
Genome Editing in C. elegans and Other Nematode Species

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Development of Gene Editing Protocols for Nematodes
2.1. Site-Restricted Editing by Mos1
2.2. Universal Editing with ZFNs, TALENs and CRISPRs
2.3. Imprecise and Precise Repair Using ssOligo and dsDNA
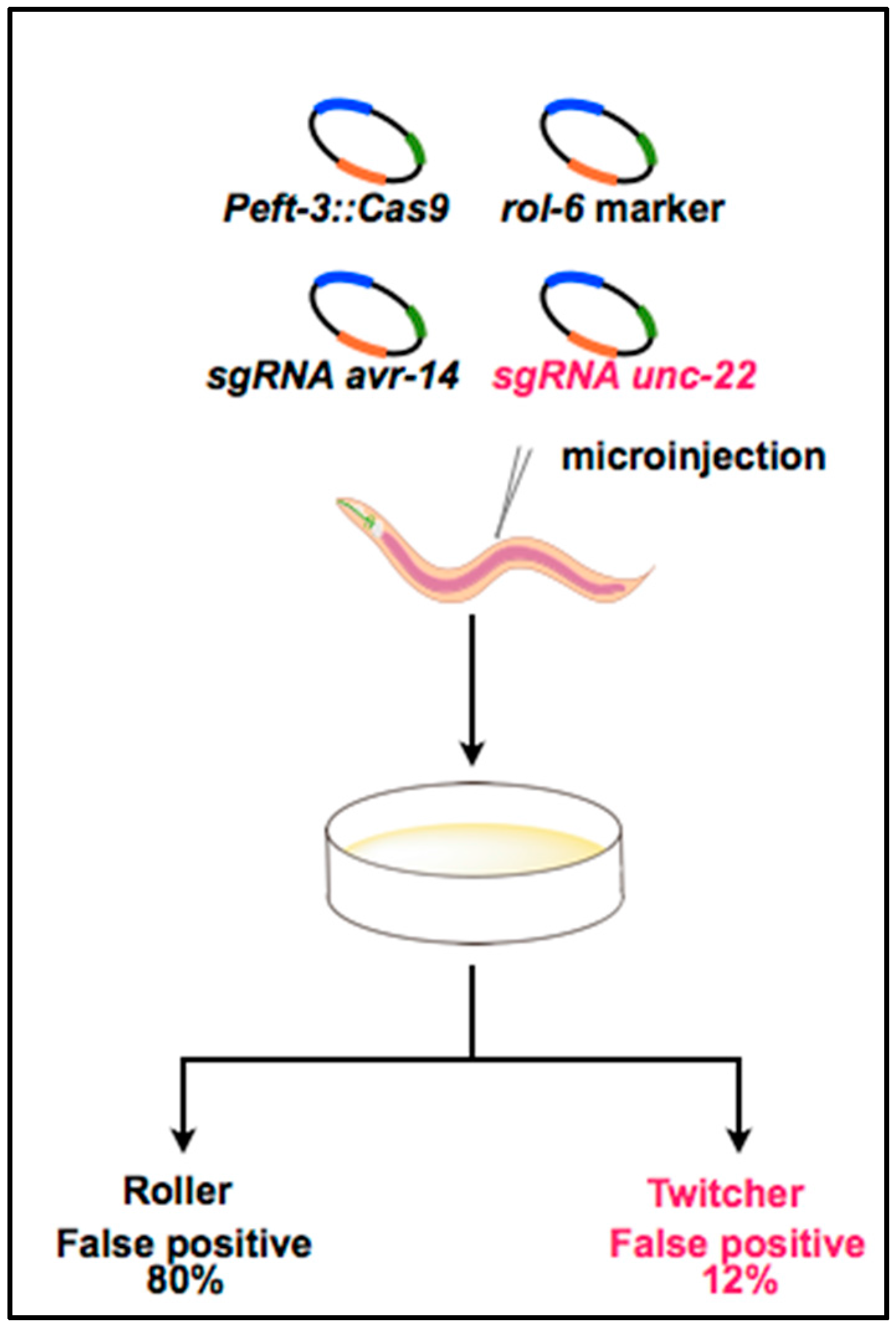
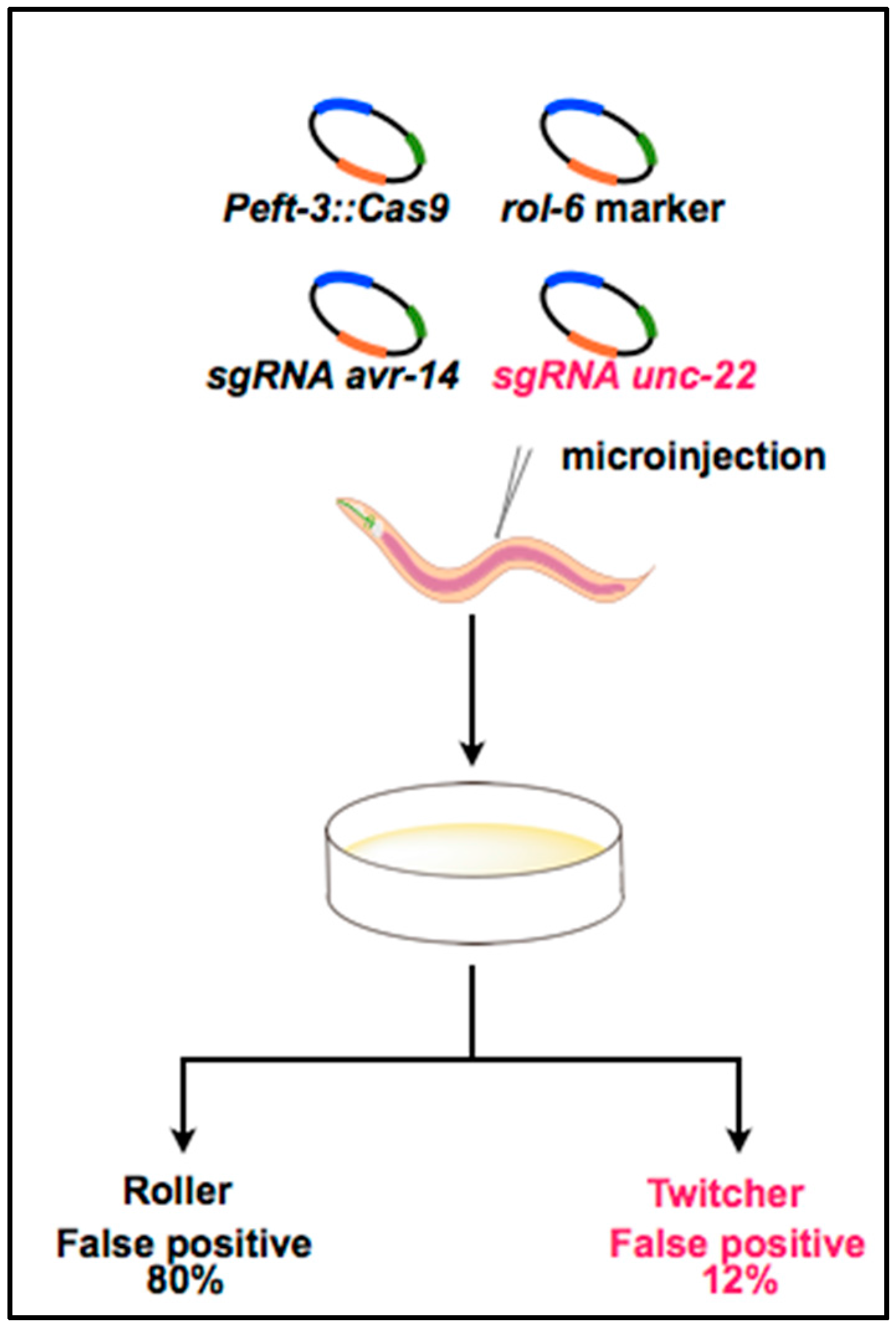
3. Optimization of the CRISPR System
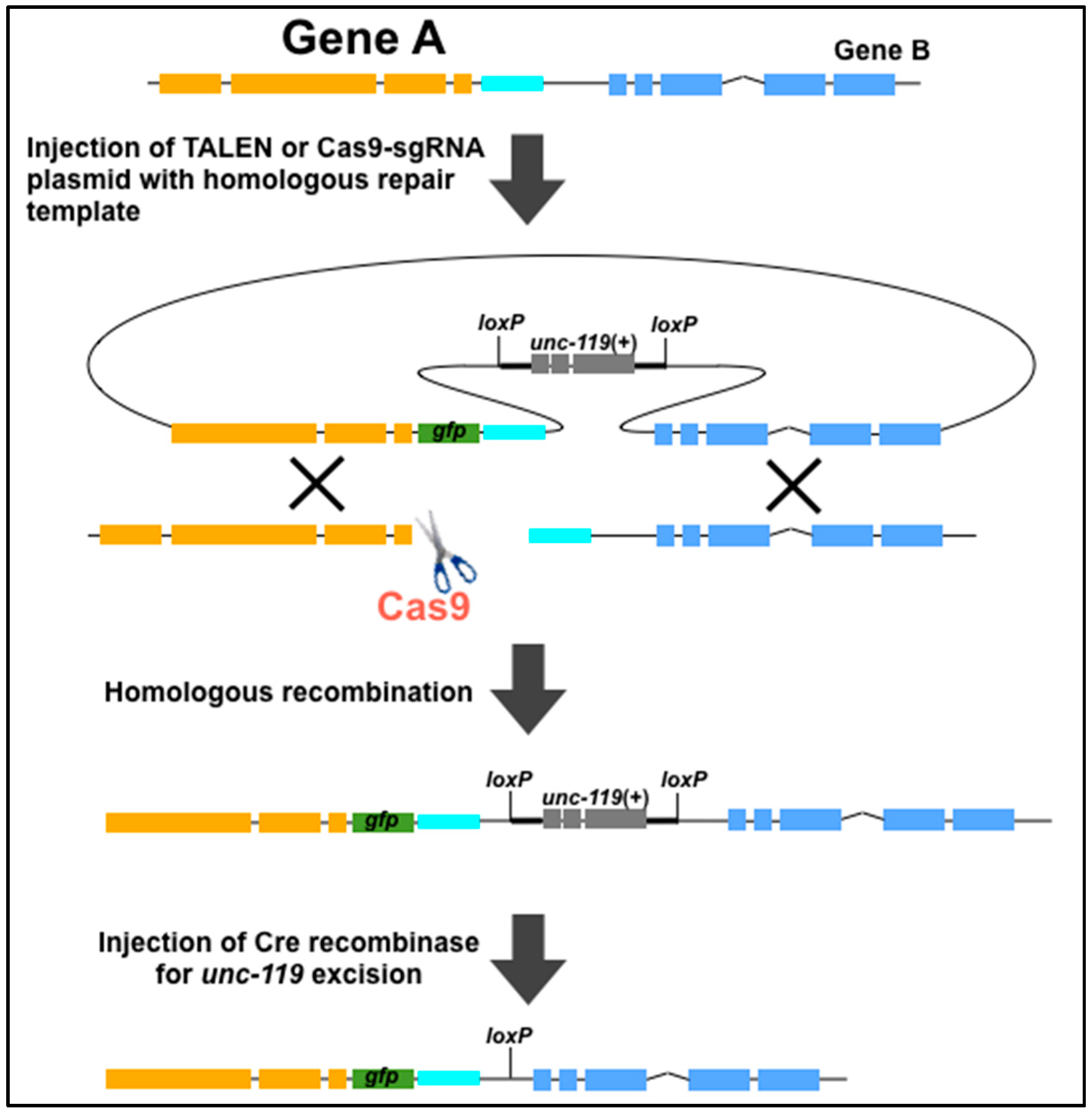
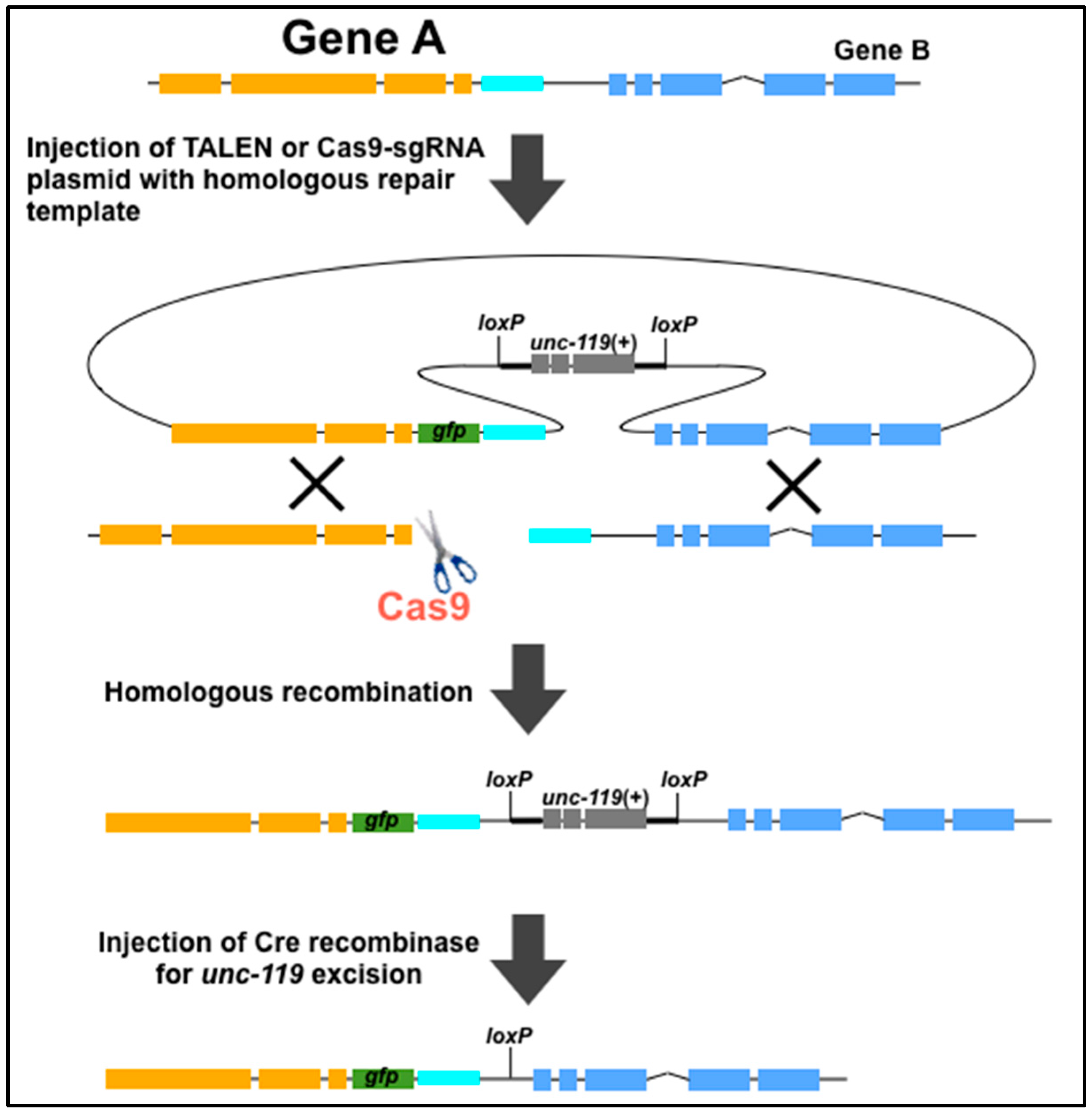
4. Use of Genome Editing Tools to Create New Methods for C. elegans
5. Important Results Achieved Using Genome Editing Techniques
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brenner, S. The genetics of Caenorhabditis elegans. Genetics 1974, 77, 71–94. [Google Scholar] [PubMed]
- Sulston, J.E.; Schierenberg, E.; White, J.G.; Thomson, J.N. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 1983, 100, 64–119. [Google Scholar] [CrossRef]
- White, J.G.; Southgate, E.; Thomson, J.N.; Brenner, S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1986, 314, 1–340. [Google Scholar] [CrossRef] [PubMed]
- Shaye, D.D.; Greenwald, I. OrthoList: A compendium of C. elegans genes with human orthologs. PLoS ONE 2011, 6, e20085. [Google Scholar] [CrossRef] [PubMed]
- Kaletta, T.; Hengartner, M.O. Finding function in novel targets: C. elegans as a model organism. Nat. Rev. Drug Discov. 2006, 5, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Culetto, E.; Sattelle, D.B. A role for Caenorhabditis elegans in understanding the function and interactions of human disease genes. Hum. Mol. Genet. 2000, 9, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Mello, C.C.; Kramer, J.M.; Stinchcomb, D.; Ambros, V. Efficient gene transfer in C. elegans: Extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991, 10, 3959–3970. [Google Scholar] [PubMed]
- Praitis, V.; Casey, E.; Collar, D.; Austin, J. Creation of low-copy integrated transgenic lines in Caenorhabditis elegans. Genetics 2001, 157, 1217–1226. [Google Scholar] [PubMed]
- Wilm, T.; Demel, P.; Koop, H.U.; Schnabel, H.; Schnabel, R. Ballistic transformation of Caenorhabditis elegans. Gene 1999, 229, 31–35. [Google Scholar] [CrossRef]
- Sarov, M.; Murray, J.I.; Schanze, K.; Pozniakovski, A.; Niu, W.; Angermann, K.; Hasse, S.; Rupprecht, M.; Vinis, E.; Tinney, M.; et al. A genome-scale resource for in vivo tag-based protein function exploration in C. elegans. Cell 2012, 150, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Robert, V.; Bessereau, J.-L. Targeted engineering of the Caenorhabditis elegans genome following Mos1-triggered chromosomal breaks. EMBO J. 2007, 26, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Frøkjaer-Jensen, C.; Davis, M.W.; Hopkins, C.E.; Newman, B.J.; Thummel, J.M.; Olesen, S.-P.; Grunnet, M.; Jorgensen, E.M. Single-copy insertion of transgenes in Caenorhabditis elegans. Nat. Genet. 2008, 40, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Morton, J.; Davis, M.W.; Jorgensen, E.M.; Carroll, D. Induction and repair of zinc-finger nuclease-targeted double-strand breaks in Caenorhabditis elegans somatic cells. Proc. Natl. Acad. Sci. USA 2006, 103, 16370–16375. [Google Scholar] [CrossRef] [PubMed]
- Wood, A.J.; Lo, T.-W.; Zeitler, B.; Pickle, C.S.; Ralston, E.J.; Lee, A.H.; Amora, R.; Miller, J.C.; Leung, E.; Meng, X.; et al. Targeted genome editing across species using ZFNs and TALENs. Science 2011, 333, 307. [Google Scholar] [CrossRef] [PubMed]
- Lo, T.-W.; Pickle, C.S.; Lin, S.; Ralston, E.J.; Gurling, M.; Schartner, C.M.; Bian, Q.; Doudna, J.A.; Meyer, B.J. Precise and heritable genome editing in evolutionarily diverse nematodes using TALENs and CRISPR/Cas9 to engineer insertions and deletions. Genetics 2013, 195, 331–348. [Google Scholar] [CrossRef] [PubMed]
- Friedland, A.E.; Tzur, Y.B.; Esvelt, K.M.; Colaiácovo, M.P.; Church, G.M.; Calarco, J.A. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat. Meth. 2013, 10, 741–743. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.; Schwartz, H.T.; Antoshechkin, I.; Sternberg, P.W. Transgene-free genome editing in Caenorhabditis elegans using CRISPR-Cas. Genetics 2013, 195, 1167–1171. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cho, S.W.; Lee, J.; Carroll, D.; Kim, J.-S.; Lee, J. Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins. Genetics 2013, 195, 1177–1180. [Google Scholar] [CrossRef] [PubMed]
- Katic, I.; Großhans, H. Targeted heritable mutation and gene conversion by Cas9-CRISPR in Caenorhabditis elegans. Genetics 2013, 195, 1173–1176. [Google Scholar] [CrossRef] [PubMed]
- Tzur, Y.B.; Friedland, A.E.; Nadarajan, S.; Church, G.M.; Calarco, J.A.; Colaiácovo, M.P. Heritable custom genomic modifications in Caenorhabditis elegans via a CRISPR-Cas9 system. Genetics 2013, 195, 1181–1185. [Google Scholar] [CrossRef] [PubMed]
- Waaijers, S.; Portegijs, V.; Kerver, J.; Lemmens, B.B.L.G.; Tijsterman, M.; van Den Heuvel, S.; Boxem, M. CRISPR/Cas9-targeted mutagenesis in Caenorhabditis elegans. Genetics 2013, 195, 1187–1191. [Google Scholar] [CrossRef] [PubMed]
- Frøkjaer-Jensen, C. Exciting prospects for precise engineering of Caenorhabditis elegans genomes with CRISPR/Cas9. Genetics 2013, 195, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Frøkjaer-Jensen, C.; Davis, M.W.; Ailion, M.; Jorgensen, E.M. Improved Mos1-mediated transgenesis in C. elegans. Nat. Meth. 2012, 9, 117–118. [Google Scholar] [CrossRef] [PubMed]
- Mussolino, C.; Cathomen, T. TALE nucleases: Tailored genome engineering made easy. Curr. Opin. Biotechnol. 2012, 23, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Moscou, M.J.; Bogdanove, A.J. A simple cipher governs DNA recognition by TAL effectors. Science 2009, 326, 1501–1501. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Shen, Y.; Chen, X.; Shifman, Y.; Ellis, R.E. Rapid creation of forward-genetics tools for C. briggsae using TALENs: Lessons for nonmodel organisms. Mol. Biol. Evol. 2014, 31, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Zhao, Y.; Guo, Y.; Stomel, J.; Stires, R.; Ellis, R.E. Co-option of alternate sperm activation programs in the evolution of self-fertile nematodes. Nat. Commun. 2014, 5, 5888. [Google Scholar] [CrossRef] [PubMed]
- Sugi, T.; Sakuma, T.; Ohtani, Y.; Yamamoto, T. Versatile strategy for isolating transcription activator-like effector nuclease-mediated knockout mutants in Caenorhabditis elegans. Dev. Growth Differ. 2014, 56, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Witte, H.; Moreno, E.; Rödelsperger, C.; Kim, J.; Kim, J.-S.; Streit, A.; Sommer, R.J. Gene inactivation using the CRISPR/Cas9 system in the nematode Pristionchus pacificus. Dev. Genes Evol. 2015, 225, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Fenk, L.A.; de Bono, M. Efficient genome editing in Caenorhabditis elegans by CRISPR-targeted homologous recombination. Nucleic Acids Res. 2013, 41, e193. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Long, L.; Xiong, K.; Yu, B.; Chang, N.; Xiong, J.-W.; Zhu, Z.; Liu, D. Heritable/conditional genome editing in C. elegans using a CRISPR-Cas9 feeding system. Cell Res. 2014, 24, 886–889. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Katic, I.; Xu, L.; Ciosk, R. CRISPR/Cas9 Genome Editing in Caenorhabditis elegans: Evaluation of templates for homology-mediated repair and knock-ins by homology-independent DNA repair. G3 (Bethesda) 2015, 5, 1649–1656. [Google Scholar] [CrossRef] [PubMed]
- Van Schendel, R.; Roerink, S.F.; Portegijs, V.; van Den Heuvel, S.; Tijsterman, M. Polymerase Θ is a key driver of genome evolution and of CRISPR/Cas9-mediated mutagenesis. Nat. Commun. 2015, 6, 7394. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, D.J.; Ward, J.D.; Reiner, D.J.; Goldstein, B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat. Meth. 2013, 10, 1028–1034. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Zhang, Z.; Ke, H.; Yue, Y.; Xue, D. Oligonucleotide-based targeted gene editing in C. elegans via the CRISPR/Cas9 system. Cell Res. 2014, 24, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Paix, A.; Wang, Y.; Smith, H.E.; Lee, C.-Y.S.; Calidas, D.; Lu, T.; Smith, J.; Schmidt, H.; Krause, M.W.; Seydoux, G. Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 Sites in Caenorhabditis elegans. Genetics 2014, 198, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xu, F.; Zhu, C.; Ji, J.; Zhou, X.; Feng, X.; Guang, S. Dual sgRNA-directed gene knockout using CRISPR/Cas9 technology in Caenorhabditis elegans. Sci. Rep. 2014, 4, 7581. [Google Scholar] [CrossRef] [PubMed]
- Norris, A.D.; Kim, H.-M.; Colaiácovo, M.P.; Calarco, J.A. Efficient genome editing in Caenorhabditis elegans with a toolkit of dual-marker selection cassettes. Genetics 2015, 201, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, D.J.; Pani, A.M.; Heppert, J.K.; Higgins, C.D.; Goldstein, B. Streamlined genome engineering with a self-excising drug selection cassette. Genetics 2015, 200, 1035–1049. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Ishidate, T.; Ghanta, K.S.; Seth, M.; Conte, D.; Shirayama, M.; Mello, C.C. A co-CRISPR strategy for efficient genome editing in Caenorhabditis elegans. Genetics 2014, 197, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Moerman, D.G.; Baillie, D.L. Genetic organization in Caenorhabditis elegans: Fine-structure analysis of the unc-22 gene. Genetics 1979, 91, 95–103. [Google Scholar] [PubMed]
- Arribere, J.A.; Bell, R.T.; Fu, B.X.H.; Artiles, K.L.; Hartman, P.S.; Fire, A.Z. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics 2014, 198, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.D. Rapid and precise engineering of the Caenorhabditis elegans genome with lethal mutation co-conversion and inactivation of NHEJ repair. Genetics 2015, 199, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Farboud, B.; Meyer, B.J. Dramatic enhancement of genome editing by CRISPR/Cas9 through improved guide RNA design. Genetics 2015, 199, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Paix, A.; Folkmann, A.; Rasoloson, D.; Seydoux, G. High Efficiency, homology-directed genome editing in Caenorhabditis elegans using CRISPR-Cas9 ribonucleoprotein complexes. Genetics 2015, 201, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Yi, P.; Wang, X.; Chai, Y.; Feng, G.; Yang, Y.; Liang, X.; Zhu, Z.; Li, W.; Ou, G. Conditional targeted genome editing using somatically expressed TALENs in C. elegans. Nat. Biotechnol. 2013, 31, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Zhang, X.; Chai, Y.; Zhu, Z.; Yi, P.; Feng, G.; Li, W.; Ou, G. Conditional knockouts generated by engineered CRISPR-Cas9 endonuclease reveal the roles of coronin in C. elegans neural development. Dev. Cell 2014, 30, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yi, P.; Ou, G. Somatic CRISPR-Cas9-induced mutations reveal roles of embryonically essential dynein chains in Caenorhabditis elegans cilia. J. Cell Biol. 2015, 208, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.; Diao, M.; Jiang, Y.; Sun, L.; Zhang, Y.; Chen, Z.; Huang, S.; Ou, G. Anillin regulates neuronal migration and neurite growth by linking RhoG to the actin cytoskeleton. Curr. Biol. 2015, 25, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ward, J.D.; Cheng, Z.; Dernburg, A.F. The auxin-inducible degradation (AID) system enables versatile conditional protein depletion in C. elegans. Development 2015, 142, 4374–4384. [Google Scholar] [CrossRef] [PubMed]
- Yumerefendi, H.; Dickinson, D.J.; Wang, H.; Zimmerman, S.P.; Bear, J.E.; Goldstein, B.; Hahn, K.; Kuhlman, B. Control of protein activity and cell fate specification via light-mediated nuclear translocation. PLoS ONE 2015, 10, e0128443. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Guo, H.; Yao, D.; Xiong, K.; Li, Y.; Liu, P.; Zhu, Z.; Liu, D. Regulation of transcriptionally active genes via the catalytically inactive Cas9 in C. elegans and D. rerio. Cell Res. 2015, 25, 638–641. [Google Scholar] [CrossRef] [PubMed]
- Crane, E.; Bian, Q.; McCord, R.P.; Lajoie, B.R.; Wheeler, B.S.; Ralston, E.J.; Uzawa, S.; Dekker, J.; Meyer, B.J. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature 2015, 523, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Ecsedi, M.; Rausch, M.; Großhans, H. The let-7 microRNA directs vulval development through a single target. Dev. Cell 2015, 32, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Miki, T.S.; Richter, H.; Rüegger, S.; Großhans, H. PAXT-1 promotes XRN2 activity by stabilizing it through a conserved domain. Mol. Cell 2014, 53, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shen, Y.; Ellis, R.E. Dependence of the sperm/oocyte decision on the nucleosome remodeling factor complex was acquired during recent Caenorhabditis briggsae evolution. Mol. Biol. Evol. 2014, 31, 2573–2585. [Google Scholar] [CrossRef] [PubMed]
- LaMunyon, C.W.; Nasri, U.; Sullivan, N.G.; Shaw, M.A.; Prajapati, G.; Christensen, M.; Elmatari, D.; Clark, J.N. A New Player in the spermiogenesis pathway of Caenorhabditis elegans. Genetics 2015, 201, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Uozumi, T.; Hamakawa, M.; Deno, Y.-K.; Nakajo, N.; Hirotsu, T. Voltage-dependent anion channel (VDAC-1) is required for olfactory sensing in Caenorhabditis elegans. Genes Cells 2015, 20, 802–816. [Google Scholar] [CrossRef] [PubMed]
- Crocker, J.; Stern, D.L. TALE-mediated modulation of transcriptional enhancers in vivo. Nat. Meth. 2013, 10, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Hsu, P.D.; Heidenreich, M.; Cong, L.; Platt, R.J.; Scott, D.A.; Church, G.M.; Zhang, F. Optical control of mammalian endogenous transcription and epigenetic states. Nature 2013, 500, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Mendenhall, E.M.; Williamson, K.E.; Reyon, D.; Zou, J.Y.; Ram, O.; Joung, J.K.; Bernstein, B.E. Locus-specific editing of histone modifications at endogenous enhancers. Nat. Biotechnol. 2013, 31, 1133–1136. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Gilbert, L.A.; Cimini, B.A.; Schnitzbauer, J.; Zhang, W.; Li, G.-W.; Park, J.; Blackburn, E.H.; Weissman, J.S.; Qi, L.S.; et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 2013, 155, 1479–1491. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sugi, T. Genome Editing in C. elegans and Other Nematode Species. Int. J. Mol. Sci. 2016, 17, 295. https://doi.org/10.3390/ijms17030295
Sugi T. Genome Editing in C. elegans and Other Nematode Species. International Journal of Molecular Sciences. 2016; 17(3):295. https://doi.org/10.3390/ijms17030295
Chicago/Turabian StyleSugi, Takuma. 2016. "Genome Editing in C. elegans and Other Nematode Species" International Journal of Molecular Sciences 17, no. 3: 295. https://doi.org/10.3390/ijms17030295
APA StyleSugi, T. (2016). Genome Editing in C. elegans and Other Nematode Species. International Journal of Molecular Sciences, 17(3), 295. https://doi.org/10.3390/ijms17030295