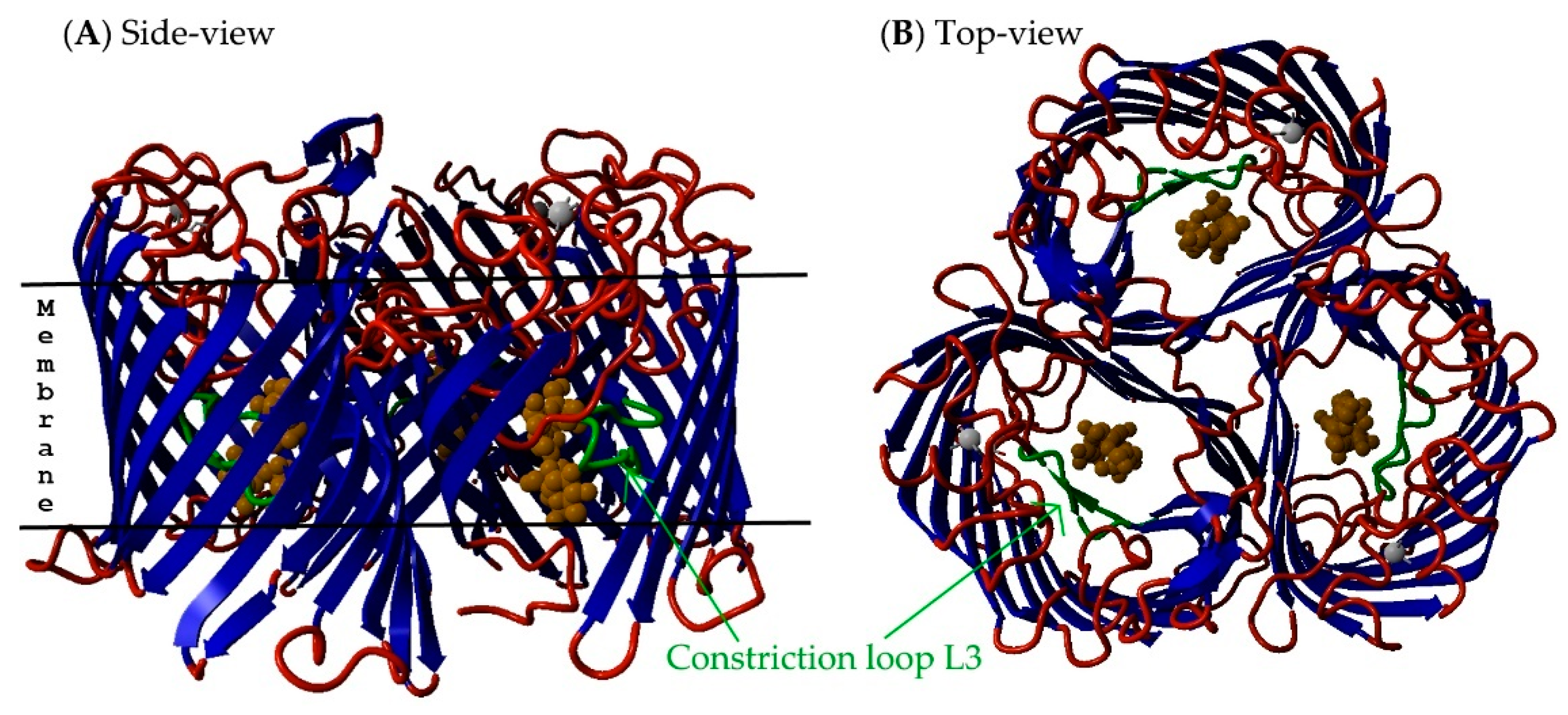
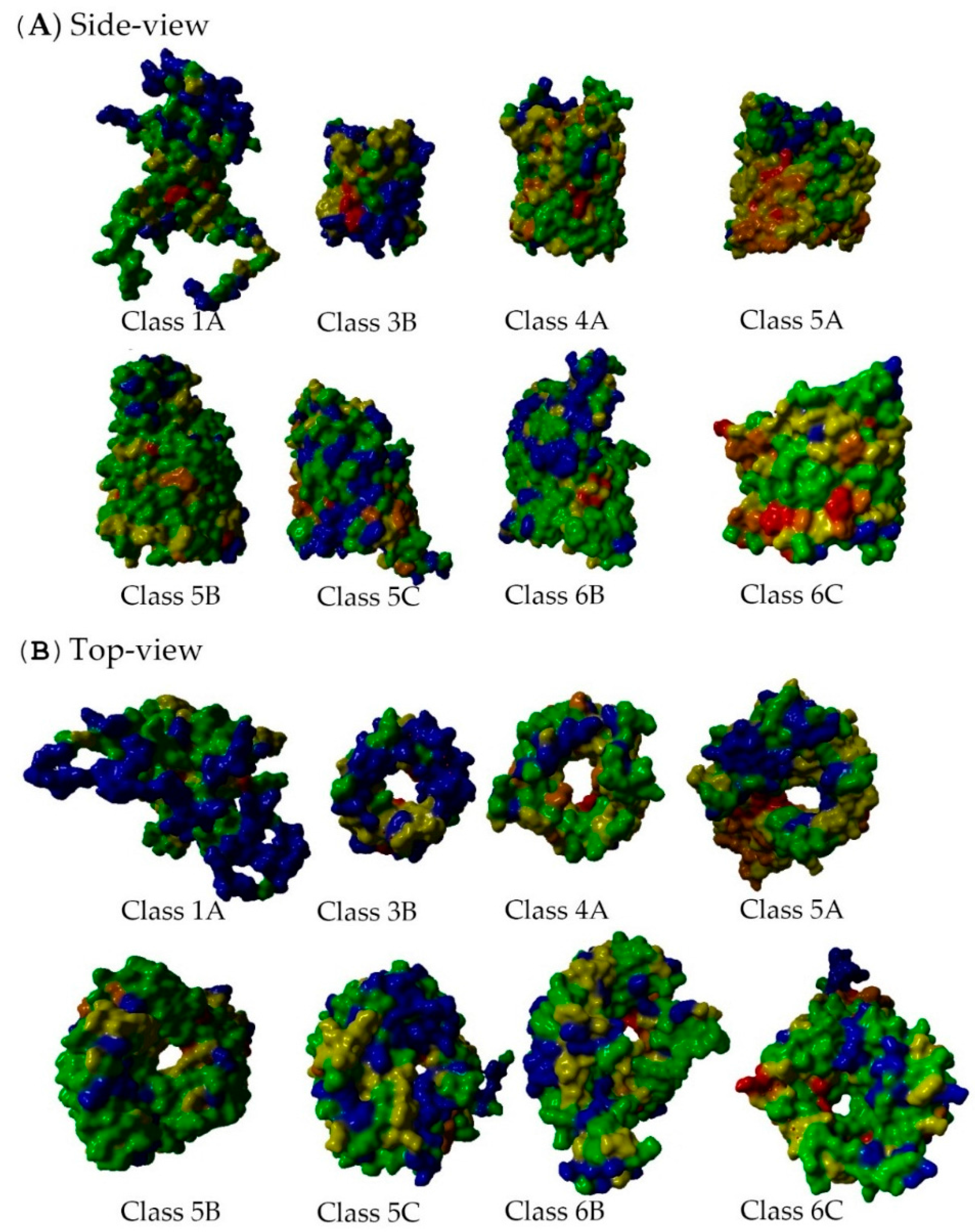
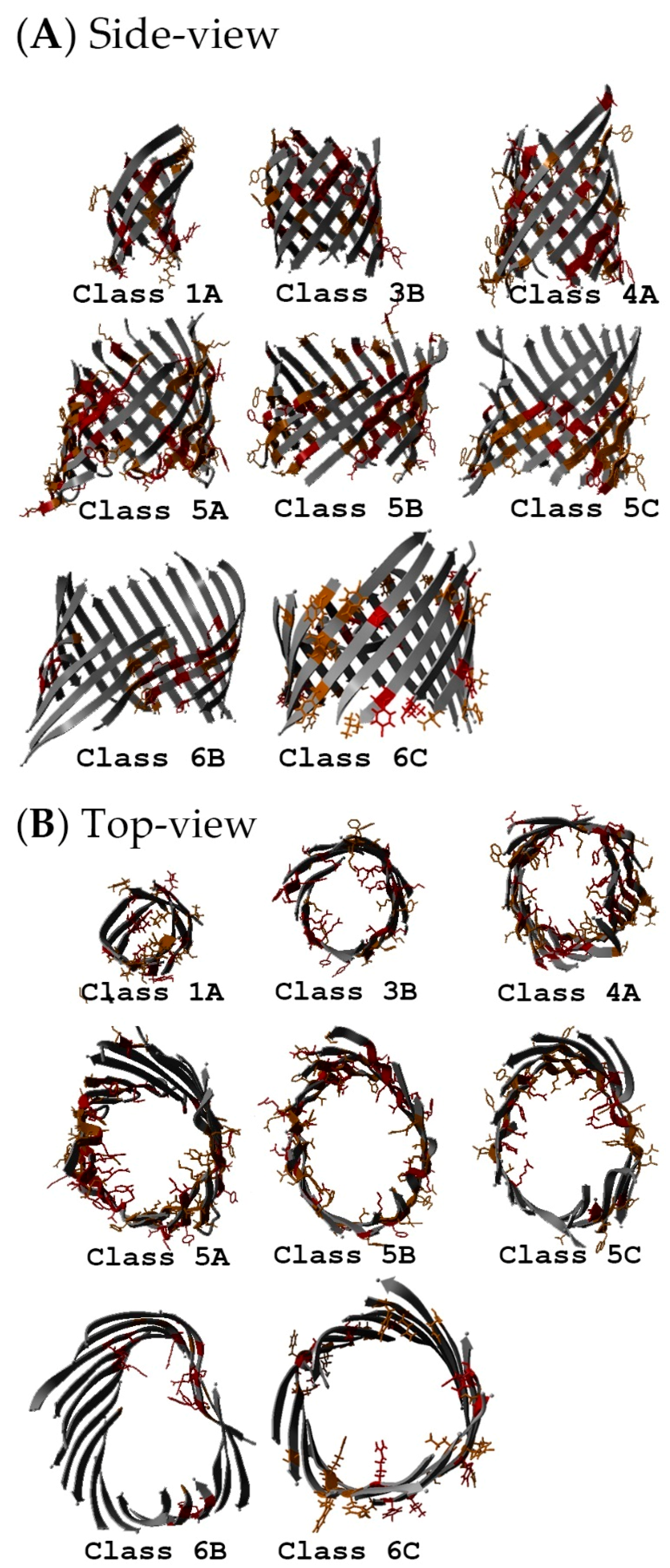
2.5. Non-Specific Porin and Specific Diffusion Channel Structure Analysis
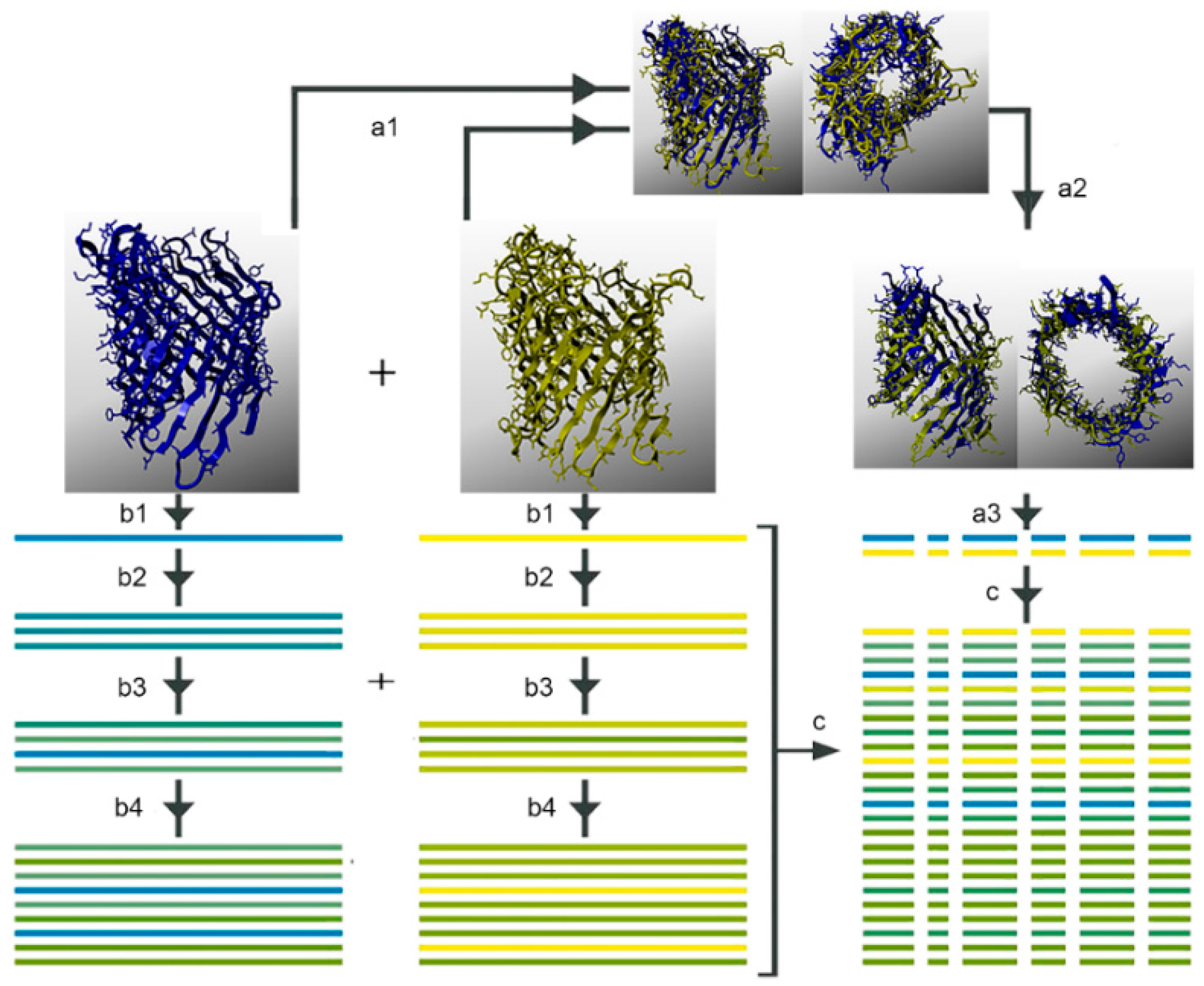
2.6. Multiple Sequence Alignments
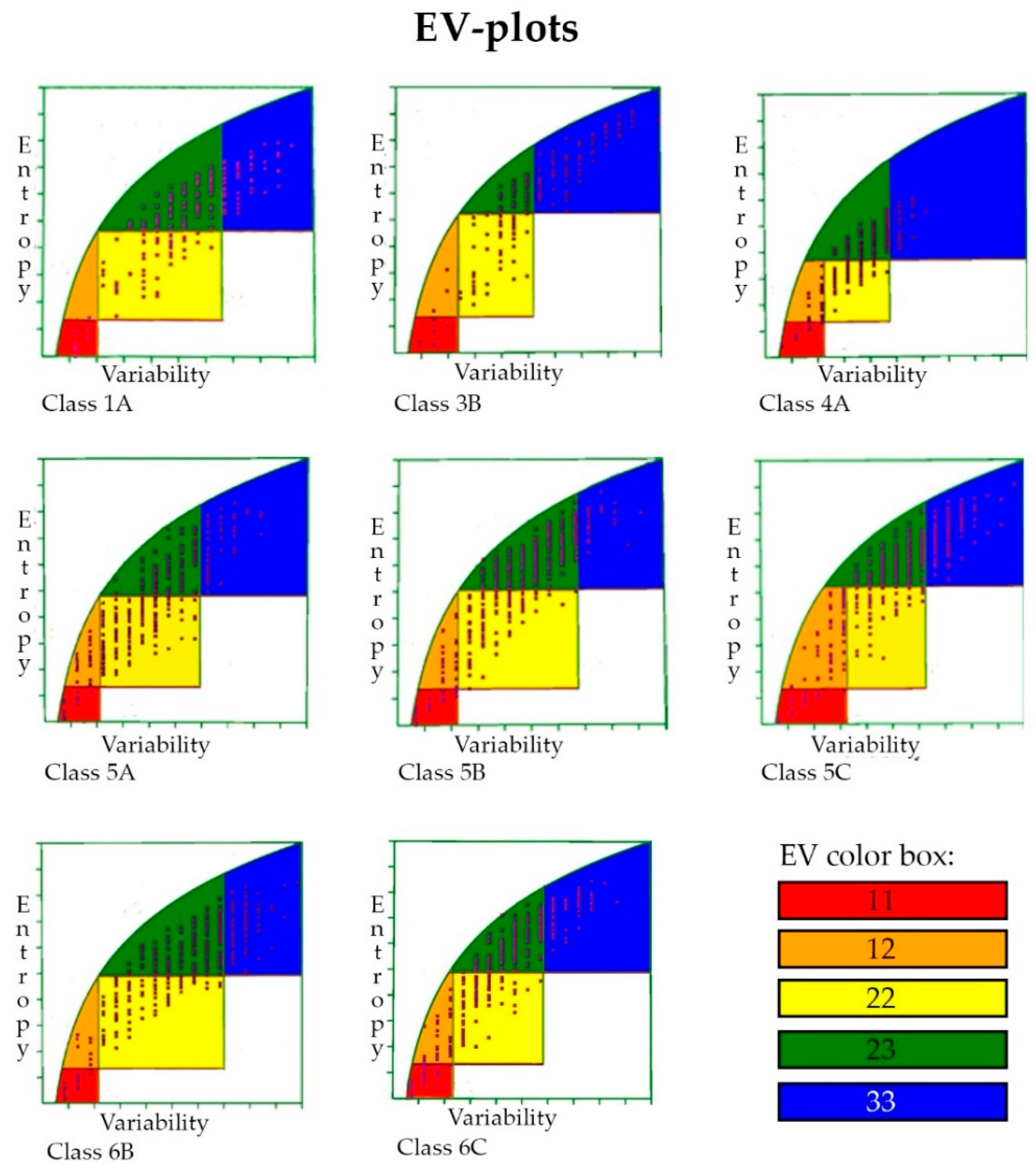
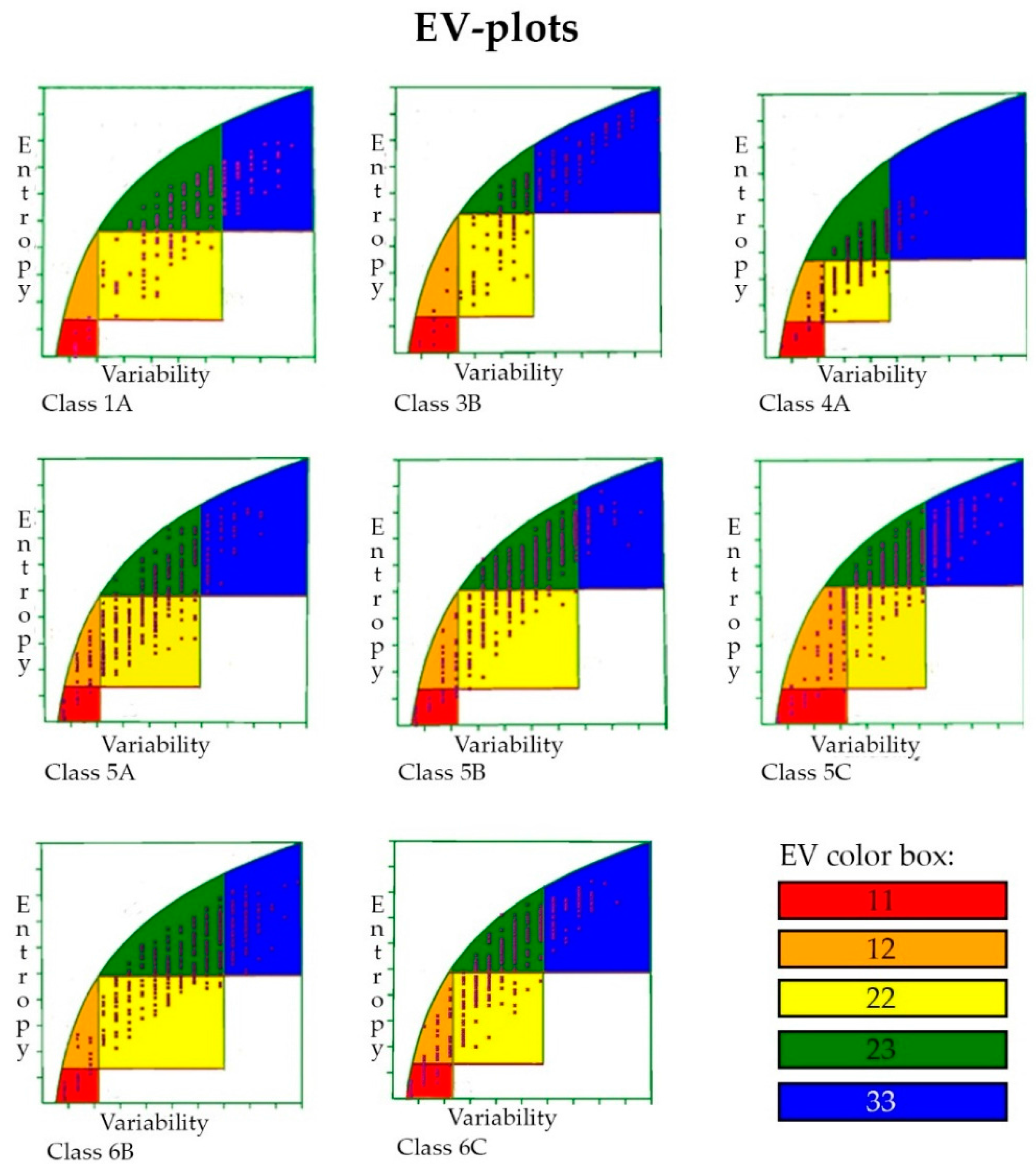
2.7. Entropy-Variability Analysis
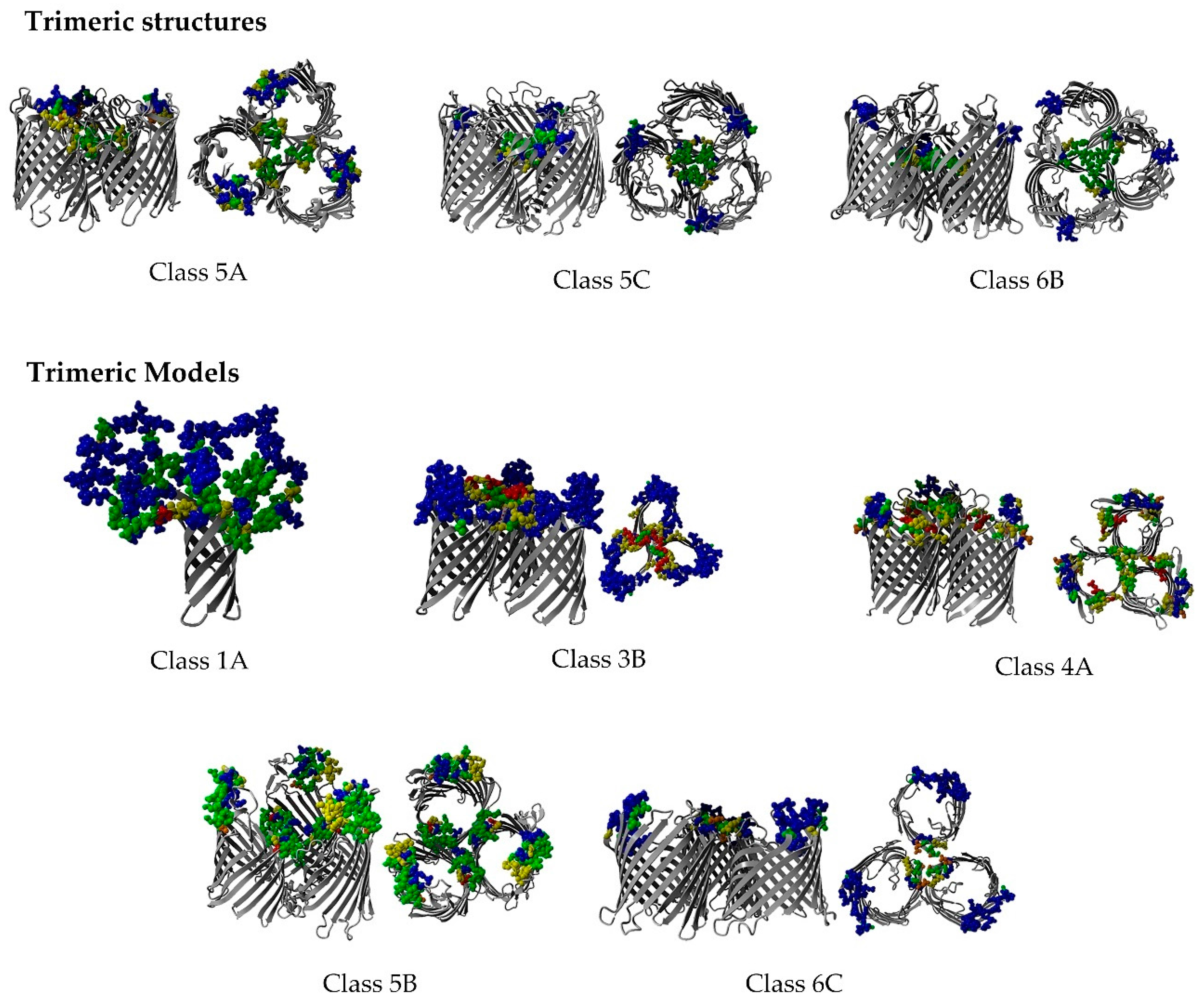
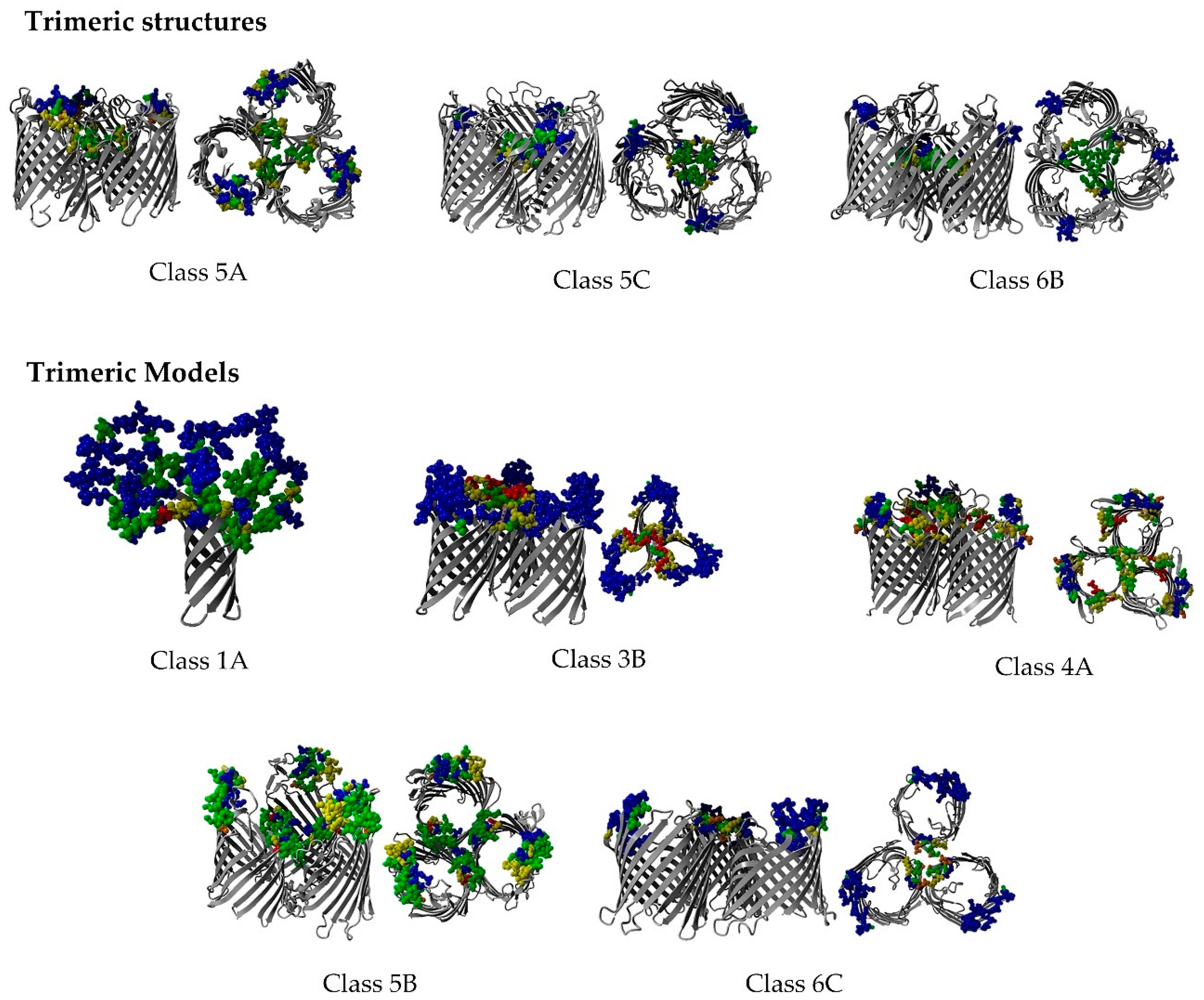
2.8. Non-Specific Porin and Specific Diffusion Channel Multimericity
3. Discussion
3.1. Data Collection and Secondary Structure Composition
3.2. Non-Specific Porin and Specific Diffusion Channel Phylogenetic Tree
3.3. Non-Specific Porin and Specific Diffusion Channel Classification System
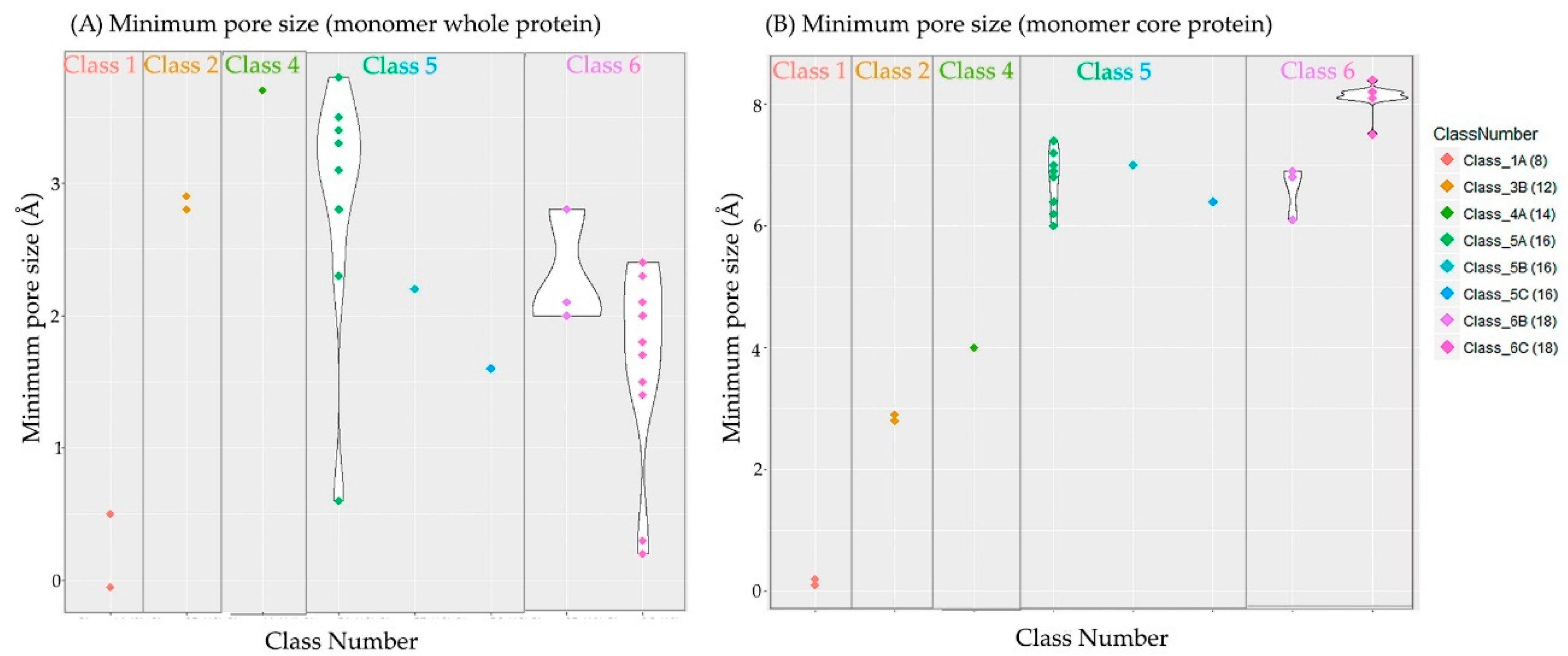
3.4. Non-Specific Porin and Specific Diffusion Channel Structure Analysis
3.5. Multiple Sequence Alignments
3.6. Entropy-Variability Analysis
3.7. Non-Specific Porin and Specific Diffusion Channel Multimericity
4. Materials and Methods
4.1. Non-Specific Porin and Specific Diffusion Channel Multimericity
4.2. Structure Analysis
4.3. Multiple Sequence Analysis
4.4. Sequence Filtering
4.5. Entropy-Variability Analysis
4.6. Phylogenetic Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AbOprD | Acinetobacter baumannii OprD |
| EcMaltoporin | E. coli maltoporin |
| EcOmp | E. coli outer membrane protein |
| EV | entropy-variability |
| EVA | entropy-variability analysis |
| GDP | general diffusion porin |
| KpOmpA | K. pneumoniae outer membrane protein A |
| MSA | multiple sequence alignment |
| NanC | N-acetylneuraminic acid-inducible outer-membrane channel |
| NMP | non-specific monomeric porin |
| NTP | non-specific trimeric porin |
| Occ | outer membrane carboxylate channel |
| OMP | outer membrane protein |
| OmpK36 | outer membrane porin of K. pneumoniae |
| OMPLA | outer membrane phospholipase A |
| Opr | outer membrane porin |
| PhoE | Phosphoporin |
| PDB | Protein Data Bank |
| RbGDP | R. blastica general diffusion porin |
| RcGDP | R. capsulatus general diffusion porin |
| RMSD | root-mean-square deviation |
| SMDC | specific monomeric diffusion channel |
| Salmonella Typhimurium | Salmonella enterica serovar Typhimurium |
| StMaltoporin | Salmonella Typhimurium maltoporin |
| StOmp | Salmonella Typhimurium outer membrane protein |
| STDC | specific trimeric diffusion channel |
Appendix A
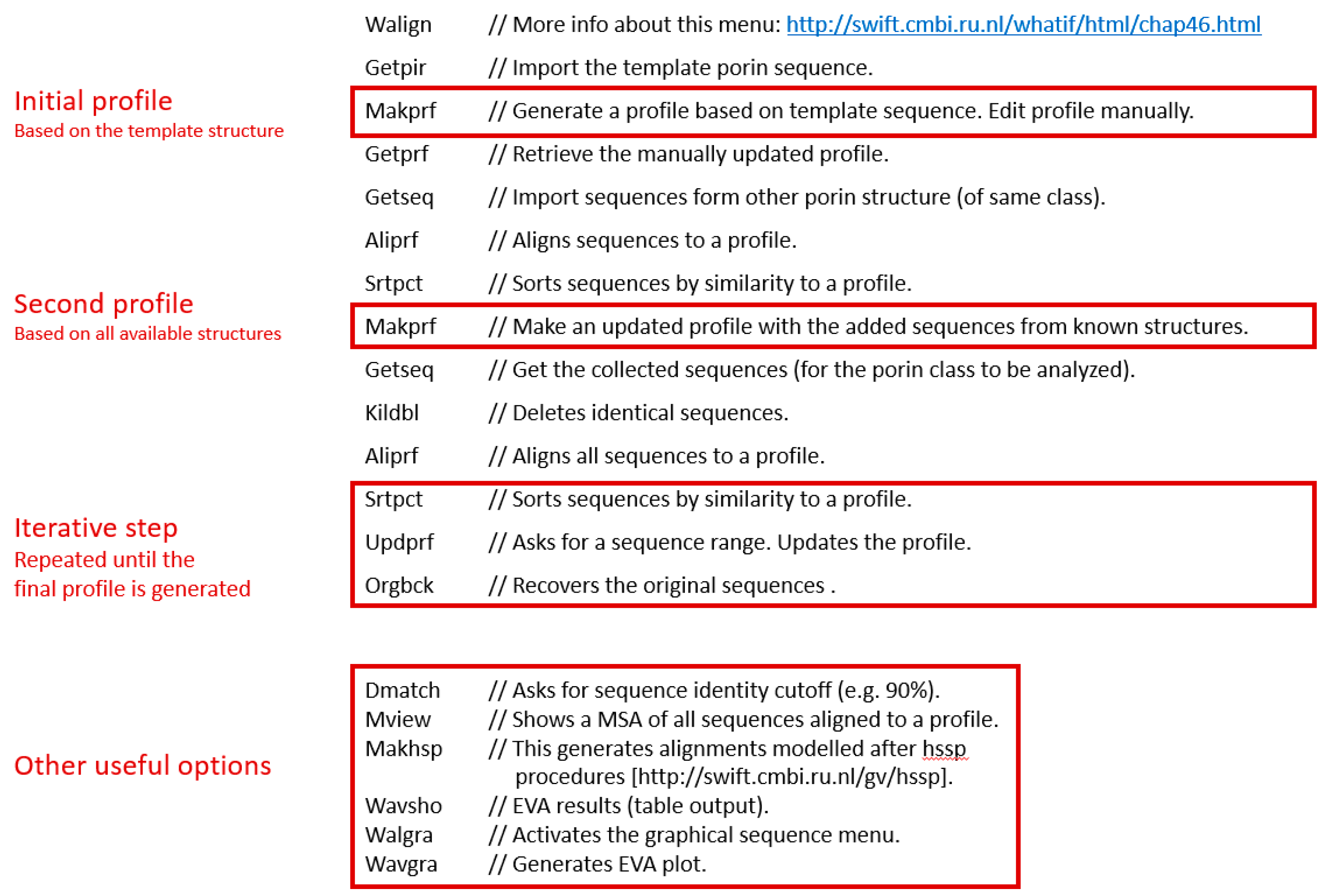
Iterative Profile Alignment Method
References
- Gupta, R.S. Origin of diderm (Gram-negative) bacteria: Antibiotic selection pressure rather than endosymbiosis likely led to the evolution of bacterial cells with two membranes. Antonie Van Leeuwenhoek 2011, 100, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Bos, M.P.; Robert, V.; Tommassen, J. Biogenesis of the gram-negative bacterial outer membrane. Annu. Rev. Microbiol. 2007, 61, 191–214. [Google Scholar] [CrossRef] [PubMed]
- Koebnik, R.; Locher, K.P.; van Gelder, P. Structure and function of bacterial outer membrane proteins: Barrels in a nutshell. Mol. Microbiol. 2000, 37, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Baslé, A.; Rummel, G.; Storici, P.; Rosenbusch, J.P.; Schirmer, T. Crystal structure of osmoporin OmpC from E. coli at 2.0 a. J. Mol. Biol. 2006, 362, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, S.; Falanga, A.; Cantisani, M.; Tarallo, R.; Della Pepa, M.E.; D'Oriano, V.; Galdiero, M. Microbe-host interactions: Structure and role of gram-negative bacterial porins. Curr. Protein Pept. Sci. 2012, 13, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Zakharov, S.D.; Eroukova, V.Y.; Rokitskaya, T.I.; Zhalnina, M.V.; Sharma, O.; Loll, P.J.; Zgurskaya, H.I.; Antonenko, Y.N.; Cramer, W.A. Colicin occlusion of OmpF and TolC channels: Outer membrane translocons for colicin import. Biophys. J. 2004, 87, 3901–3911. [Google Scholar] [CrossRef] [PubMed]
- Housden, N.G.; Wojdyla, J.A.; Korczynska, J.; Grishkovskaya, I.; Kirkpatrick, N.; Brzozowski, A.M.; Kleanthous, C. Directed epitope delivery across the Escherichia coli outer membrane through the porin OmpF. Proc. Natl. Acad. Sci. USA 2010, 107, 21412–21417. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Huang, S.; Zhang, Q. Outer membrane proteins: Key players for bacterial adaptation in host niches. Microb. Infect. 2002, 4, 325–331. [Google Scholar] [CrossRef]
- Achouak, W.; Heulin, T.; Pages, J.M. Multiple facets of bacterial porins. FEMS Microbiol. Lett. 2001, 199, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Pages, J.M.; James, C.E.; Winterhalter, M. The porin and the permeating antibiotic: A selective diffusion barrier in gram-negative bacteria. Nat. Rev. Microbiol. 2008, 6, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Forst, D.; Welte, W.; Wacker, T.; Diederichs, K. Structure of the sucrose-specific porin ScrY from Salmonella typhimurium and its complex with sucrose. Nat. Struct. Biol. 1998, 5, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, T. General and specific porins from bacterial outer membranes. J. Struct. Biol. 1998, 121, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Faraldo-Gómez, J.D.; Sansom, M.S. Acquisition of siderophores in gram-negative bacteria. Nat. Rev. Mol. Cell Biol. 2003, 4, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H. Porins and specific channels of bacterial outer membranes. Mol. Microbiol. 1992, 6, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Welte, W.; Nestel, U.; Wacker, T.; Diederichs, K. Structure and function of the porin channel. Kidney Int. 1995, 48, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Aguilella, V.M.; Queralt-Martín, M.; Alcaraz, A. Bacterial porins. In Electrophysiology of Unconventional Channels and Pores; Delcour, H.A., Ed.; Springer International Publishing: Cham, Albania, 2015; pp. 101–121. [Google Scholar]
- Jap, B.K.; Walian, P.J. Structure and functional mechanism of porins. Physiol. Rev. 1996, 76, 1073–1088. [Google Scholar] [PubMed]
- Cox, G.; Wright, G.D. Intrinsic antibiotic resistance: Mechanisms, origins, challenges and solutions. Int. J. Med. Microbiol. 2013, 303, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Freixeiro, P.; Dieguez-Casal, E.; Costoya, L.; Marzoa, J.; Ferreiros, C.M.; Criado, M.T.; Sanchez, S. High resolution clear native electrophoresis (hrCNE) allows a detailed analysis of the heterotrimeric structure of recombinant neisseria meningitidis porins inserted into liposomes. J. Proteome Res. 2013, 12, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Schulz, G.E. The structure of bacterial outer membrane proteins. Biochim. Biophys. Acta 2002, 1565, 308–317. [Google Scholar] [CrossRef]
- Smith, S.G.; Mahon, V.; Lambert, M.A.; Fagan, R.P. A molecular swiss army knife: OmpA structure, function and expression. FEMS Microbiol. Lett. 2007, 273, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, B. Structural basis for outer membrane sugar uptake in pseudomonads. J. Biol. Chem. 2012, 287, 41044–41052. [Google Scholar] [CrossRef] [PubMed]
- Wirth, C.; Condemine, G.; Boiteux, C.; Berneche, S.; Schirmer, T.; Peneff, C.M. NanC crystal structure, a model for outer-membrane channels of the acidic sugar-specific KdgM porin family. J. Mol. Biol. 2009, 394, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M. Facilitating Folding of Outer Membrane Proteins, Roles of the Periplasmic Chaperone Skp and the Outer Membrane Lipoprotein Bamd; Universität Kassel: Kassel, Germany, 2014. [Google Scholar]
- Whitelegge, J. Gas-phase structure of the E. coli OmpA dimer. Structure 2014, 22, 666–667. [Google Scholar] [CrossRef] [PubMed]
- Marcoux, J.; Politis, A.; Rinehart, D.; Marshall, D.P.; Wallace, M.I.; Tamm, L.K.; Robinson, C.V. Mass spectrometry defines the C-terminal dimerization domain and enables modeling of the structure of full-length OmpA. Structure 2014, 22, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Eren, E.; Vijayaraghavan, J.; Liu, J.; Cheneke, B.R.; Touw, D.S.; Lepore, B.W.; Indic, M.; Movileanu, L.; van den Berg, B. Substrate specificity within a family of outer membrane carboxylate channels. PLoS Biol. 2012, 10, e1001242. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Eren, E.; Vijayaraghavan, J.; Cheneke, B.R.; Indic, M.; van den Berg, B.; Movileanu, L. OccK channels from Pseudomonas aeruginosa exhibit diverse single-channel electrical signatures but conserved anion selectivity. Biochemistry 2012, 51, 2319–2330. [Google Scholar] [CrossRef] [PubMed]
- Moraes, T.F.; Bains, M.; Hancock, R.E.; Strynadka, N.C. An arginine ladder in OprP mediates phosphate-specific transfer across the outer membrane. Nat. Struct. Mol. Biol. 2007, 14, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; Dutzler, R.; Rizkallah, P.J.; Rosenbusch, J.P.; Schirmer, T. Channel specificity: Structural basis for sugar discrimination and differential flux rates in maltoporin. J. Mol. Biol. 1997, 272, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.; Paiva, P.B.; Paiva, A.C.; Vriend, G. Identification of functionally conserved residues with the use of entropy-variability plots. Proteins 2003, 52, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Sillitoe, I.; Lewis, T.E.; Cuff, A.; Das, S.; Ashford, P.; Dawson, N.L.; Furnham, N.; Laskowski, R.A.; Lee, D.; Lees, J.G.; et al. CATH: Comprehensive structural and functional annotations for genome sequences. Nucleic Acids Res. 2015, 43, D376–D381. [Google Scholar] [CrossRef] [PubMed]
- Saier, M.H., Jr.; Reddy, V.S.; Tamang, D.G.; Vastermark, A. The transporter classification database. Nucleic Acids Res. 2014, 42, D251–D258. [Google Scholar] [CrossRef] [PubMed]
- Tsirigos, K.D.; Bagos, P.G.; Hamodrakas, S.J. OMPdb: A database of β-barrel outer membrane proteins from gram-negative bacteria. Nucleic Acids Res. 2011, 39, D324–D331. [Google Scholar] [CrossRef] [PubMed]
- Fox, N.K.; Brenner, S.E.; Chandonia, J.M. Scope: Structural classification of proteins-extended, integrating scop and astral data and classification of new structures. Nucleic Acids Res. 2014, 42, D304–D309. [Google Scholar] [CrossRef] [PubMed]
- Smart, O.S.; Neduvelil, J.G.; Wang, X.; Wallace, B.A.; Sansom, M.S. HOLE: A program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 1996, 14, 354–360. [Google Scholar] [CrossRef]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with yasara nova—A self-parameterizing force field. Proteins 2002, 47, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Vriend, G. What if: A molecular modeling and drug design program. J. Mol. Graph. 1990, 8, 52–56. [Google Scholar] [CrossRef]
- Phale, P.S.; Philippsen, A.; Kiefhaber, T.; Koebnik, R.; Phale, V.P.; Schirmer, T.; Rosenbusch, J.P. Stability of trimeric OmpF porin: The contributions of the latching loop l2. Biochemistry 1998, 37, 15663–15670. [Google Scholar] [CrossRef] [PubMed]
- Tamm, L.K.; Hong, H.; Liang, B. Folding and assembly of β-barrel membrane proteins. Biochim. Biophys. Acta 2004, 1666, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Fairman, J.W.; Noinaj, N.; Buchanan, S.K. The structural biology of β-barrel membrane proteins: A summary of recent reports. Curr. Opin. Struct. Biol. 2011, 21, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Subbarao, G.V.; van den Berg, B. Crystal structure of the monomeric porin OmpG. J. Mol. Biol. 2006, 360, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Mari, S.A.; Köster, S.; Bippes, C.A.; Yildiz, O.; Kühlbrandt, W.; Muller, D.J. Ph-induced conformational change of the β-barrel-forming protein OmpG reconstituted into native E. coli lipids. J. Mol. Biol. 2010, 396, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Rokitskaya, T.I.; Kotova, E.A.; Naberezhnykh, G.A.; Khomenko, V.A.; Gorbach, V.I.; Firsov, A.M.; Zelepuga, E.A.; Antonenko, Y.N.; Novikova, O.D. Single channel activity of OmpF-like porin from yersinia pseudotuberculosis. Biochim. Biophys. Acta 2016, 1858, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Rostovtseva, T.K.; Nestorovich, E.M.; Bezrukov, S.M. Partitioning of differently sized poly(ethylene glycol)s into OmpF porin. Biophys. J. 2002, 82, 160–169. [Google Scholar] [CrossRef]
- Besya, A.B.; Mobasheri, H.; Ejtehadi, M.R. Gating and conduction of nano-channel forming proteins: A computational approach. J. Biomol. Struct. Dyn. 2013, 31, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Tamber, S.; Ochs, M.M.; Hancock, R.E. Role of the novel OprD family of porins in nutrient uptake in Pseudomonas aeruginosa. J. Bacteriol. 2006, 188, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Niramitranon, J.; Sansom, M.S.; Pongprayoon, P. Why do the outer membrane proteins OmpF from E. coli and OprP from P. aeruginosa prefer trimers? Simulation studies. J. Mol. Graph. Model. 2016, 65, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.S.; Re, S.; Wu, E.L.; Qi, Y.; Klebba, P.E.; Widmalm, G.; Yeom, M.S.; Sugita, Y.; Im, W. Dynamics and interactions of OmpF and LPS: Influence on pore accessibility and ion permeability. Biophys. J. 2016, 110, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Hancock, R.E. The role of specific surface loop regions in determining the function of the imipenem-specific pore protein oprd of Pseudomonas aeruginosa. J. Bacteriol. 1996, 178, 3085–3090. [Google Scholar] [PubMed]
- Folkertsma, S.; van Noort, P.; Van Durme, J.; Joosten, H.J.; Bettler, E.; Fleuren, W.; Oliveira, L.; Horn, F.; de Vlieg, J.; Vriend, G. A family-based approach reveals the function of residues in the nuclear receptor ligand-binding domain. J. Mol. Biol. 2004, 341, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.; Paiva, P.B.; Paiva, A.C.; Vriend, G. Sequence analysis reveals how g protein-coupled receptors transduce the signal to the g protein. Proteins 2003, 52, 553–560. [Google Scholar] [CrossRef] [PubMed]
- LeClerc, J.E.; Li, B.; Payne, W.L.; Cebula, T.A. High mutation frequencies among Escherichia coli and salmonella pathogens. Science 1996, 274, 1208–1211. [Google Scholar] [CrossRef] [PubMed]
- Easton, D. Bacterial outer membrane porins—Are they decoy agents. Aust. J. Med. Sci. 2005, 26, 1038–1643. [Google Scholar]
- Iordanov, I.; Renault, M.; Reat, V.; Bosshart, P.D.; Engel, A.; Saurel, O.; Milon, A. Dynamics of Klebsiella pneumoniae OmpA transmembrane domain: The four extracellular loops display restricted motion behavior in micelles and in lipid bilayers. Biochim. Biophys. Acta 2012, 1818, 2344–2353. [Google Scholar] [CrossRef] [PubMed]
- Power, M.L.; Ferrari, B.C.; Littlefield-Wyer, J.; Gordon, D.M.; Slade, M.B.; Veal, D.A. A naturally occurring novel allele of Escherichia coli outer membrane protein A reduces sensitivity to bacteriophage. Appl. Environ. Microbiol. 2006, 72, 7930–7932. [Google Scholar] [CrossRef] [PubMed]
- Pezeshki, S.; Chimerel, C.; Bessonov, A.N.; Winterhalter, M.; Kleinekathofer, U. Understanding ion conductance on a molecular level: An all-atom modeling of the bacterial porin OmpF. Biophys. J. 2009, 97, 1898–1906. [Google Scholar] [CrossRef] [PubMed]
- Eppens, E.F.; Saint, N.; van Gelder, P.; van Boxtel, R.; Tommassen, J. Role of the constriction loop in the gating of outer membrane porin phoe of Escherichia coli. FEBS Lett. 1997, 415, 317–320. [Google Scholar] [CrossRef]
- Sugawara, E.; Nikaido, H. Pore-forming activity of OmpA protein of Escherichia coli. J. Biol. Chem. 1992, 267, 2507–2511. [Google Scholar] [PubMed]
- Pautsch, A.; Schulz, G.E. Structure of the outer membrane protein A transmembrane domain. Nat. Struct. Biol. 1998, 5, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Pautsch, A.; Schulz, G.E. High-resolution structure of the OmpA membrane domain. J. Mol. Biol. 2000, 298, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Kleinschmidt, J.H. Membrane protein folding on the example of outer membrane protein A of Escherichia coli. Cell. Mol. Life Sci. 2003, 60, 1547–1558. [Google Scholar] [CrossRef] [PubMed]
- Dieguez-Casal, E.; Freixeiro, P.; Costoya, L.; Criado, M.T.; Ferreiros, C.; Sanchez, S. High resolution clear native electrophoresis is a good alternative to blue native electrophoresis for the characterization of the Escherichia coli membrane complexes. J. Microbiol. Methods 2014, 102, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Stenberg, F.; Chovanec, P.; Maslen, S.L.; Robinson, C.V.; Ilag, L.L.; von Heijne, G.; Daley, D.O. Protein complexes of the escherichia coli cell envelope. J. Biol. Chem. 2005, 280, 34409–34419. [Google Scholar] [CrossRef] [PubMed]
- Dovling Kaspersen, J.; Moestrup Jessen, C.; Stougaard Vad, B.; Skipper Sorensen, E.; Kleiner Andersen, K.; Glasius, M.; Pinto Oliveira, C.L.; Otzen, D.E.; Pedersen, J.S. Low-resolution structures of OmpA*DDM protein-detergent complexes. Chembiochem 2014, 15, 2113–2124. [Google Scholar] [CrossRef] [PubMed]
- Patel, G.J.; Kleinschmidt, J.H. The lipid bilayer-inserted membrane protein BamA of Escherichia coli facilitates insertion and folding of outer membrane protein A from its complex with Skp. Biochemistry 2013, 52, 3974–3986. [Google Scholar] [CrossRef] [PubMed]
- Manchur, M.A.; Kikumoto, M.; Kanao, T.; Takada, J.; Kamimura, K. Characterization of an OmpA-like outer membrane protein of the acidophilic iron-oxidizing bacterium, acidithiobacillus ferrooxidans. Extremophiles 2011, 15, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Nagano, K.; Read, E.K.; Murakami, Y.; Masuda, T.; Noguchi, T.; Yoshimura, F. Trimeric structure of major outer membrane proteins homologous to OmpA in Porphyromonas gingivalis. J. Bacteriol. 2005, 187, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Andersen, K.K.; Vad, B.S.; Otzen, D.E. OmpA can form folded and unfolded oligomers. Biochim. Biophys. Acta 2013, 1834, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Hutter, C.A.; Lehner, R.; Wirth, C.; Condemine, G.; Peneff, C.; Schirmer, T. Structure of the oligogalacturonate-specific KdgM porin. Acta Crystallogr. 2014, 70, 1770–1778. [Google Scholar] [CrossRef] [PubMed]
- Pellinen, T.; Ahlfors, H.; Blot, N.; Condemine, G. Topology of the erwinia chrysanthemi oligogalacturonate porin KdgM. Biochem. J. 2003, 372, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Blot, N.; Berrier, C.; Hugouvieux-Cotte-Pattat, N.; Ghazi, A.; Condemine, G. The oligogalacturonate-specific porin KdgM of erwinia chrysanthemi belongs to a new porin family. J. Biol. Chem. 2002, 277, 7936–7944. [Google Scholar] [CrossRef] [PubMed]
- Dunton, T.A.; Goose, J.E.; Gavaghan, D.J.; Sansom, M.S.; Osborne, J.M. The free energy landscape of dimerization of a membrane protein, NanC. PLoS Comput. Biol. 2014, 10, e1003417. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, O.; Vinothkumar, K.R.; Goswami, P.; Kühlbrandt, W. Structure of the monomeric outer-membrane porin OmpG in the open and closed conformation. EMBO J. 2006, 25, 3702–3713. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, T.; Chisholm, C.; Chen, M.; Tamm, L.K. NMR-based conformational ensembles explain pH-gated opening and closing of OmpG channel. J. Am. Chem. Soc. 2013, 135, 15101–15113. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Tamm, L.K. Structure of outer membrane protein G by solution nmr spectroscopy. Proc. Natl. Acad. Sci. USA 2007, 104, 16140–16145. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, D.A.; Cheung, J.; Ito, C.; Sugawara, E.; Nikaido, H.; Misra, R. Biochemistry and regulation of a novel Escherichia coli K-12 porin protein, OmpG, which produces unusually large channels. J. Bacteriol. 1998, 180, 4452–4459. [Google Scholar] [PubMed]
- Schiltz, E.; Kreusch, A.; Nestel, U.; Schulz, G.E. Primary structure of porin from Rhodobacter capsulatus. Eur. J. Biochem. FEBS 1991, 199, 587–594. [Google Scholar] [CrossRef]
- Balasubramaniam, D.; Arockiasamy, A.; Kumar, P.D.; Sharma, A.; Krishnaswamy, S. Asymmetric pore occupancy in crystal structure of OmpF porin from Salmonella typhi. J. Struct. Biol. 2012, 178, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Dutzler, R.; Rummel, G.; Alberti, S.; Hernandez-Alles, S.; Phale, P.; Rosenbusch, J.; Benedi, V.; Schirmer, T. Crystal structure and functional characterization of OmpK36, the osmoporin of Klebsiella pneumoniae. Structure 1999, 7, 425–434. [Google Scholar] [CrossRef]
- Weiss, M.S.; Schulz, G.E. Structure of porin refined at 1.8 Å resolution. J. Mol. Biol. 1992, 227, 493–509. [Google Scholar] [CrossRef]
- Efremov, R.G.; Sazanov, L.A. Structure of Escherichia coli OmpF porin from lipidic mesophase. J. Struct. Biol. 2012, 178, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Zachariae, U.; Kluhspies, T.; De, S.; Engelhardt, H.; Zeth, K. High resolution crystal structures and molecular dynamics studies reveal substrate binding in the porin Omp32. J. Biol. Chem. 2006, 281, 7413–7420. [Google Scholar] [CrossRef] [PubMed]
- Cowan, S.W.; Schirmer, T.; Rummel, G.; Steiert, M.; Ghosh, R.; Pauptit, R.A.; Jansonius, J.N.; Rosenbusch, J.P. Crystal structures explain functional properties of two E. coli porins. Nature 1992, 358, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Kreusch, A.; Neubuser, A.; Schiltz, E.; Weckesser, J.; Schulz, G.E. Structure of the membrane channel porin from Rhodopseudomonas blastica at 2.0 Å resolution. Protein Sci. 1994, 3, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Adewoye, L.O.; Tschetter, L.; O’Neil, J.; Worobec, E.A. Channel specificity and secondary structure of the glucose-inducible porins of Pseudomonas spp. J. Bioenerg. Biomembr. 1998, 30, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, R.; Basu, B.; Godbole, A.; Mathew, M.K.; Apte, S.K.; Phale, P.S. Repression of the glucose-inducible outer-membrane protein OprB during utilization of aromatic compounds and organic acids in Pseudomonas putida CSV86. Microbiology 2011, 157, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Pongprayoon, P.; Beckstein, O.; Wee, C.L.; Sansom, M.S. Simulations of anion transport through OprP reveal the molecular basis for high affinity and selectivity for phosphate. Proc. Natl. Acad. Sci. USA 2009, 106, 21614–21618. [Google Scholar] [CrossRef] [PubMed]
- Angus, B.L.; Hancock, R.E. Outer membrane porin proteins F, P, and D1 of Pseudomonas aeruginosa and PhoE of Escherichia coli: Chemical cross-linking to reveal native oligomers. J. Bacteriol. 1983, 155, 1042–1051. [Google Scholar] [PubMed]
- Hancock, R.E.; Egli, C.; Benz, R.; Siehnel, R.J. Overexpression in Escherichia coli and functional analysis of a novel PPi-selective porin, oprO, from Pseudomonas aeruginosa. J. Bacteriol. 1992, 174, 471–476. [Google Scholar] [PubMed]
- Meyer, J.E.; Hofnung, M.; Schulz, G.E. Structure of maltoporin from salmonella typhimurium ligated with a nitrophenyl-maltotrioside. J. Mol. Biol. 1997, 266, 761–775. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Mohammad, M.M.; Patel, D.R.; Movileanu, L.; van den Berg, B. Structural insight into OprD substrate specificity. Nat. Struct. Mol. Biol. 2007, 14, 1108–1109. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Mohammad, M.M.; Movileanu, L.; van den Berg, B. Crystal structure of the outer membrane protein OpdK from Pseudomonas aeruginosa. Structure 2008, 16, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Catel-Ferreira, M.; Nehme, R.; Molle, V.; Aranda, J.; Bouffartigues, E.; Chevalier, S.; Bou, G.; Jouenne, T.; Dé, E. Deciphering the function of the outer membrane protein OprD homologue of Acinetobacter baumannii. Antimicrob. Agent Chemother. 2012, 56, 3826–3832. [Google Scholar] [CrossRef] [PubMed]
- Ishida, H.; Garcia-Herrero, A.; Vogel, H.J. The periplasmic domain of Escherichia coli outer membrane protein A can undergo a localized temperature dependent structural transition. Biochim. Biophys. Acta 2014, 1838, 3014–3024. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.; LeDoyt, M.; Karanian, C.; Luthra, A.; Koszelak-Rosenblum, M.; Malkowski, M.G.; Puthenveetil, R.; Vinogradova, O.; Radolf, J.D. Bipartite topology of treponema pallidum repeat proteins C/D and I: Outer membrane insertion, trimerization, and porin function require a C-terminal β-barrel domain. J. Biol. Chem. 2015, 290, 12313–12331. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Zakharov, S.; Hasan, S.S.; Ryan, C.M.; Whitelegge, J.P.; Cramer, W.A. Structure-function of cyanobacterial outer-membrane protein, slr1270: Homolog of Escherichia coli drug export/colicin import protein, TolC. FEBS Lett. 2014, 588, 3793–3801. [Google Scholar] [CrossRef] [PubMed]
- Baaden, M.; Meier, C.; Sansom, M.S. A molecular dynamics investigation of mono and dimeric states of the outer membrane enzyme OMPLA. J. Mol. Biol. 2003, 331, 177–189. [Google Scholar] [CrossRef]
- Adamian, L.; Naveed, H.; Liang, J. Lipid-binding surfaces of membrane proteins: Evidence from evolutionary and structural analysis. Biochim. Biophys. Acta 2011, 1808, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Naveed, H.; Jimenez-Morales, D.; Tian, J.; Pasupuleti, V.; Kenney, L.J.; Liang, J. Engineered oligomerization state of OmpF protein through computational design decouples oligomer dissociation from unfolding. J. Mol. Biol. 2012, 419, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Konagurthu, A.S.; Whisstock, J.C.; Stuckey, P.J.; Lesk, A.M. Mustang: A multiple structural alignment algorithm. Proteins 2006, 64, 559–574. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Goujon, M.; McWilliam, H.; Li, W.; Valentin, F.; Squizzato, S.; Paern, J.; Lopez, R. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 2010, 38, W695–W699. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, R.K.; Joosten, H.J.; Van Berkel, W.J.; Leferink, N.G.; Rooijen, E.; Ittmann, E.; van Zimmeren, F.; Jochens, H.; Bornscheuer, U.; Vriend, G.; et al. 3DM: Systematic analysis of heterogeneous superfamily data to discover protein functionalities. Proteins 2010, 78, 2101–2113. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Whelan, S.; Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 2001, 18, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Hordijk, W.; Gascuel, O. Improving the efficiency of SPR moves in phylogenetic tree search methods based on maximum likelihood. Bioinformatics 2005, 21, 4338–4347. [Google Scholar] [CrossRef] [PubMed]
- Criss, A.K.; Seifert, H.S. A bacterial siren song: Intimate interactions between neisseria and neutrophils. Nat. Rev. Microbiol. 2012, 10, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Stanley, A.M.; Chuawong, P.; Hendrickson, T.L.; Fleming, K.G. Energetics of outer membrane phospholipase A (OMPLA) dimerization. J. Mol. Biol. 2006, 358, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Bishop, R.E. Structural biology of membrane-intrinsic β-barrel enzymes: Sentinels of the bacterial outer membrane. Biochim. Biophys. Acta 2008, 1778, 1881–1896. [Google Scholar] [CrossRef] [PubMed]
- Voorintholt, R.; Kosters, M.T.; Vegter, G.; Vriend, G.; Hol, W.G. A very fast program for visualizing protein surfaces, channels and cavities. J. Mol. Graph. 1989, 7, 243–245. [Google Scholar] [CrossRef]
- Rasmussen, A.A.; Eriksen, M.; Gilany, K.; Udesen, C.; Franch, T.; Petersen, C.; Valentin-Hansen, P. Regulation of ompa mrna stability: The role of a small regulatory RNA in growth phase-dependent control. Mol. Microbiol. 2005, 58, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Confer, A.W.; Ayalew, S. The OmpA family of proteins: Roles in bacterial pathogenesis and immunity. Vet. Microbiol. 2013, 163, 207–222. [Google Scholar] [CrossRef] [PubMed]
- Datta, D.; Vaidehi, N.; Floriano, W.B.; Kim, K.S.; Prasadarao, N.V.; Goddard, W.A., 3rd. Interaction of E. coli outer-membrane protein a with sugars on the receptors of the brain microvascular endothelial cells. Proteins 2003, 50, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Maruvada, R.; Kim, K.S. Extracellular loops of the Eschericia coli outer membrane protein A contribute to the pathogenesis of meningitis. J. Infect. Dis. 2011, 203, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Mittal, R.; Krishnan, S.; Gonzalez-Gomez, I.; Prasadarao, N.V. Deciphering the roles of outer membrane protein A extracellular loops in the pathogenesis of Escherichia coli k1 meningitis. J. Biol. Chem. 2011, 286, 2183–2193. [Google Scholar] [CrossRef] [PubMed]
- Koebnik, R. Structural and functional roles of the surface-exposed loops of the β-barrel membrane protein OmpA from Escherichia coli. J. Bacteriol. 1999, 181, 3688–3694. [Google Scholar] [PubMed]
- Andersen, C.; Bachmeyer, C.; Täuber, H.; Benz, R.; Wang, J.; Michel, V.; Newton, S.M.; Hofnung, M.; Charbit, A. In vivo and in vitro studies of major surface loop deletion mutants of the Escherichia coli k-12 maltoporin: Contribution to maltose and maltooligosaccharide transport and binding. Mol. Microbiol. 1999, 32, 851–867. [Google Scholar] [CrossRef] [PubMed]
- Gouaux, E. Roll out the barrel. Nat. Struct. Biol. 1998, 5, 931–932. [Google Scholar] [CrossRef] [PubMed]
- Sidorova, O.V.; Khomenko, V.A.; Portnyagina, O.Y.; Likhatskaya, G.N.; Vakorina, T.I.; Kim, N.Y.; Chistyulin, D.K.; Solov’eva, T.F.; Novikova, O.D. Mutant OmpF porins of yersinia pseudotuberculosis with deletions of external loops: Structure-functional and immunochemical properties. Biochem. Biophys. Res. Commun. 2014, 445, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Stenkova, A.M.; Isaeva, M.P.; Shubin, F.N.; Rasskazov, V.A.; Rakin, A.V. Trends of the major porin gene (OmpF) evolution: Insight from the genus yersinia. PLoS ONE 2011, 6, e20546. [Google Scholar] [CrossRef] [PubMed]
- Modi, N.; Bárcena-Uribarri, I.; Bains, M.; Benz, R.; Hancock, R.E.; Kleinekathöfer, U. Role of the central arginine r133 toward the ion selectivity of the phosphate specific channel OprP: Effects of charge and solvation. Biochemistry 2013, 52, 5522–5532. [Google Scholar] [CrossRef] [PubMed]
- Siehnel, R.J.; Egli, C.; Hancock, R.E. Polyphosphate-selective porin OprO of Pseudomonas aeruginosa: Expression, purification and sequence. Mol. Microbiol. 1992, 6, 2319–2326. [Google Scholar] [CrossRef] [PubMed]
- Dumas, F.; Koebnik, R.; Winterhalter, M.; Van Gelder, P. Sugar transport through maltoporin of Escherichia coli. Role of polar tracks. J. Biol. Chem. 2000, 275, 19747–19751. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Rothenberg, E. Interaction of bacteriophage λ with its E. coli receptor, LamB. Viruses 2012, 4, 3162–3178. [Google Scholar] [CrossRef] [PubMed]
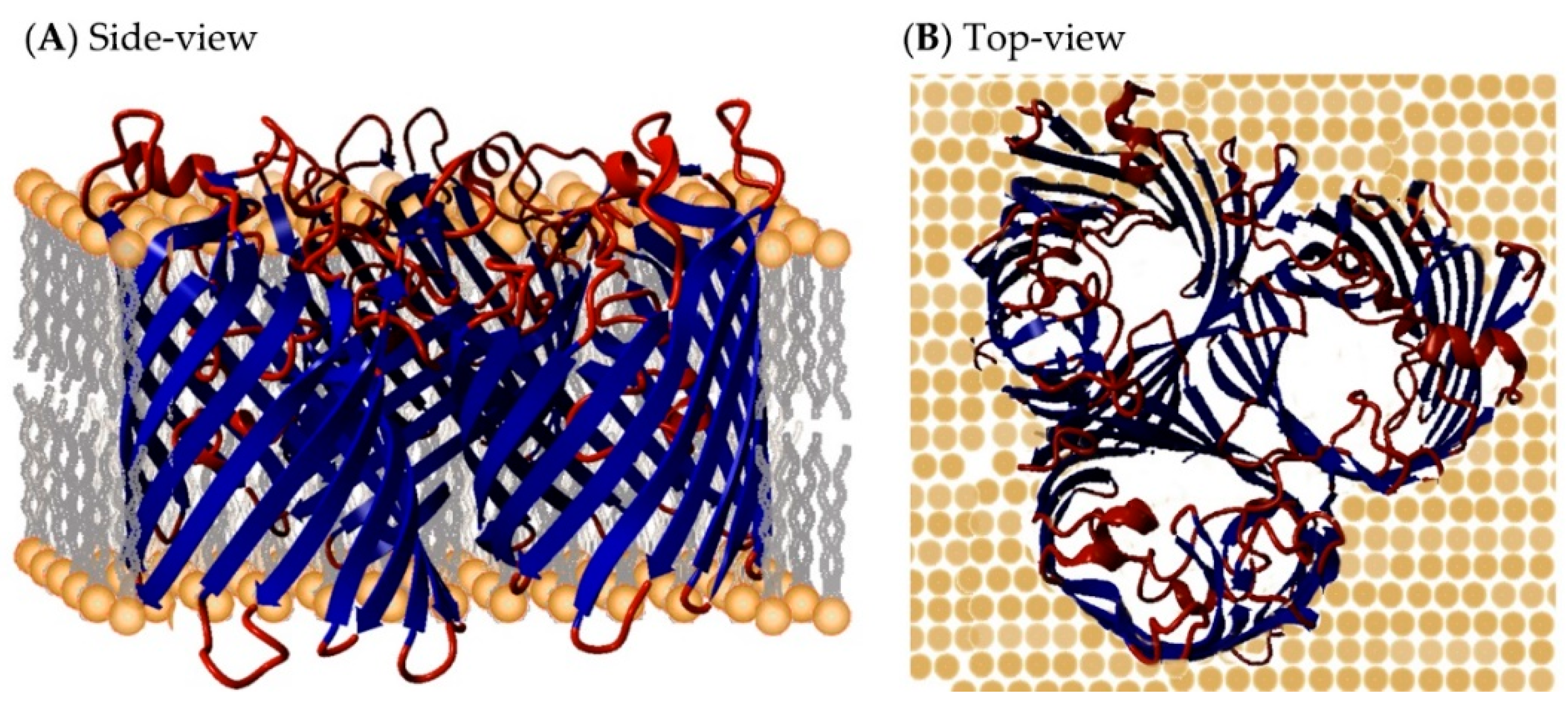
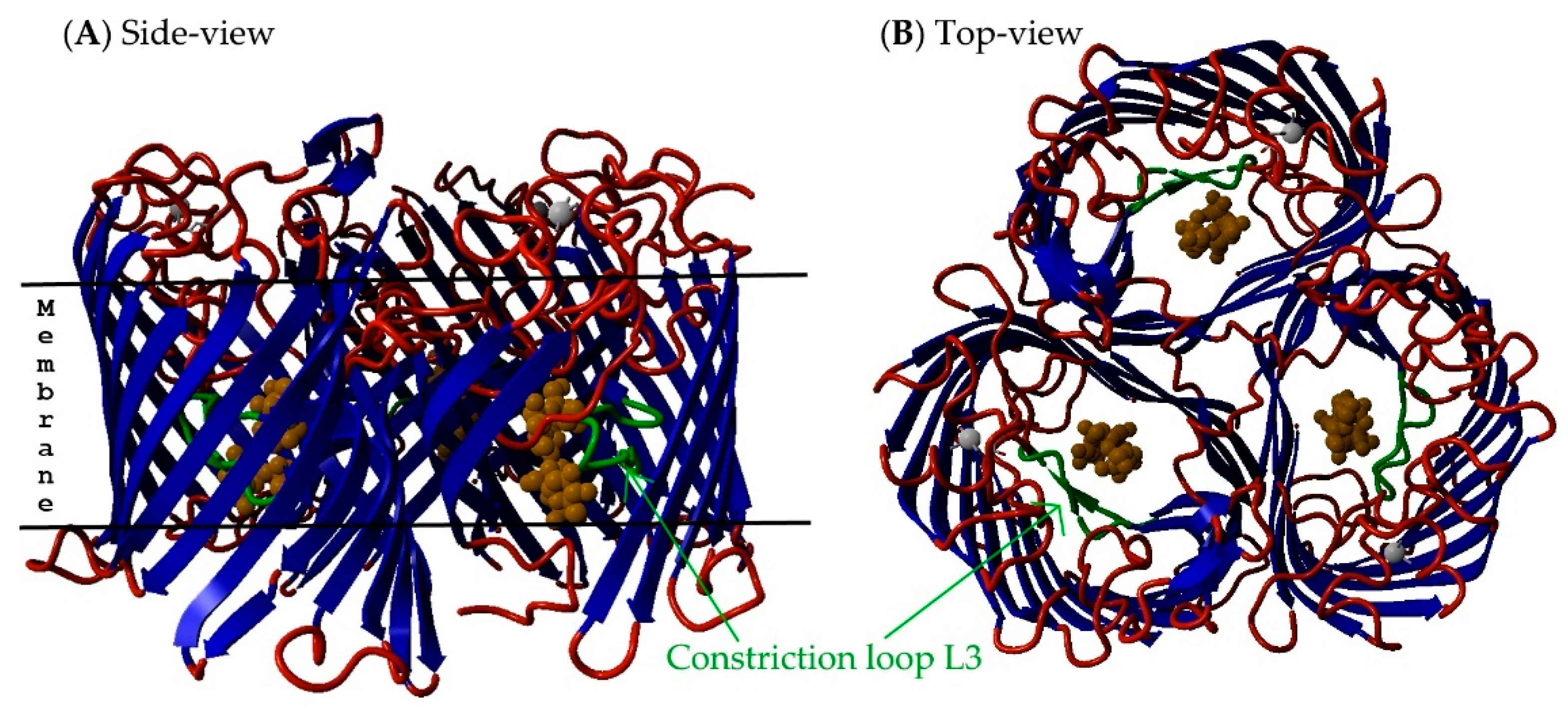


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
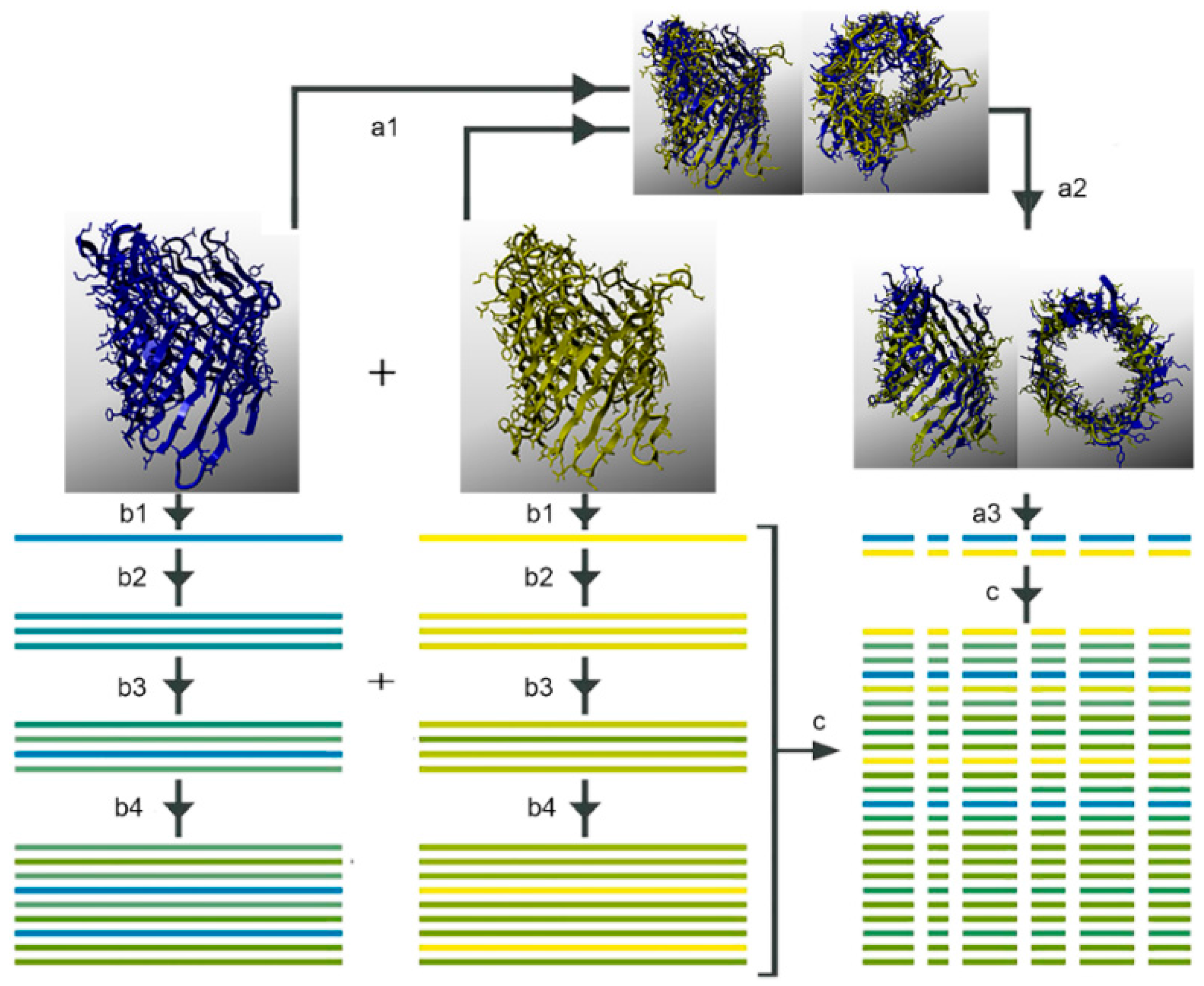{kind=link}
{kind=link}
| Groups | Name | Bacteriae | Size | PDB ID | Resolution (Å) |
|---|---|---|---|---|---|
| Monomeric proteins | |||||
| NMP | EcOmpA | Escherichia coli | 8 | 1QJP | 1.7 |
| NMP | KpOmpA | Klebsiella pneumonia | 8 | 2K0L | – |
| NMP | OmpG | Escherichia coli | 14 | 2IWV | 2.3 |
| SMDC | NanC | Escherichia coli | 12 | 2WJR | 1.8 |
| SMDC | KdgM | Dickeya dadantii | 12 | 4FQE | 1.9 |
| SMDC | OprB | Pseudomonas putida | 16 | 4GEY | 2.7 |
| SMDC | OccD | Pseudomonas putida/Pseudomonas fluorescens | 18 | 3SYS, 3SZD, 3SZV, 3T0S, 3T20, 3T24, 4FRT, 4FRX, 4FT6, 4FSO, 4FSP, 3JTY, 3SY7, 3SY9, 3SYB | Average: 2.4 (1.5–3.2) |
| Trimeric proteins | |||||
| NTP | EcOmpC | Escherichia coli | 16 | 2J1N | 2.0 |
| NTP | StOmpC | Salmonella Typhimurium | 16 | 3UPG | 3.2 |
| NTP | OmpK36 | Klebsiella pneumonia | 16 | 1OSM | 3.2 |
| NTP | StOmpF | Salmonella Typhimurium | 16 | 3NSG | 2.8 |
| NTP | EcOmpF | Escherichia coli | 16 | 4GCS | 1.9 |
| NTP | PhoE | Escherichia coli | 16 | 1PHO | 3.0 |
| NTP | RcGDP | Rhodobacter capsulatus | 16 | 2POR | 1.8 |
| NTP | RbGDP | Rhodopseudomonas blastica | 16 | 1PRN | 2.0 |
| NTP | Omp32 | Delftia acidovorans | 16 | 2FGQ | 1.5 |
| STDC | EcMaltoporin | Escherichia coli | 18 | 1AF6 | 2.4 |
| STDC | StMaltoporin | Salmonella Typhimurium | 18 | 2MPR | 2.4 |
| STDC | ScrY | Salmonella Typhimurium | 18 | 1OH2 | 2.4 |
| STDC | OprP | Pseudomonas aeruginosa | 16 | 2O4V | 1.9 |
| Porin Group | Average Length | Barrel (%) | Periplasmic Loop (%) | Extracellular Loop (%) |
|---|---|---|---|---|
| NMP | 219 | 57 | 12 | 30 |
| SMDC | 385 | 56 | 14 | 28 |
| NTP | 325 | 57 | 11 | 30 |
| STDC | 417 | 54 | 11 | 34 |
| Class | Subclass | Protein Structures | |||
|---|---|---|---|---|---|
| Number | Size | Number | Name | Protein Name | PDB ID Template (Other) |
| 1 | 8 | 1A | Non-specific, petite porin | OmpA | 2K0L (1QJP) |
| 2 | 10 | 2A | Non-specific, mini porin | – | – |
| 3 | 12 | 3A | Non-specific, small porin | – | – |
| 3B | Oligogalacturonate-specific, small channel | KdgM and NanC | 4FQE (2WJR) | ||
| 4 | 14 | 4A | Non-specific, intermediate porin | OmpG | 2IWV |
| 5 | 16 | 5A | Non-specific, medium porin | OmpC, OmpK36, OmpF, PhoE, Omp32, RcGDP and RbGDP | 2J1N (1PRN, 3UPG, 1OSM, 3NSG, 4GCS, 2POR, 2FGQ, and 1PHO) |
| 5B | Sugar-specific, medium channel | OprB | 4GEY | ||
| 5C | Phosphate-specific, medium channel | OprP | 2O4V | ||
| 6 | 18 | 6A | Non-specific, large porin | – | – |
| 6B | Sugar-specific, large channel | Maltoporin and ScrY | 2MPR (1AF6 and 1OH2) | ||
| 6C | Carboxyl-specific, large channel | Occ Channels | 3SZV (3SYS, 3SZD, 3T0S, 3T20, 3T24, 4FRT, 4FRX, 4FT6, 4FSO, 4FSP. 3JTY, 3SY7, 3SY9, and 3SYB) | ||
| Subclasses | Resolution | % Residues Superposed | % Sequence Identity | Pore Size | |||
|---|---|---|---|---|---|---|---|
| Number | Name | RMSD (Å) | Mustang | Mustang | Clustal Ω | Core | Whole |
| 1A | Non-specific, petite porin | 1.6 | 98.7 | 96.1 | 93.6 | ~ 0 | 0.1 |
| 3B | Oligogalacturonate-specific, small channel | 1.4 | 95.0 | 25.6 | 28.1 | 2.9 | 2.9 |
| 4A | Non-specific, intermediate porin | – | – | – | – | 3.7 | 4.0 |
| 5A | Non-specific, medium porin | 1.4 | 90.7 | 40.2 | 41.3 | 2.8 | 6.9 |
| 5B | Sugar-specific, medium channel | – | – | – | – | 2.2 | 7.0 |
| 5C | Phosphate-specific, medium channel | – | – | – | – | 1.6 | 6.4 |
| 6B | Sugar-specific, large channel | 0.9 | 95.6 | 50.3 | 49.5 | 2.1 | 6.9 |
| 6C | Carboxyl-specific, large channel | 0.9 | 96.2 | 46.4 | 45.7 | 1.8 | 7.8 |
| Average | 1.2 | 95.2 | 52.0 | 51.6 | 2.4 | 7.5 | |
| Subclass Number | Subclass Name | Number of Sequences Used in the MSA |
|---|---|---|
| 1A | Non-specific, petite porin | 389 |
| 3B | Oligogalacturonate-specific, small channel | 246 |
| 4A | Non-specific, intermediate porin | 50 |
| 5A | Non-specific, medium porin | 725 |
| 5B | Sugar-specific, medium channel | 319 |
| 5C | Phosphate-specific, medium channel | 180 |
| 6B | Sugar-specific, large channel | 663 |
| 6C | Carboxyl-specific, large channel | 1394 |
| Class Number | Class Name | Function | Monomer | Dimer | Oligomer |
|---|---|---|---|---|---|
| 1A | Non-specific, petite porin | Abundant multifunctional porin; host evasion [22] | EcOmpA [3,41,59,60,61,62,63,64,65], KpOmpA [56] | EcOmpA [26,27,66,67] | FopA [68], Pgm6/7 [69], EcOmpA [70] |
| 3B | Oligogalacturonate-specific, small channel | Oligogalacturonate-specific channel [71] | KdgM [71,72,73], NanC [24] | NanC [74] | – |
| 4A | Non-specific, intermediate porin | pH-dependent rare rescue porin [75] | EcOmpG [44,75,76,77,78] | EcOmpG [44] | – |
| 5A | Non-specific, medium porin | Classical porins or general diffusion porin (GDP) [11,79] | – | – | GDP [3,4,16,80,81,82,83,84,85,86] |
| 5B | Sugar-specific, medium channel | Oligosaccharide specific channel [23] | OprB [23,87]. | – | OprB [88] |
| 5C | Phosphate-specific, medium channel | Phosphate specific channel [89] | – | – | OprP [30,90], OprO/P heterotrimer [91] |
| 6B | Sugar-specific, large channel | Sugar specific channel [31] | – | – | EcMaltoporin [31], StMaltoporin [92], ScrY [12] |
| 6C | Carboxyl-specific, large channel | Small water-soluble specific channel (Occ channels) [29] | Occ [28,29], OccD1 (OprD) [93]. | – | OccK1 (OpdK) [94], AbOprD [95] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vollan, H.S.; Tannæs, T.; Vriend, G.; Bukholm, G. In Silico Structure and Sequence Analysis of Bacterial Porins and Specific Diffusion Channels for Hydrophilic Molecules: Conservation, Multimericity and Multifunctionality. Int. J. Mol. Sci. 2016, 17, 599. https://doi.org/10.3390/ijms17040599
Vollan HS, Tannæs T, Vriend G, Bukholm G. In Silico Structure and Sequence Analysis of Bacterial Porins and Specific Diffusion Channels for Hydrophilic Molecules: Conservation, Multimericity and Multifunctionality. International Journal of Molecular Sciences. 2016; 17(4):599. https://doi.org/10.3390/ijms17040599
Chicago/Turabian StyleVollan, Hilde S., Tone Tannæs, Gert Vriend, and Geir Bukholm. 2016. "In Silico Structure and Sequence Analysis of Bacterial Porins and Specific Diffusion Channels for Hydrophilic Molecules: Conservation, Multimericity and Multifunctionality" International Journal of Molecular Sciences 17, no. 4: 599. https://doi.org/10.3390/ijms17040599
APA StyleVollan, H. S., Tannæs, T., Vriend, G., & Bukholm, G. (2016). In Silico Structure and Sequence Analysis of Bacterial Porins and Specific Diffusion Channels for Hydrophilic Molecules: Conservation, Multimericity and Multifunctionality. International Journal of Molecular Sciences, 17(4), 599. https://doi.org/10.3390/ijms17040599