
Single-Base Pair Genome Editing in Human Cells by Using Site-Specific Endonucleases

Abstract
:1. Introduction
2. Seamless Single-Base Pair Editing and Related Techniques
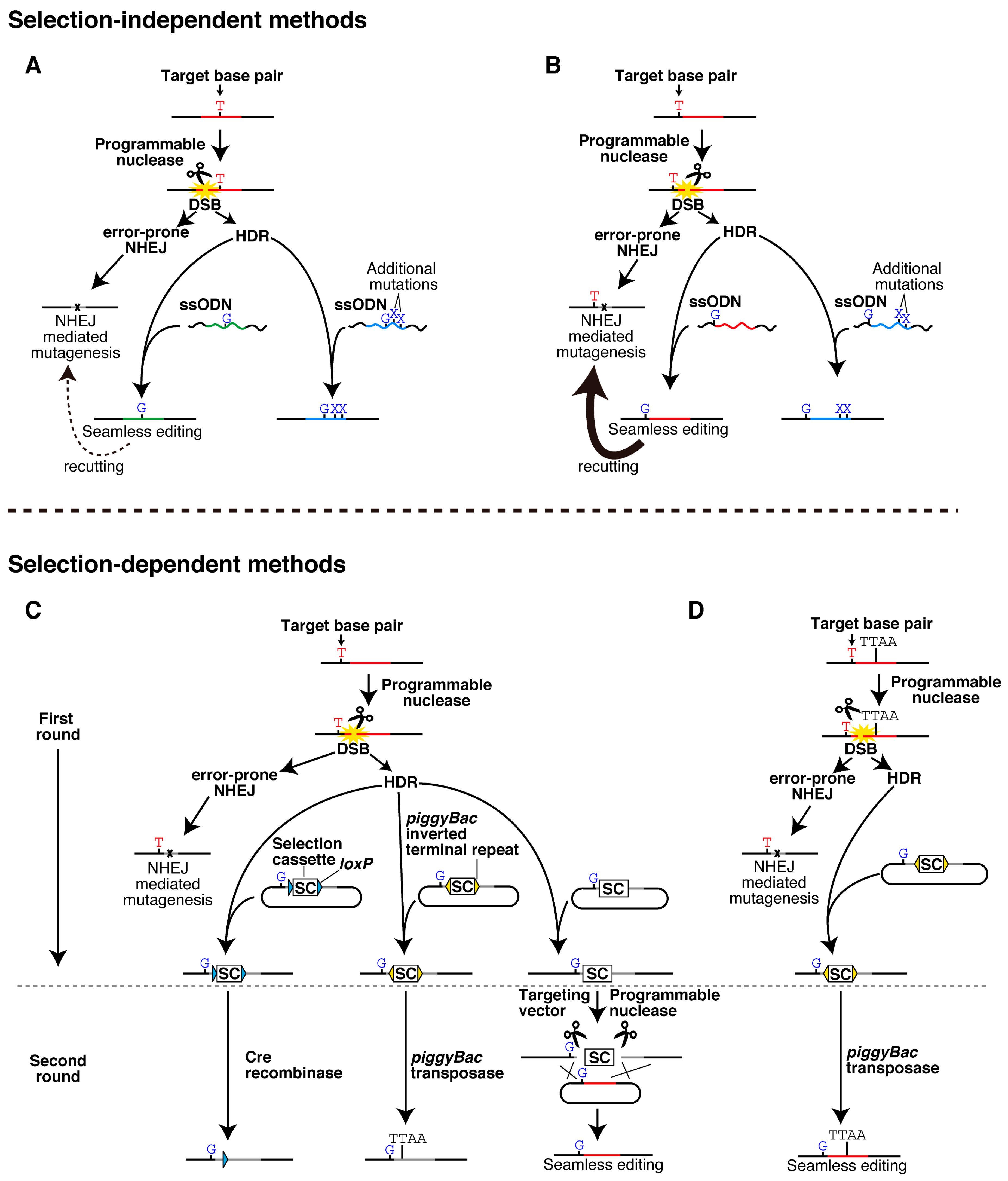

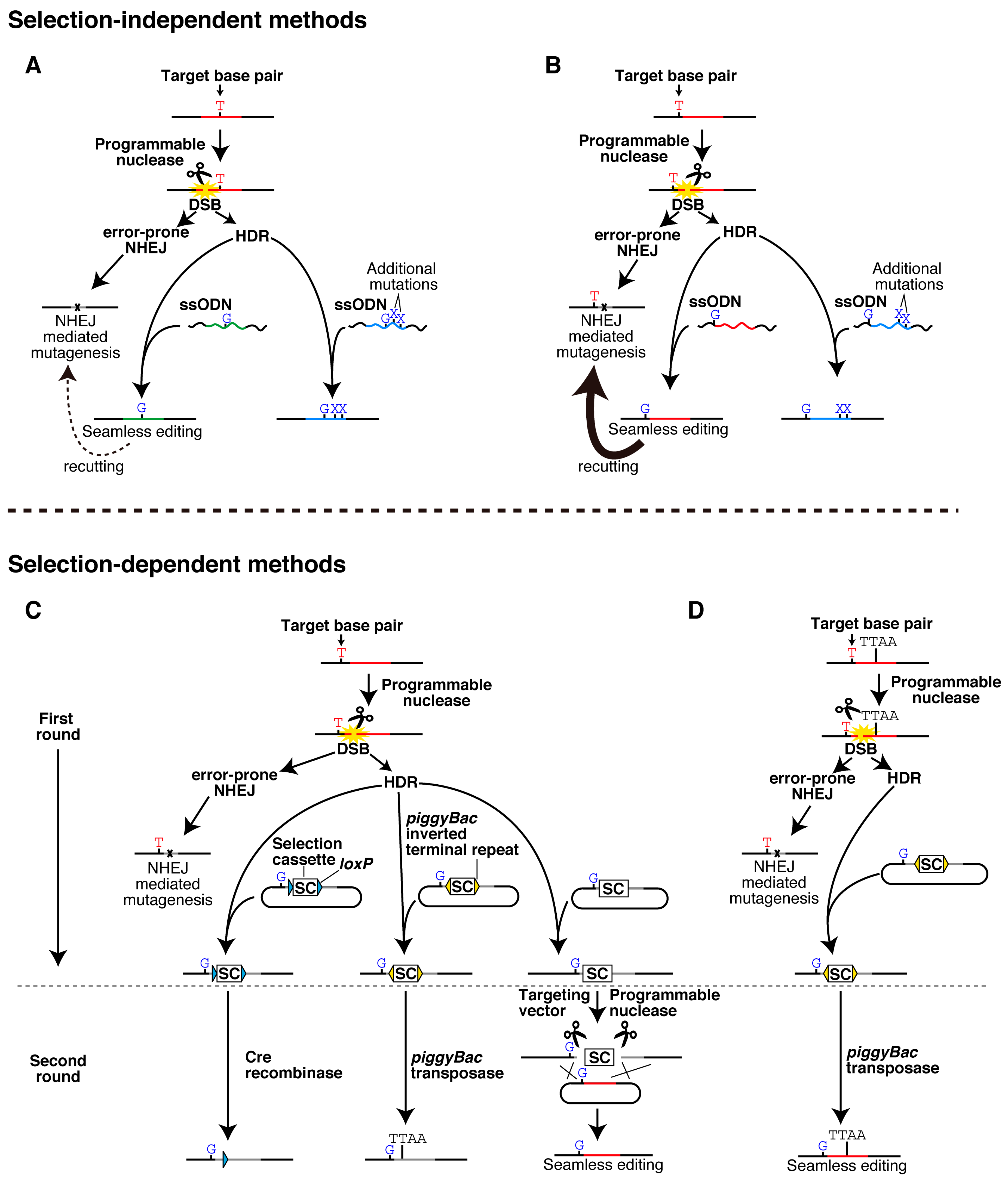{kind=link}
| Programmable Nuclease | Host Cell | Target | Template | Edits Introduced | Reference |
|---|---|---|---|---|---|
| ZFN | K562 | IL2Rγ | plasmid | 1 bp substitution | [14] |
| CD4+ T | |||||
| ZFN | K562 | RSK2 | ssODN | Substitution of 6 bp in and out of ZFN recognition site | [16] |
| ZFN | ES | SNCA | ssODN | 1 bp substitution | [17] |
| iPS | plasmid | ||||
| TALEN | iPS | CCR5 | ssODN | 2 bp substitutions | [18] |
| CRISPR | |||||
| TALEN | iPS | AKT2 | ssODN | 2 bp substitutions | [6] |
| TALEN | iPS | PHOX2B | ssODN | 1 bp substitution | [19] |
| PRKAG2 |
| Programmable Nuclease | Excision Method | Host Cell | Target | Edits Introduced | Reference |
|---|---|---|---|---|---|
| ZFN | Cre/loxP | ES iPS | SNCA | 1 bp substitution and loxP site insertion | [17] |
| ZFN | piggyBac | iPS | A1AT | 1 bp substitution of interest and 2 to generate TTAA site | [20] |
| CRISPR | piggyBac | iPS | HBB | 1 bp substitution and 4 bp insertion * | [15] |
| TALEN | piggyBac | iPS | HBB | 3 bp substitutions | [21] |
| TALEN | TALEN | HCT116 | Interge region (upstream of BUBR1) | 1 bp substitution | [5] |
2.1. Selection-Independent Methods
2.2. Selection-Dependent Methods
2.2.1. Cre/loxP-Mediated Excision
2.2.2. piggyBac-Mediated Excision
2.2.3. Excision by a Programmable Nuclease
3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Carroll, D. Genome engineering with targetable nucleases. Annu. Rev. Biochem. 2014, 83, 409–439. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. CRISPR, the disruptor. Nature 2015, 522, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, H.; Miyamoto, T.; Kanai, A.; Hosoba, K.; Sakuma, T.; Kudo, Y.; Asami, K.; Ogawa, A.; Watanabe, A.; Kajii, T.; et al. TALEN-mediated single-base-pair editing identification of an intergenic mutation upstream of BUB1B as causative of PCS (MVA) syndrome. Proc. Natl. Acad. Sci. USA 2014, 111, 1461–1466. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Lee, Y.-K.; Schaefer, E.A.K.; Peters, D.T.; Veres, A.; Kim, K.; Kuperwasser, N.; Motola, D.L.; Meissner, T.B.; Hendriks, W.T.; et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell 2013, 12, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Welter, D.; MacArthur, J.; Morales, J.; Burdett, T.; Hall, P.; Junkins, H.; Klemm, A.; Flicek, P.; Manolio, T.; Hindorff, L.; et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014, 42, D1001–D1006. [Google Scholar] [CrossRef] [PubMed]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- Shlyueva, D.; Stampfel, G.; Stark, A. Transcriptional enhancers: From properties to genome-wide predictions. Nat. Rev. Genet. 2014, 15, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Visser, M.; Kayser, M.; Palstra, R.-J. HERC2 rs12913832 modulates human pigmentation by attenuating chromatin-loop formation between a long-range enhancer and the OCA2 promoter. Genome Res. 2012, 22, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Ward, L.D.; Kellis, M. Interpreting noncoding genetic variation in complex traits and human disease. Nat. Biotechnol. 2012, 30, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K.; Strong, A.; Frank-Kamenetsky, M.; Lee, N.E.; Ahfeldt, T.; Sachs, K.V.; Li, X.; Li, H.; Kuperwasser, N.; Ruda, V.M.; et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature 2010, 466, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Urnov, F.D.; Miller, J.C.; Lee, Y.-L.; Beausejour, C.M.; Rock, J.M.; Augustus, S.; Jamieson, A.C.; Porteus, M.H.; Gregory, P.D.; Holmes, M.C. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 2005, 435, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Ye, L.; Chang, J.C.; Beyer, A.I.; Wang, J.; Muench, M.O.; Kan, Y.W. Seamless gene correction of β-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac. Genome Res. 2014, 24, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Pruett-Miller, S.M.; Huang, Y.; Gjoka, M.; Duda, K.; Taunton, J.; Collingwood, T.N.; Frodin, M.; Davis, G.D. High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat. Methods 2011, 8, 753–755. [Google Scholar] [CrossRef] [PubMed]
- Soldner, F.; Laganière, J.; Cheng, A.W.; Hockemeyer, D.; Gao, Q.; Alagappan, R.; Khurana, V.; Golbe, L.I.; Myers, R.H.; Lindquist, S.; et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset parkinson point mutations. Cell 2011, 146, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Guell, M.; Byrne, S.; Yang, J.L.; De Los Angeles, A.; Mali, P.; Aach, J.; Kim-Kiselak, C.; Briggs, A.W.; Rios, X.; et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Res. 2013, 41, 9049–9061. [Google Scholar] [CrossRef] [PubMed]
- Miyaoka, Y.; Chan, A.H.; Judge, L.M.; Yoo, J.; Huang, M.; Nguyen, T.D.; Lizarraga, P.P.; So, P.-L.; Conklin, B.R. Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nat. Methods 2014, 11, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Yusa, K.; Rashid, S.T.; Strick-Marchand, H.; Varela, I.; Liu, P.-Q.; Paschon, D.E.; Miranda, E.; Ordóñez, A.; Hannan, N.R.F.; Rouhani, F.J.; et al. Targeted gene correction of α1-antitrypsin deficiency in induced pluripotent stem cells. Nature 2011, 478, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Zhao, H. Seamless correction of the sickle cell disease mutation of the HBB gene in human induced pluripotent stem cells using TALENs. Biotechnol. Bioeng. 2014, 111, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, R.; Lombardo, A.; Arens, A.; Miller, J.C.; Genovese, P.; Kaeppel, C.; Nowrouzi, A.; Bartholomae, C.C.; Wang, J.; Friedman, G.; et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat. Biotechnol. 2011, 29, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Klug, A. The Discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu. Rev. Biochem. 2010, 79, 213–231. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Bikard, D.; Cox, D.; Zhang, F.; Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Meng, X.; Zhu, L.J.; Lawson, N.D.; Wolfe, S.A. Zinc finger protein-dependent and -independent contributions to the in vivo off-target activity of zinc finger nucleases. Nucleic Acids Res. 2010, 16, 17589–17610. [Google Scholar] [CrossRef] [PubMed]
- Guilinger, J.P.; Pattanayak, V.; Reyon, D.; Tsai, S.Q.; Sander, J.D.; Joung, J.K.; Liu, D.R. Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nat. Methods 2014, 1–91. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hu, Y.C.; Markoulaki, S.; Welstead, G.G.; Cheng, A.W.; Shivalila, C.S.; Pyntikova, T.; Dadon, D.B.; Voytas, D.F.; Bogdanove, A.J.; et al. TALEN-mediated editing of the mouse Y chromosome. Nat. Biotechnol. 2013, 31, 530–532. [Google Scholar] [CrossRef] [PubMed]
- Yusa, K. Seamless genome editing in human pluripotent stem cells using custom endonuclease-based gene targeting and the piggyBac transposon. Nat. Protoc. 2013, 8, 2061–2078. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A. Cre recombinase: the universal reagent for genome tailoring. Genesis 2000, 26, 99–109. [Google Scholar] [CrossRef]
- Semprini, S.; Troup, T.J.; Kotelevtseva, N.; King, K.; Davis, J.R.E.; Mullins, L.J.; Chapman, K.E.; Dunbar, D.R.; Mullins, J.J. Cryptic loxP sites in mammalian genomes: Genome-wide distribution and relevance for the efficiency of BAC/PAC recombineering techniques. Nucleic Acids Res. 2007, 35, 1402–1410. [Google Scholar] [CrossRef] [PubMed]
- Harno, E.; Cottrell, E.C.; White, A. Metabolic Pitfalls of CNS Cre-Based Technology. Cell Metab. 2013, 18, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.J.; Ciszczon, T.; Elick, T.; Bauser, C. Precise excision of TTAA-specific lepidopteran transposons piggyBac (IFP2) and tagalong (TFP3) from the baculovirus genome in cell lines from two species of Lepidoptera. Insect Mol. Biol. 1996, 5, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lin, C.; Lu, D.; Ning, Z.; Cox, T.; Melvin, D.; Wang, X.; Bradley, A.; Liu, P. Chromosomal transposition of PiggyBac in mouse embryonic stem cells. Proc. Natl. Acad. Sci. USA 2008, 105, 9290–9295. [Google Scholar] [CrossRef] [PubMed]
- Lacoste, A.; Berenshteyn, F.; Brivanlou, A.H. An efficient and reversible transposable system for gene delivery and lineage-specific differentiation in human embryonic stem cells. Cell Stem Cell 2009, 5, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Nakade, S.; Tsubota, T.; Sakane, Y.; Kume, S.; Sakamoto, N.; Obara, M.; Daimon, T.; Sezutsu, H.; Yamamoto, T.; Sakuma, T.; et al. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nat. Commun. 2014, 5, 5560. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.C.; Holmes, M.C.; Wang, J.; Guschin, D.Y.; Lee, Y.-L.; Rupniewski, I.; Beausejour, C.M.; Waite, A.J.; Wang, N.S.; Kim, K.A.; et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat. Biotechnol. 2007, 25, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Szczepek, M.; Brondani, V.; Büchel, J.; Serrano, L.; Segal, D.J.; Cathomen, T. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat. Biotechnol. 2007, 25, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Doyon, Y.; Vo, T.D.; Mendel, M.C.; Greenberg, S.G.; Wang, J.; Xia, D.F.; Miller, J.C.; Urnov, F.D.; Gregory, P.D.; Holmes, M.C. Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. Nat. Methods 2010, 8, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.-Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Topkar, V.V.; Nguyen, N.T.; Zheng, Z.; Gonzales, A.P.W.; Li, Z.; Peterson, R.T.; Yeh, J.-R.J.; et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 2015, 523, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 2014, 156, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.; Niewoehner, O.; Duerst, A.; Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 2014, 513, 569–573. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ochiai, H. Single-Base Pair Genome Editing in Human Cells by Using Site-Specific Endonucleases. Int. J. Mol. Sci. 2015, 16, 21128-21137. https://doi.org/10.3390/ijms160921128
Ochiai H. Single-Base Pair Genome Editing in Human Cells by Using Site-Specific Endonucleases. International Journal of Molecular Sciences. 2015; 16(9):21128-21137. https://doi.org/10.3390/ijms160921128
Chicago/Turabian StyleOchiai, Hiroshi. 2015. "Single-Base Pair Genome Editing in Human Cells by Using Site-Specific Endonucleases" International Journal of Molecular Sciences 16, no. 9: 21128-21137. https://doi.org/10.3390/ijms160921128
APA StyleOchiai, H. (2015). Single-Base Pair Genome Editing in Human Cells by Using Site-Specific Endonucleases. International Journal of Molecular Sciences, 16(9), 21128-21137. https://doi.org/10.3390/ijms160921128