
Microarray Analysis on Gene Regulation by Estrogen, Progesterone and Tamoxifen in Human Endometrial Stromal Cells

Abstract
:1. Introduction
2. Results
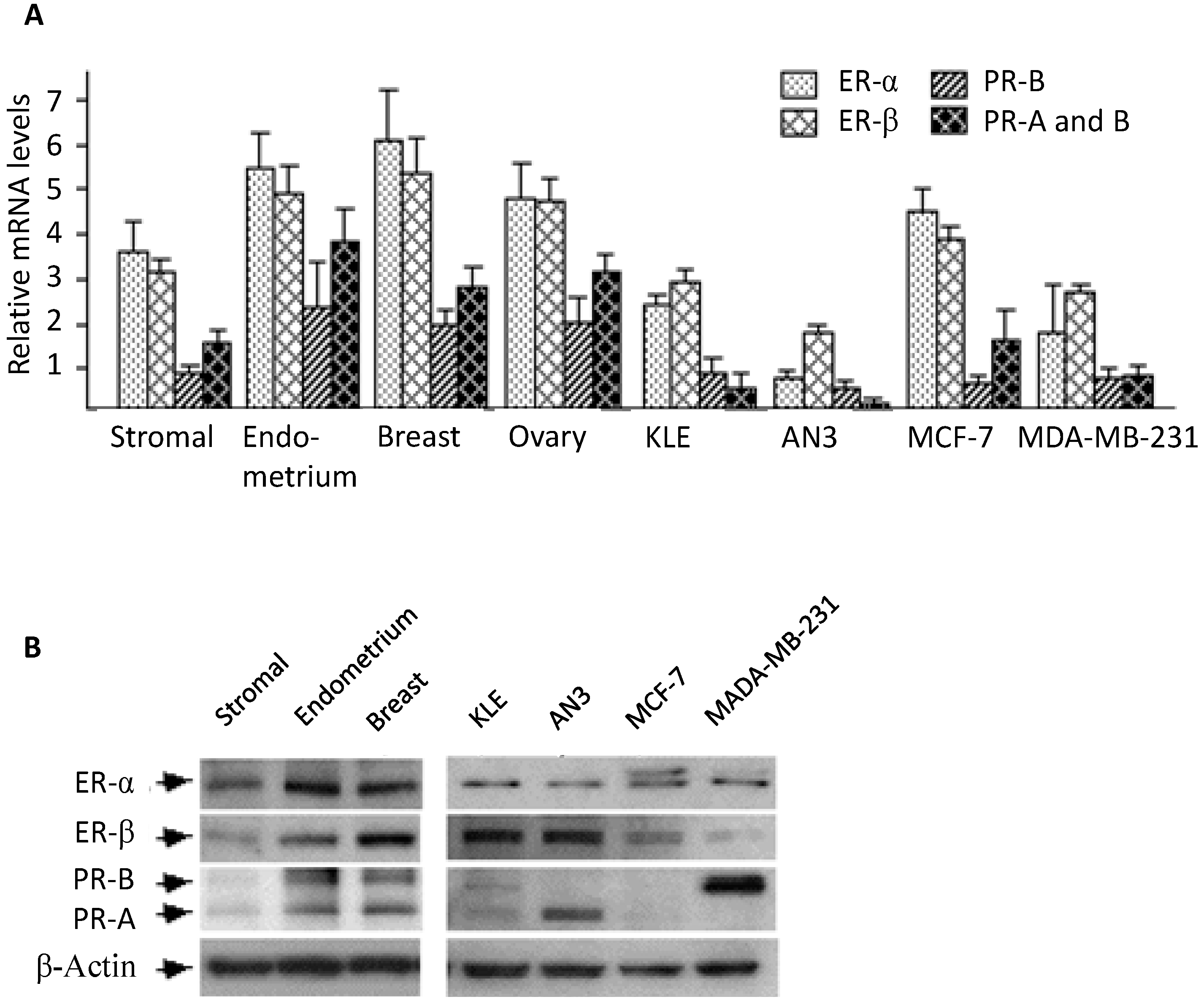
2.1. Expression of Estrogen and Progesterone Receptors in Stromal Cell Cultures
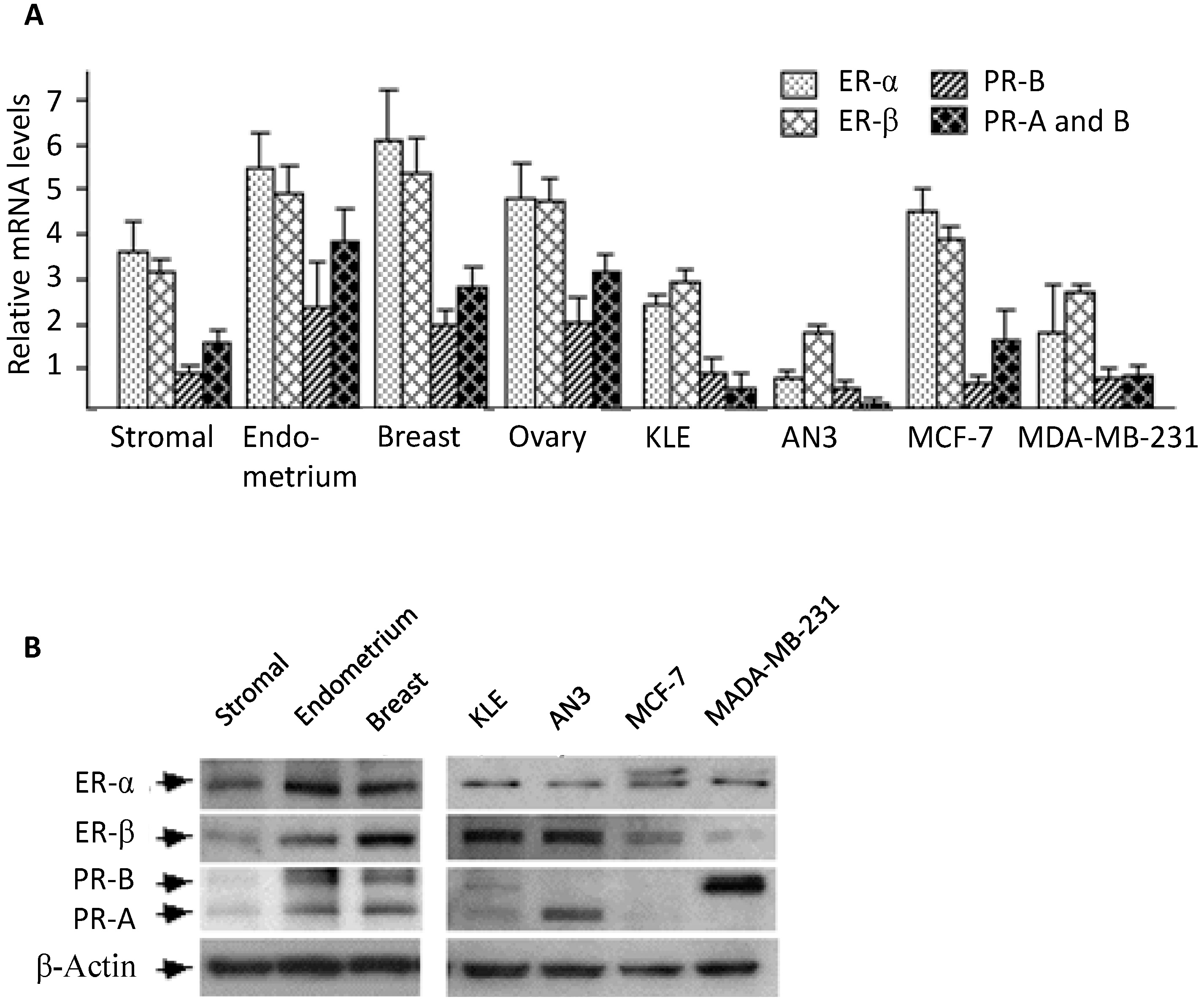
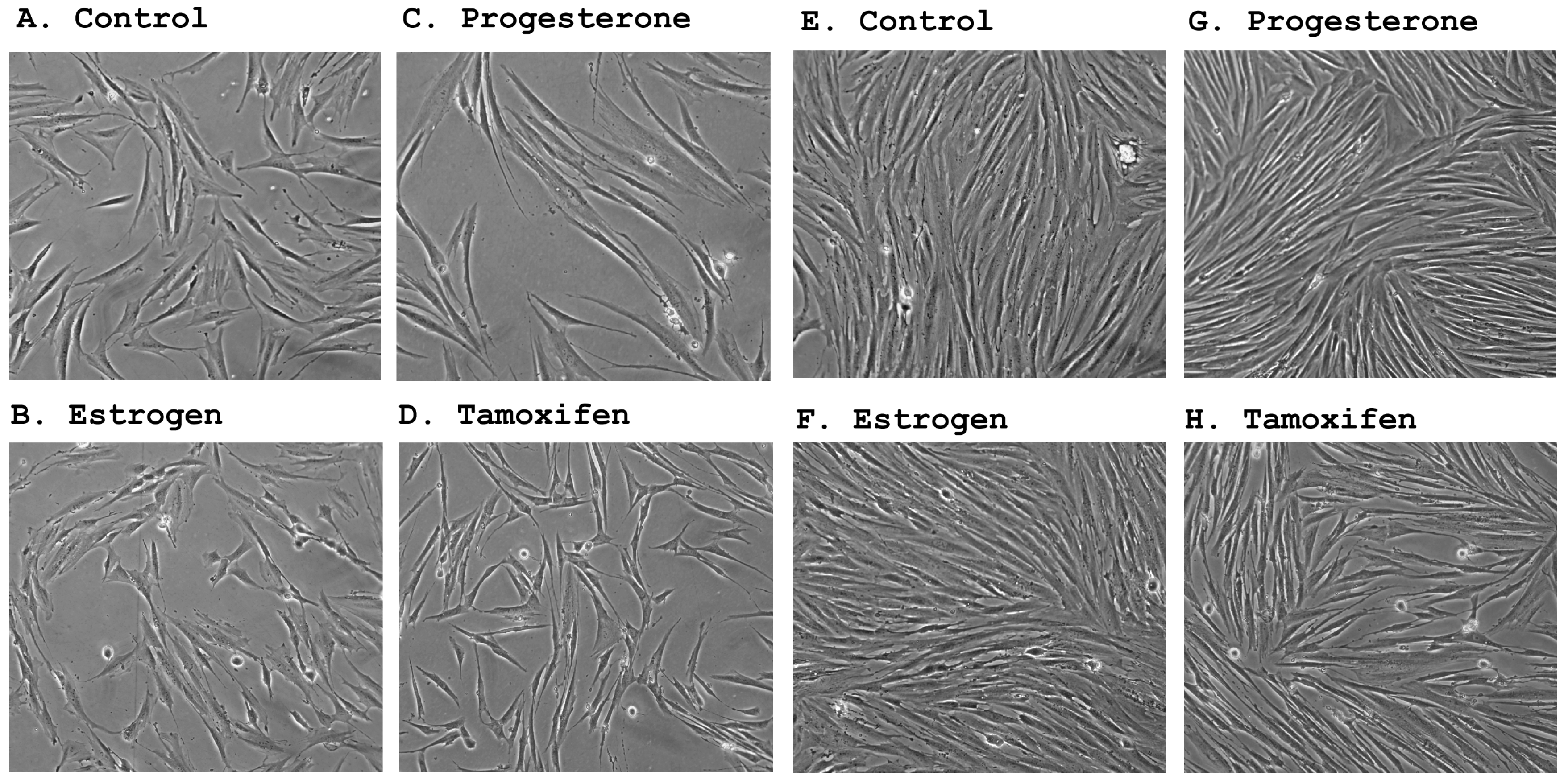
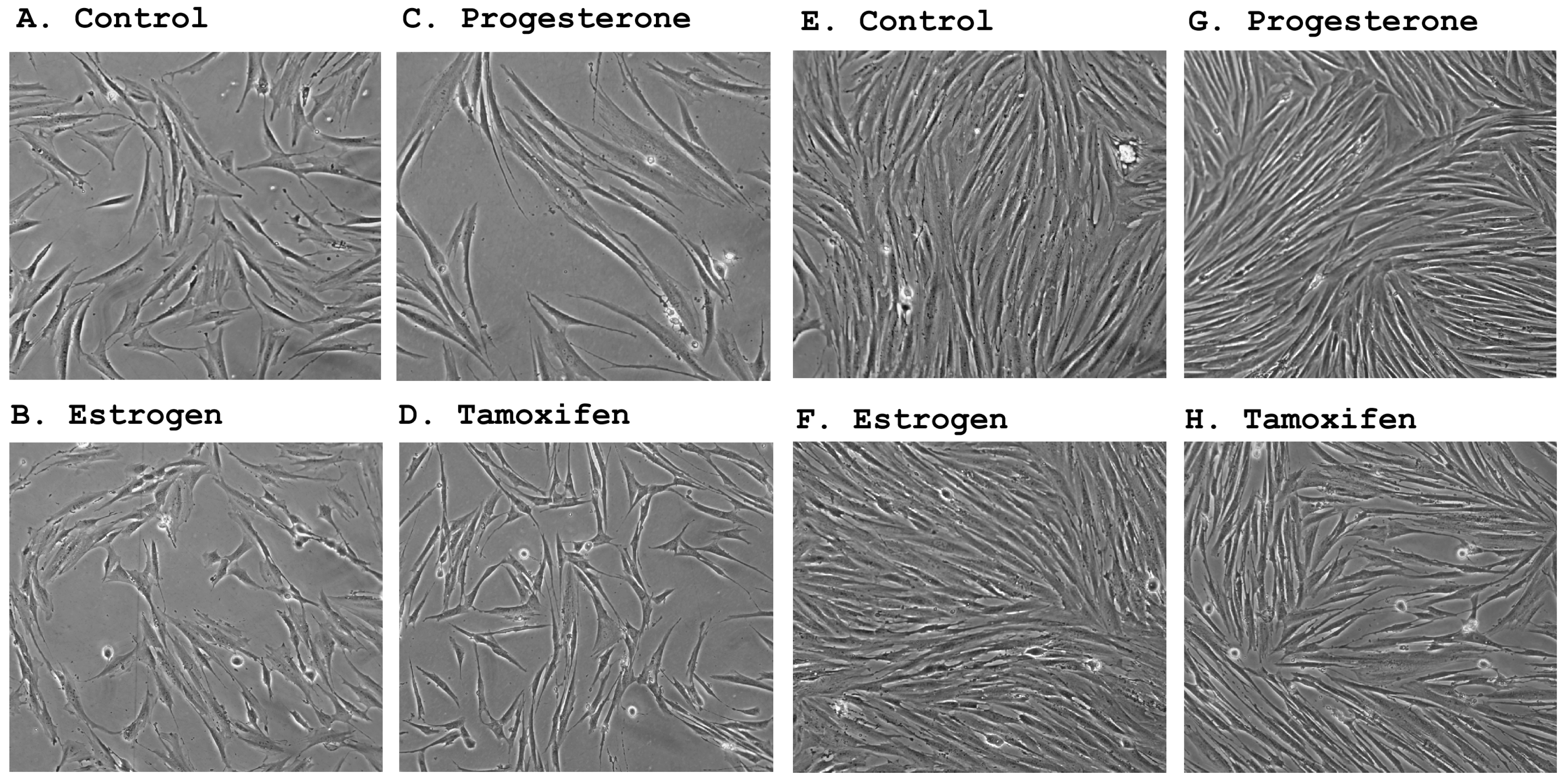
2.2. Morphological Changes Following Hormonal Treatment
2.3. Estrogen and Progesterone Induce Different Gene Expression

{kind=link}
{kind=link}
{kind=link}
| Gene Name | Description | E2/Ctl |
|---|---|---|
| Structural | ||
| CNTN4 | contactin 4 | 0.08 |
| TRAR1 | trace amine receptor 1 | 0.10 |
| COL14A1 | collagen, type XIV, α1 (undulin) | 0.14 |
| SFXN5 | sideroflexin 5 | 0.15 |
| PAPLN | papilin, proteoglycan-like sulfated glycoprotein | 0.17 |
| CDRT1 | CMT1A duplicated region transcript 1 | 0.29 |
| SHPRH | SNF2 histone linker PHD RING helicase | 3.10 |
| OR2W1 | olfactory receptor, family 2, subfamily W, member 1 | 3.49 |
| CANP | cancer-associated nucleoprotein | 3.66 |
| COL9A1 | collagen, type IX, α1 | 6.06 |
| HIST2H4 | histone 2, H4 | 6.28 |
| SESN3 | sestrin 3 | 6.34 |
| TUBB1 | tubulin, β1 | 9.88 |
| CLECSF12 | C-type (calcium dependent, carbohydrate-recognition domain) lectin, superfamily member 12 | 23.48 |
| Enzymes | ||
| SMA4 | SMA4 | 0.16 |
| TLL1 | tolloid-like 1 | 0.21 |
| ANG | angiogenin, ribonuclease, RNase A family, 5 | 0.22 |
| B3GALT4 | UDP-Gal:β GlcNAc β 1,3-galactosyltransferase, polypeptide 4 | 0.24 |
| PDE11A | phosphodiesterase 11A | 0.25 |
| ABCG4 | ATP-binding cassette, sub-family G (WHITE), member 4 | 0.26 |
| PFKFB2 | 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 2 | 0.26 |
| CYP7B1 | cytochrome P450, family 7, subfamily B, polypeptide 1 | 0.29 |
| ENPP1 | ectonucleotide pyrophosphatase/phosphodiesterase 1 | 3.21 |
| TIGD6 | tigger transposable element derived 6 | 3.54 |
| MMRN1 | multimerin 1 | 5.24 |
| RTN4IP1 | reticulon 4 interacting protein 1 | 9.90 |
| PTPRZ1 | protein tyrosine phosphatase, receptor-type, Z polypeptide 1 | 14.49 |
| Transcription Factors | ||
| ZNF608 | zinc finger protein 608 | 0.13 |
| TSPAN-2 | tetraspan 2 | 0.21 |
| ZNF75 | zinc finger protein 75 (D8C6) | 0.24 |
| ZIC4 | Zic family member 4 | 0.27 |
| ZNF423 | zinc finger protein 423 | 0.31 |
| POGZ | pogo transposable element with ZNF domain | 0.33 |
| MXI1 | MAX interactor 1 | 0.33 |
| CLDN23 | claudin 23 | 3.12 |
| ZNF197 | zinc finger protein 197 | 3.65 |
| TTTY15 | testis-specific transcript, Y-linked 15 | 5.84 |
| Signaling | ||
| IL12B | interleukin 12B (natural killer cell stimulatory factor 2, cytotoxic lymphocyte maturation factor 2, p40) | 0.07 |
| STARS | striated muscle activator of Rho-dependent signaling | 0.13 |
| CFC1 | cripto, FRL-1, cryptic family 1 | 0.16 |
| GPR133 | G protein-coupled receptor 133 | 0.28 |
| OLFML2A | olfactomedin-like 2A | 0.30 |
| PTTG2 | pituitary tumor-transforming 2 | 3.04 |
| EDG7 | endothelial differentiation, lysophosphatidic acid G-protein-coupled receptor, 7 | 3.06 |
| BAGE | B melanoma antigen | 3.08 |
| MKLN1 | muskelin 1, intracellular mediator containing kelch motifs | 3.24 |
| Cell cycle and Apoptosis | ||
| ID1 | inhibitor of DNA binding 1, dominant negative helix-loop-helix protein | 3.82 |
| CHAF1B | chromatin assembly factor 1, subunit B (p60) | 4.09 |
| IL8 | interleukin 8 | 5.18 |
| ID3 | inhibitor of DNA binding 3, dominant negative helix-loop-helix protein | 6.51 |
| Novel Genes | ||
| FLJ31842 | hypothetical protein FLJ31842 | 0.06 |
| FLJ13544 | hypothetical protein FLJ13544 | 0.18 |
| LOC338773 | hypothetical protein LOC338773 | 0.24 |
| LOC200169 | hypothetical protein LOC200169 | 7.28 |
| MGC39830 | hypothetical protein MGC39830 | 8.20 |
| Gene Name | Description | Prog/Ctl |
|---|---|---|
| Structural | ||
| LMNB1 | lamin B1 | 0.18 |
| JUP | junction plakoglobin | 0.18 |
| SYNPO | synaptopodin | 0.20 |
| ACTG2 | actin, γ2, smooth muscle, enteric | 0.23 |
| ARHGDIB | Rho GDP dissociation inhibitor (GDI) β | 0.27 |
| COL11A1 | collagen, type XI, α1 | 0.28 |
| ANLN | anillin, actin binding protein (scraps homolog, Drosophila) | 0.32 |
| ITGA2 | integrin, α2 (CD49B, α2 subunit of VLA-2 receptor) | 3.19 |
| STCH | stress 70 protein chaperone, microsome-associated, 60 kDa | 3.42 |
| ADFP | adipose differentiation-related protein | 3.71 |
| TUBE1 | tubulin, epsilon 1 | 4.44 |
| NPC1 | Niemann-Pick disease, type C1 | 5.63 |
| Enzymes | ||
| SMA4 | SMA4 | 0.06 |
| CYP1B1 | cytochrome P450, family 1, subfamily B, polypeptide 1 | 0.13 |
| MEST | mesoderm specific transcript homolog (mouse) | 0.13 |
| PLK4 | polo-like kinase 4 (Drosophila) | 0.22 |
| BRIP1 | BRCA1 interacting protein C-terminal helicase 1 | 0.24 |
| RNASEL | ribonuclease L (2',5'-oligoisoadenylate synthetase-dependent) | 0.31 |
| ASNS | asparagine synthetase | 3.01 |
| NNMT | nicotinamide N-methyltransferase | 3.43 |
| HS3ST3A1 | heparan sulfate (glucosamine) 3-O-sulfotransferase 3A1 | 3.72 |
| LIAS | lipoic acid synthetase | 7.07 |
| PRKY | protein kinase, Y-linked | 9.49 |
| JAK2 | Janus kinase 2 (a protein tyrosine kinase) | 10.82 |
| Transcription Factors | ||
| SOX4 | SRY (sex determining region Y)-box 4 | 0.15 |
| ZNF323 | zinc finger protein 323 | 0.17 |
| FOXL2 | forkhead box L2 | 0.33 |
| TBX2 | T-box 2 | 3.01 |
| E2F7 | E2F transcription factor 7 | 3.04 |
| DDIT4 | DNA-damage-inducible transcript 4 | 3.31 |
| TGIF | TGFB-induced factor (TALE family homeobox) | 3.44 |
| FOXD1 | forkhead box D1 | 3.78 |
| ZNF197 | zinc finger protein 197 | 3.84 |
| DSCR1 | Down syndrome critical region gene 1 | 3.88 |
| ID3 | inhibitor of DNA binding 3, dominant negative helix-loop-helix protein | 5.25 |
| DDIT3 | DNA-damage-inducible transcript 3 | 8.18 |
| TCF21 | transcription factor 21 | 13.97 |
| Signaling | ||
| TACSTD1 | tumor-associated calcium signal transducer 1 | 0.21 |
| PIR51 | RAD51 associated protein 1 | 0.26 |
| FGF9 | fibroblast growth factor 9 (glia-activating factor) | 0.29 |
| RHOB | ras homolog gene family, member B | 0.31 |
| IL6 | interleukin 6 (interferon, β2) | 3.28 |
| VEGF | vascular endothelial growth factor | 3.39 |
| HSPBAP1 | HSPB (heat shock 27 kDa) associated protein 1 | 3.47 |
| AREG | amphiregulin (schwannoma-derived growth factor) | 3.67 |
| GDF15 | growth differentiation factor 15 | 4.49 |
| GADD45A | growth arrest and DNA-damage-inducible, α | 5.02 |
| RIS1 | Ras-induced senescence 1 | 6.32 |
| Cell cycle and Apoptosis | ||
| CDC25A | cell division cycle 25A | 0.13 |
| SKP2 | S-phase kinase-associated protein 2 (p45) | 0.24 |
| CCNE2 | cyclin E2 | 0.29 |
| CDKN2B | cyclin-dependent kinase inhibitor 2B (p15, inhibits CDK4) | 3.12 |
| SH3MD2 | SH3 multiple domains 2 | 3.29 |
| BTG1 | B-cell translocation gene 1, anti-proliferative | 3.50 |
| Novel Genes | ||
| FLJ11795 | hypothetical protein FLJ11795 | 0.04 |
| LOC283112 | hypothetical protein LOC283112 | 0.05 |
| LOC145741 | hypothetical LOC145741 | 7.70 |
| FLJ11011 | hypothetical protein FLJ11011 | 7.95 |
| MGC39830 | hypothetical protein MGC39830 | 8.94 |
| Gene Name | Description | E2/Ctl | Prog/Ctl |
|---|---|---|---|
| Structural | |||
| COL4A3BP | collagen, type IV, α3 (Goodpasture antigen) binding protein | 0.62 | 2.05 |
| MYLIP | myosin regulatory light chain interacting protein | 0.63 | 2.56 |
| LAMA1 | laminin, α1 | 0.69 | 2.17 |
| TUBE1 | tubulin, epsilon 1 | 0.71 | 4.44 |
| HIST1H2AC | histone 1, H2ac | 0.84 | 2.71 |
| CHS1 | Chediak-Higashi syndrome 1 | 1.09 | 2.14 |
| H2AFX | H2A histone family, member X | 1.69 | 0.47 |
| KIF20A | kinesin family member 20A | 2.23 | 0.44 |
| TMPO | thymopoietin | 2.24 | 0.61 |
| OIP5 | Opa-interacting protein 5 | 2.26 | 0.52 |
| HCAP-G | chromosome condensation protein G | 2.41 | 0.65 |
| NEK2 | NIMA (never in mitosis gene a)-related kinase 2 | 2.45 | 0.58 |
| MAD2L1 * | MAD2 mitotic arrest deficient-like 1 (yeast) | 2.56 | 0.47 |
| Enzymes | |||
| SCD | stearoyl-CoA desaturase (δ-9-desaturase) | 0.43 | 1.96 |
| HMGCR | 3-hydroxy-3-methylglutaryl-Coenzyme A reductase | 0.44 | 1.97 |
| ASNS | asparagine synthetase | 0.66 | 3.01 |
| EIF2AK3 | eukaryotic translation initiation factor 2-α kinase 3 | 0.81 | 2.58 |
| PRIM2A | primase, polypeptide 2A, 58 kDa | 1.54 | 0.47 |
| FEN1 | flap structure-specific endonuclease 1 | 1.93 | 0.44 |
| POLA2 | polymerase (DNA-directed), α (70 kD) | 2.17 | 0.84 |
| TOP2A * | topoisomerase (DNA) II α 170 kDa | 2.26 | 0.41 |
| DNA2L * | DNA2 DNA replication helicase 2-like (yeast) | 2.43 | 0.37 |
| POLE2 * | polymerase (DNA directed), epsilon 2 (p59 subunit) | 2.52 | 0.48 |
| PLK4 * | polo-like kinase 4 (Drosophila) | 2.56 | 0.22 |
| TOPK | T-LAK cell-originated protein kinase | 2.77 | 0.51 |
| Transcription Factors | |||
| DDIT4 * | DNA-damage-inducible transcript 4 | 0.35 | 3.31 |
| DDIT4L | DNA-damage-inducible transcript 4-like | 0.69 | 2.01 |
| ATF4 | activating transcription factor 4 (tax-responsive enhancer element B67) | 0.69 | 2.26 |
| DSCR3 | Down syndrome critical region gene 3 | 0.82 | 2.10 |
| ATF3 | activating transcription factor 3 | 0.85 | 3.65 |
| MAFF | v-maf musculoaponeurotic fibrosarcoma oncogene homolog F (avian) | 0.87 | 2.45 |
| NFIL3 | nuclear factor, interleukin 3 regulated | 0.88 | 3.74 |
| TGIF | TGFB-induced factor (TALE family homeobox) | 0.90 | 3.44 |
| OLIG1 | oligodendrocyte transcription factor 1 | 2.16 | 0.85 |
| TCF19 | transcription factor 19 (SC1) | 2.43 | 0.83 |
| PTTG1 | pituitary tumor-transforming 1 | 2.48 | 0.67 |
| FOXM1 | forkhead box M1 | 2.53 | 0.82 |
| MYBL1 | v-myb myeloblastosis viral oncogene homolog (avian)-like 1 | 2.60 | 0.66 |
| Signaling | |||
| STARS * | striated muscle activator of Rho-dependent signaling | 0.13 | 2.25 |
| RAB33A * | RAB33A, member RAS oncogene family | 0.42 | 3.00 |
| NGFB | nerve growth factor, β polypeptide | 0.58 | 2.69 |
| GDF15 | growth differentiation factor 15 | 0.67 | 4.49 |
| MAP4K3 | mitogen-activated protein kinase kinase kinase kinase 3 | 0.75 | 2.04 |
| VEGF | vascular endothelial growth factor | 0.85 | 3.39 |
| MTSS1 | metastasis suppressor 1 | 0.90 | 3.72 |
| GADD45A | growth arrest and DNA-damage-inducible, α | 0.92 | 5.02 |
| DNAJB9 | DnaJ (Hsp40) homolog, subfamily B, member 9 | 0.95 | 6.25 |
| SHCBP1 | SHC SH2-domain binding protein 1 | 2.05 | 0.41 |
| MYCBP | c-myc binding protein | 2.06 | 0.73 |
| ECT2 | epithelial cell transforming sequence 2 oncogene | 2.35 | 0.65 |
| PIR51 | RAD51 associated protein 1 | 2.45 | 0.26 |
| Cell cycle and Apoptosis | |||
| KLF4 | Kruppel-like factor 4 (gut) | 0.51 | 2.93 |
| CDKN2B | cyclin-dependent kinase inhibitor 2B (p15, inhibits CDK4) | 0.52 | 2.92 |
| CCNE2 * | cyclin E2 | 2.00 | 0.29 |
| FANCG | Fanconi anemia, complementation group G | 2.14 | 0.70 |
| CDC2 | cell division cycle 2, G1 to S and G2 to M | 2.15 | 0.53 |
| CDCA2 * | cell division cycle associated 2 | 2.17 | 0.48 |
| MTB * | more than blood homolog | 2.23 | 0.49 |
| CCNA2 | cyclin A2 | 2.31 | 0.53 |
| CCNB1 * | cyclin B1 | 2.32 | 0.48 |
| CDCA3 * | cell division cycle associated 3 | 2.34 | 0.45 |
| CDKN3 | cyclin-dependent kinase inhibitor 3 (CDK2-associated dual specificity phosphatase) | 2.40 | 0.69 |
| CCNB2 | cyclin B2 | 2.45 | 0.55 |
| CDC20 | CDC20 cell division cycle 20 homolog (S. cerevisiae) | 3.29 | 0.58 |
| Novel Genes | |||
| FLJ20366 * | hypothetical protein FLJ20366 | 0.38 | 2.73 |
| LOC153346 * | hypothetical protein LOC153346 | 0.46 | 2.71 |
| FLJ20105 * | hypothetical protein FLJ20105 | 2.77 | 0.33 |
| FLJ10719 | hypothetical protein FLJ10719 | 3.22 | 0.52 |
| FLJ13273 | hypothetical protein FLJ13273 | 4.82 | 0.67 |
| Gene Name | Description | E2/Ctl | Tam/Ctl |
|---|---|---|---|
| Structural | |||
| CRABP2 | cellular retinoic acid binding protein 2 | 0.33 | 0.49 |
| KCTD7 | potassium channel tetramerisation domain containing 7 | 0.38 | 0.39 |
| FLRT2 | fibronectin leucine rich transmembrane protein 2 | 0.39 | 0.47 |
| DAAM2 | dishevelled associated activator of morphogenesis 2 | 0.46 | 0.48 |
| CHAF1A | chromatin assembly factor 1, subunit A (p150) | 2.00 | 2.44 |
| CNAP1 | chromosome condensation-related SMC-associated protein 1 | 2.01 | 2.45 |
| KNTC2 | kinetochore associated 2 | 2.02 | 2.92 |
| ITGA2 | integrin, α2 (CD49B, α2 subunit of VLA-2 receptor) | 2.16 | 2.86 |
| OIP5 | Opa-interacting protein 5 | 2.26 | 2.47 |
| RAMP | RA-regulated nuclear matrix-associated protein | 2.40 | 3.09 |
| HCAP-G | chromosome condensation protein G | 2.41 | 2.96 |
| NEK2 | NIMA (never in mitosis gene a)-related kinase 2 | 2.45 | 2.46 |
| MAD2L1 | MAD2 mitotic arrest deficient-like 1 (yeast) | 2.56 | 2.22 |
| PODXL | podocalyxin-like | 2.69 | 3.95 |
| Enzymes | |||
| SMA4 | SMA4 | 0.16 | 0.42 |
| MMP11 | matrix metalloproteinase 11 (stromelysin 3) | 0.48 | 0.41 |
| RRM2 | ribonucleotide reductase M2 polypeptide | 2.09 | 2.35 |
| RNASEH2A | ribonuclease H2, large subunit | 2.12 | 2.60 |
| POLA2 | polymerase (DNA-directed), α (70 kD) | 2.17 | 3.08 |
| TOP2A | topoisomerase (DNA) II α 170 kDa | 2.31 | 2.60 |
| POLE2 | polymerase (DNA directed), epsilon 2 (p59 subunit) | 2.52 | 3.01 |
| CDKN3 | cyclin-dependent kinase inhibitor 3 (CDK2-associated dual specificity phosphatase) | 2.62 | 2.64 |
| HAS2 | hyaluronan synthase 2 | 2.72 | 4.27 |
| MMP1 | matrix metalloproteinase 1 (interstitial collagenase) | 2.73 | 8.91 |
| PLK1 | polo-like kinase 1 (Drosophila) | 2.75 | 3.86 |
| BUB1 | BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) | 2.81 | 2.48 |
| Transcription Factors | |||
| POGZ | pogo transposable element with ZNF domain | 0.33 | 0.39 |
| KLF4 | Kruppel-like factor 4 (gut) | 0.42 | 0.48 |
| PBXIP1 | pre-B-cell leukemia transcription factor interacting protein 1 | 0.44 | 0.49 |
| RFC4 | replication factor C (activator 1) 4, 37 kDa | 2.03 | 2.69 |
| TCF19 | transcription factor 19 (SC1) | 2.43 | 2.16 |
| PTTG1 | pituitary tumor-transforming 1 | 2.48 | 2.95 |
| FOXM1 | forkhead box M1 | 2.53 | 3.39 |
| MYBL1 | v-myb myeloblastosis viral oncogene homolog (avian)-like 1 | 2.60 | 2.60 |
| ID1 | inhibitor of DNA binding 1, dominant negative helix-loop-helix protein | 3.82 | 4.37 |
| Signaling | |||
| DDIT4 | DNA-damage-inducible transcript 4 | 0.35 | 0.26 |
| RASSF2 | Ras association (RalGDS/AF-6) domain family 2 | 0.44 | 0.38 |
| RASGRP1 | RAS guanyl releasing protein 1 (calcium and DAG-regulated) | 0.50 | 0.37 |
| FBXO5 | F-box protein 5 | 2.20 | 2.16 |
| RANBP1 | RAN binding protein 1 | 2.21 | 2.11 |
| ECT2 | epithelial cell transforming sequence 2 oncogene | 2.35 | 2.09 |
| PIR51 | RAD51 associated protein 1 | 2.45 | 2.34 |
| RGS4 | regulator of G-protein signalling 4 | 2.94 | 3.81 |
| IL8 | interleukin 8 | 5.18 | 2.00 |
| Cell cycle and Apoptosis | |||
| TRIB3 | tribbles homolog 3 (Drosophila) | 0.47 | 0.35 |
| GTSE1 | G-2 and S-phase expressed 1 | 2.07 | 2.42 |
| FANCG | Fanconi anemia, complementation group G | 2.14 | 2.23 |
| CDC2 | cell division cycle 2, G1 to S and G2 to M | 2.20 | 2.12 |
| MTB | more than blood homolog | 2.23 | 2.20 |
| CCNA2 | cyclin A2 | 2.31 | 2.70 |
| CCNB1 | cyclin B1 | 2.32 | 2.71 |
| CCNB2 | cyclin B2 | 2.45 | 2.83 |
| CDCA1 | cell division cycle associated 1 | 2.61 | 3.56 |
| FANCE | Fanconi anemia, complementation group E | 2.95 | 2.62 |
| Novel Genes | |||
| LOC338773 | hypothetical protein LOC338773 | 0.24 | 0.43 |
| KIAA1164 | hypothetical protein KIAA1164 | 0.31 | 0.34 |
| DKFZP434L142 | hypothetical protein DKFZp434L142 | 0.39 | 0.34 |
| FLJ10719 | hypothetical protein FLJ10719 | 3.22 | 4.02 |
| FLJ13273 | hypothetical protein FLJ13273 | 4.82 | 3.96 |
| Gene Name | Description | E2/Ctl | Tam/Ctl |
|---|---|---|---|
| Structural | |||
| ARRDC2 | arrestin domain containing 2 | 0.58 | 2.34 |
| SLC16A6 | solute carrier family 16 (monocarboxylic acid transporters), member 6 | 0.77 | 2.33 |
| SYCP2 | synaptonemal complex protein 2 | 2.48 | 0.84 |
| Enzymes | |||
| PIN1 | protein (peptidyl-prolyl cis/trans isomerase) NIMA-interacting 1 | 0.72 | 2.21 |
| Transcription Factors | |||
| OLIG1 | oligodendrocyte transcription factor 1 | 2.16 | 0.86 |
| Signaling | |||
| NOV | nephroblastoma overexpressed gene | 0.83 | 2.00 |
| Novel Genes | |||
| FLJ38993 | hypothetical protein FLJ38993 | 0.77 | 2.01 |
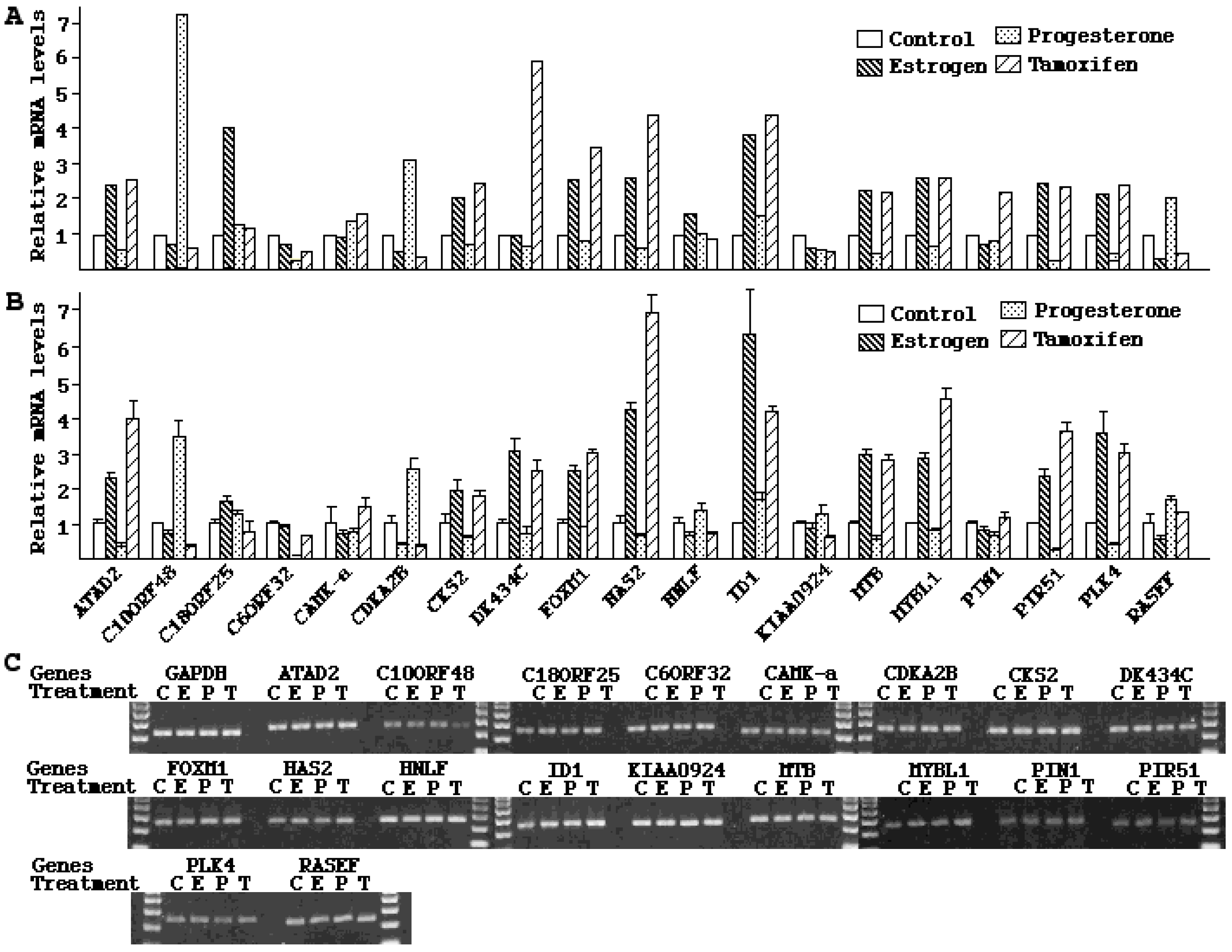
2.4. Validation of Microarray Results
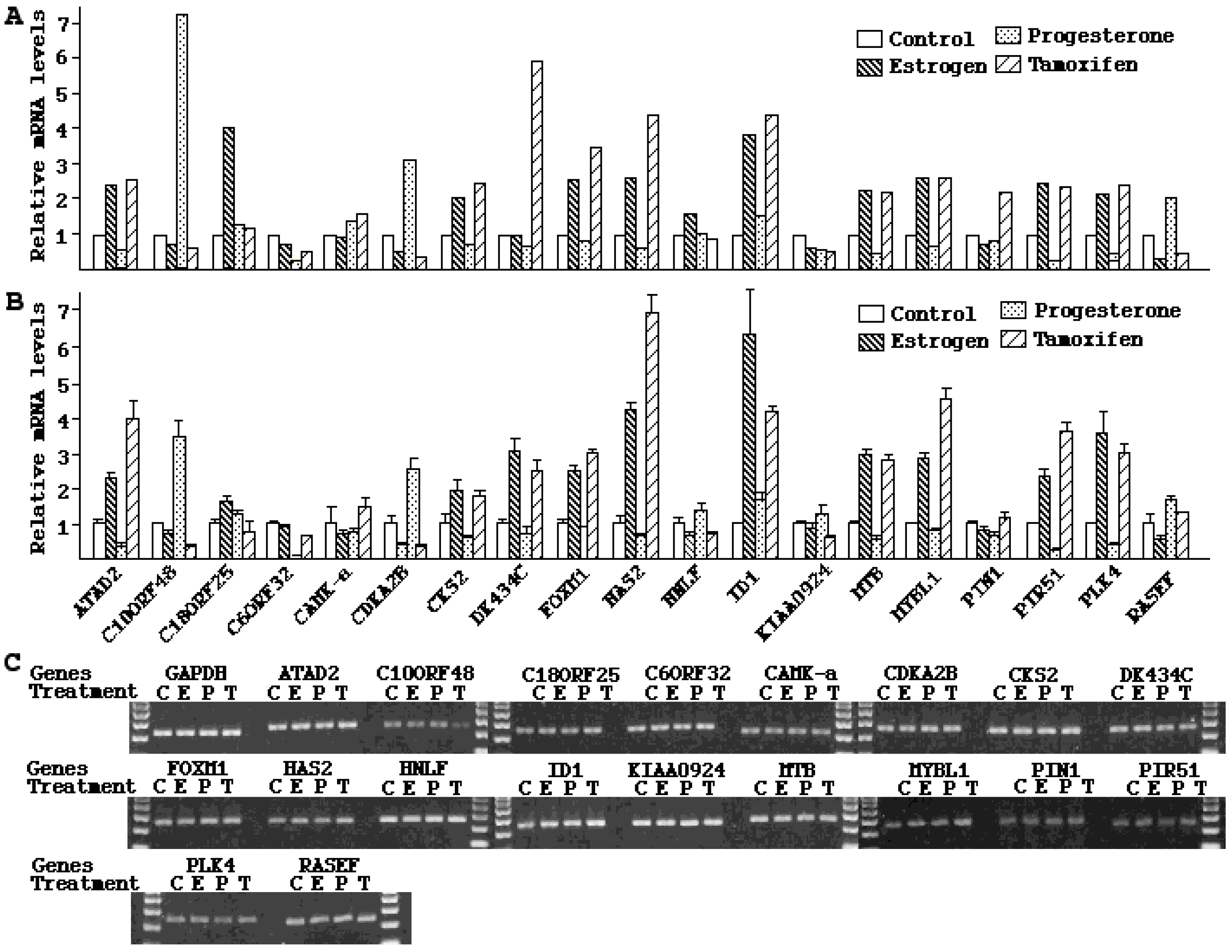
3. Discussion
4. Material and Methods
4.1. Tissue, Cell Lines, and Reagents
4.2. Cell Treatment, RNA Isolation, and Quantitative Real-Time PCR
| Gene Name | Description | 5' Primer | 3' Primer | Size (bp) |
|---|---|---|---|---|
| ER-α | Estrogen receptor α | aattcagataatcgacgccag | gtgtttcaacattctccctcctc | 344 |
| ER-β | Estrogen receptor β | tgcggaacctcaaaagagtc | cttcacacgaccagactcca | 206 |
| PR-AB | Progesterone receptor A and B | atgagccggtccgggtgcaag | gccacccagagcccgaggttt | 243 |
| PR-B | Progesterone receptor B | gactgagagcttcacagtat | tctcctaactcggggagttct | 187 |
| ATAD2 | ATPase family, AAA domain containing 2 | gattatcttccgcaggacca | gttgcattggatcaacatcg | 255 |
| C10ORF48 | chromosome 10 open reading frame 48 | gggtcaatagtgcagccagt | tgcgcttactgttactgcaaa | 247 |
| C18ORF25 | chromosome 18 open reading frame 25 | gtaggggccagactgaatga | agtgtccccagctttttcaa | 250 |
| C6ORF32 | chromosome 6 open reading frame 32 | aggagaaaatgccactgtcg | tcctctgggtcttcctcctt | 250 |
| CAMK-a | calcium/calmodulin-dependent protein kinase II | acgagaagctgagcccctac | ttgggggagttagacaccag | 221 |
| CDKN2B | cyclin-dependent kinase inhibitor 2B (p15, inhibits CDK4) | tcgtttgcttttcagggttt | cctcctccactttgtcctca | 248 |
| CKS2 | CDC28 protein kinase regulatory subunit 2 | ggagtggaggagacttggtg | cagctcatgcacaggtatgg | 236 |
| DK434C | DKFZP434C245 protein | taagctgtgggacaagagca | ttgagtcactggaggctgtg | 248 |
| FOXM1 | forkhead box M1 | cgtggattgaggaccacttt | gattcggtcgtttctgctgt | 249 |
| HAS2 | hyaluronan synthase 2 | agagcactgggacgaagtgt | atgcactgaacacacccaaa | 245 |
| HNLF | putative NFkB activating protein HNLF | agaagcgctgtttcatcgag | gccatcctggtagaattgga | 253 |
| ID1 | inhibitor of DNA binding 1, dominant negative helix-loop-helix protein | cccattctgtttcagccagt | agccgttcatgtcgtagagc | 245 |
| KIAA0924 | KIAA0924 protein | atcgctcattttgaggttgc | gcagaggacagggcagtaaa | 246 |
| MTB | more than blood homolog | tgcgggaggttctgagttac | ggaccatcgggtaaggatct | 261 |
| MYBL1 | v-myb myeloblastosis viral oncogene homolog (avian)-like 1 | gtccgaaacgttggtctgtt | gaccttccgacgcattgtag | 248 |
| PIN1 | protein (peptidyl-prolyl cis/trans isomerase) NIMA-interacting 1 | tgccaccgtcacacagtatt | gagtctgcctccagcacct | 253 |
| PIR51 | RAD51 associated protein 1 | ttctggaaggcagtgatggt | gagcagagtccaccgaagtc | 243 |
| PLK4 | polo-like kinase 4 (Drosophila) | gccaaggaccttattcacca | ttatttgggagtggctgacc | 251 |
| RASEF | RAS and EF hand domain containing | atcaaccttgtggagccaag | ctgaggtcactgagggcttc | 245 |
4.3. Western Blot Analysis
4.4. Microarray Hybridization
4.5. Microarray Data Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fadare, O.; Zheng, W. Histologic dating of the endometrium: Accuracy, reproducibility, and practical value. Adv. Anat. Pathol. 2005, 12, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Bhartiya, D.; Chowdhury, S.R.; Bajpai, V.K. Stromal cell interaction and relevance to predecidual events and menstruation. Hum. Reprod. 1996, 11, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Minagawa, Y.; Kigawa, J.; Ishihara, H.; Itamochi, H.; Terakawa, N.; Nagami, M. Quantitative analysis of endometrial stromal cells including behavioral changes related to the menstrual cycle in smear specimens. Gynecol. Obstet. Investig. 1995, 39, 39–42. [Google Scholar] [CrossRef]
- Ferenczy, A.; Bertrand, G.; Gelfand, M.M. Proliferation kinetics of human endometrium during the normal menstrual cycle. Am. J. Obstet. Gynecol. 1979, 133, 859–867. [Google Scholar] [PubMed]
- Gurpide, E.; Fleming, H.; Fridman, O.; Hausknecht, V.; Holinka, C. Receptors, enzymes, and hormonal responses of endometrial cells. Prog. Clin. Biol. Res. 1981, 74, 427–446. [Google Scholar] [PubMed]
- Chan, R.W.; Schwab, K.E.; Gargett, C.E. Clonogenicity of human endometrial epithelial and stromal cells. Biol. Reprod. 2004, 70, 1738–1750. [Google Scholar] [CrossRef] [PubMed]
- Spencer, T.E.; Bazer, F.W. Biology of progesterone action during pregnancy recognition and maintenance of pregnancy. Front. Biosci. J. Virtual Libr. 2002, 7, D1879–D1898. [Google Scholar] [CrossRef]
- Vereide, A.B.; Kaino, T.; Sager, G.; Arnes, M.; Orbo, A. Effect of levonorgestrel IUD and oral medroxyprogesterone acetate on glandular and stromal progesterone receptors (PRA and PRB), and estrogen receptors (ER-α and ER-β) in human endometrial hyperplasia. Gynecol. Oncol. 2006, 101, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Gentilini, D.; Vigano, P.; Somigliana, E.; Vicentini, L.M.; Vignali, M.; Busacca, M.; di Blasio, A.M. Endometrial stromal cells from women with endometriosis reveal peculiar migratory behavior in response to ovarian steroids. Fertil. Steril. 2010, 93, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Meden, H.; Meyer-Rath, D.; Schauer, A.; Kuhn, W. Endometrial stromal sarcoma of the uterus. Anti-Cancer Drugs 1991, 2, 35–37. [Google Scholar] [CrossRef] [PubMed]
- Gadducci, A.; Romanini, A. Adjuvant chemotherapy in early stage uterine sarcomas: An open question. Eur. J. Gynaecol. Oncol. 2001, 22, 352–357. [Google Scholar] [PubMed]
- McCluggage, W.G. Malignant biphasic uterine tumours: Carcinosarcomas or metaplastic carcinomas? J. Clin. Pathol. 2002, 55, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Zelmanowicz, A.; Hildesheim, A.; Sherman, M.E.; Sturgeon, S.R.; Kurman, R.J.; Barrett, R.J.; Berman, M.L.; Mortel, R.; Twiggs, L.B.; Wilbanks, G.D.; et al. Evidence for a common etiology for endometrial carcinomas and malignant mixed mullerian tumors. Gynecol. Oncol. 1998, 69, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.M.; Weiss, N.S.; Daling, J.R.; Gammon, M.D.; Liff, J.M.; Watt, J.; Lynch, C.F.; Newcomb, P.A.; Armstrong, B.K.; Thompson, W.D. Exogenous sex hormone use, correlates of endogenous hormone levels, and the incidence of histologic types of sarcoma of the uterus. Cancer 1996, 77, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Saga, Y.; Ohwada, M.; Kohno, T.; Takayashiki, N.; Suzuki, M. High-grade endometrial stromal sarcoma after treatment with tamoxifen in a patient treated for breast cancer. Int. J. Gynecol. Cancer 2003, 13, 690–692. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.W. The role of stromal-epithelial interaction in normal and malignant growth. Cancer Surv. 1995, 23, 33–42. [Google Scholar] [PubMed]
- Pavone-Macaluso, M.; Carruba, G.; Castagnetta, L. Steroid receptors in prostate cancer tissues and cells: Pathophysiology, problems in methodology, clinical value and controversial questions. Arch. Esp. Urol. 1994, 47, 189–201. [Google Scholar] [PubMed]
- Moinfar, F.; Man, Y.G.; Arnould, L.; Bratthauer, G.L.; Ratschek, M.; Tavassoli, F.A. Concurrent and independent genetic alterations in the stromal and epithelial cells of mammary carcinoma: Implications for tumorigenesis. Cancer Res. 2000, 60, 2562–2566. [Google Scholar] [PubMed]
- Arnold, J.T.; Lessey, B.A.; Seppala, M.; Kaufman, D.G. Effect of normal endometrial stroma on growth and differentiation in Ishikawa endometrial adenocarcinoma cells. Cancer Res. 2002, 62, 79–88. [Google Scholar] [PubMed]
- Park, D.W.; Ryu, H.S.; Choi, D.S.; Park, Y.H.; Chang, K.H.; Min, C.K. Localization of matrix metalloproteinases on endometrial cancer cell invasion in vitro. Gynecol. Oncol. 2001, 82, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Tsutsumi, A.; Imai, M.; Nakajima, T.; Yasuda, K.; Kanzaki, H. Estrogen and selective estrogen receptor modulators regulate vascular endothelial growth factor and soluble vascular endothelial growth factor receptor 1 in human endometrial stromal cells. Fertil. Steril. 2010, 93, 2680–2686. [Google Scholar] [CrossRef] [PubMed]
- Hachisuga, T.; Miyakawa, T.; Tsujioka, H.; Horiuchi, S.; Emoto, M.; Kawarabayashi, T. K-RAS mutation in tamoxifen-related endometrial polyps. Cancer 2003, 98, 1890–1897. [Google Scholar] [CrossRef] [PubMed]
- Guleria, K.; Agarwal, N.; Mishra, K.; Gulati, R.; Mehendiratta, A. Evaluation of endometrial steroid receptors and cell mitotic activity in women using copper intrauterine device: Can Cu-T prevent endometrial cancer? J. Obstet. Gynaecol. Res. 2004, 30, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, V.; Castilla, J.A.; Martinez, L.; Herruzo, A.; Concha, A.; Fontes, J.; Mendoza, N.; Garcia-Pena, M.L.; Mendoza, J.L.; Magan, R.; et al. Expression of transcription factors in endometrium during natural cycles. J. Assist. Reprod. Genet. 2003, 20, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Sereepapong, W.; Chotnopparatpattara, P.; Taneepanichskul, S.; Markham, R.; Russell, P.; Fraser, I.S. Endometrial progesterone and estrogen receptors and bleeding disturbances in depot medroxyprogesterone acetate users. Hum. Reprod. 2004, 19, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Cooke, P.S.; Buchanan, D.L.; Young, P.; Setiawan, T.; Brody, J.; Korach, K.S.; Taylor, J.; Lubahn, D.B.; Cunha, G.R. Stromal estrogen receptors mediate mitogenic effects of estradiol on uterine epithelium. Proc. Natl. Acad. Sci. USA 1997, 94, 6535–6540. [Google Scholar] [CrossRef] [PubMed]
- Hyder, S.M.; Stancel, G.M.; Chiappetta, C.; Murthy, L.; Boettger-Tong, H.L.; Makela, S. Uterine expression of vascular endothelial growth factor is increased by estradiol and tamoxifen. Cancer Res. 1996, 56, 3954–3960. [Google Scholar] [PubMed]
- Dimitriadis, E.; Robb, L.; Salamonsen, L.A. Interleukin 11 advances progesterone-induced decidualization of human endometrial stromal cells. Mol. Hum. Reprod. 2002, 8, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Reis, F.M.; Maia, A.L.; Ribeiro, M.F.; Spritzer, P.M. Progestin modulation of C-FOS and prolactin gene expression in the human endometrium. Fertil. Steril. 1999, 71, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Maruyama, T.; Sakurai, R.; Masuda, H.; Yamamoto, Y.; Shimizu, A.; Kishi, I.; Asada, H.; Yamagoe, S.; Yoshimura, Y. Involvement of histone acetylation in ovarian steroid-induced decidualization of human endometrial stromal cells. J. Biol. Chem. 2003, 278, 16675–16682. [Google Scholar] [CrossRef] [PubMed]
- Yasmeen, A.; Berdel, W.E.; Serve, H.; Muller-Tidow, C. E- and A-type cyclins as markers for cancer diagnosis and prognosis. Expert Rev. Mol. Diagn. 2003, 3, 617–633. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Li, S.Q.; Wang, X.; Muhlrad, S.; Bjartell, A.; Wolgemuth, D.J. Elevated levels and distinct patterns of expression of A-type cyclins and their associated cyclin-dependent kinases in male germ cell tumors. Int. J. Cancer 2004, 108, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Faivre, J.; Frank-Vaillant, M.; Poulhe, R.; Mouly, H.; Jessus, C.; Brechot, C.; Sobczak-Thepot, J. Centrosome overduplication, increased ploidy and transformation in cells expressing endoplasmic reticulum-associated cyclin A2. Oncogene 2002, 21, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- Badie, C.; Bourhis, J.; Sobczak-Thepot, J.; Haddada, H.; Chiron, M.; Janicot, M.; Janot, F.; Tursz, T.; Vassal, G. p53-dependent G2 arrest associated with a decrease in cyclins A2 and B1 levels in a human carcinoma cell line. Br. J. Cancer 2000, 82, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.C.; Mor, G.; Lim, C.; Zheng, W.; Parkash, V.; Schwartz, P.E. Low-grade endometrial stromal sarcoma: Hormonal aspects. Gynecol. Oncol. 2003, 90, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Lentz, S.S. Endocrine therapy of endometrial cancer. Cancer Treat. Res. 1998, 94, 89–106. [Google Scholar] [PubMed]
- Podratz, K.C.; O’Brien, P.C.; Malkasian, G.D., Jr.; Decker, D.G.; Jefferies, J.A.; Edmonson, J.H. Effects of progestational agents in treatment of endometrial carcinoma. Obstet. Gynecol. 1985, 66, 106–110. [Google Scholar] [PubMed]
- Nishitani, H.; Lygerou, Z. DNA replication licensing. Front. Biosci. J. Virtual Libr. 2004, 9, 2115–2132. [Google Scholar] [CrossRef]
- Cortes, F.; Pastor, N. Induction of endoreduplication by topoisomerase II catalytic inhibitors. Mutagenesis 2003, 18, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Kesti, T.; McDonald, W.H.; Yates, J.R., 3rd; Wittenberg, C. Cell cycle-dependent phosphorylation of the DNA polymerase epsilon subunit, Dpb2, by the Cdc28 cyclin-dependent protein kinase. J. Biol. Chem. 2004, 279, 14245–14255. [Google Scholar] [CrossRef] [PubMed]
- Mizushina, Y.; Xu, X.; Matsubara, K.; Murakami, C.; Kuriyama, I.; Oshige, M.; Takemura, M.; Kato, N.; Yoshida, H.; Sakaguchi, K. Pyridoxal 5'-phosphate is a selective inhibitor in vivo of DNA polymerase α and epsilon. Biochem. Biophys. Res. Commun. 2003, 312, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.P.; Rushlow, W.J.; Chakraborty, C.; Lala, P.K. Differential gene expression in premalignant human trophoblast: Role of IGFBP-5. Int. J. Cancer 2001, 94, 674–684. [Google Scholar] [CrossRef] [PubMed]
- Paige, L.A.; Christensen, D.J.; Gron, H.; Norris, J.D.; Gottlin, E.B.; Padilla, K.M.; Chang, C.Y.; Ballas, L.M.; Hamilton, P.T.; McDonnell, D.P.; et al. Estrogen receptor (ER) modulators each induce distinct conformational changes in ER-α and ER-β. Proc. Natl. Acad. Sci. USA 1999, 96, 3999–4004. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.C.; Skafar, D.F. From ligand structure to biological activity: Modified estratrienes and their estrogenic and antiestrogenic effects in MCF-7 cells. Steroids 2004, 69, 401–418. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.P. The role of coactivators and corepressors in the biology and mechanism of action of steroid hormone receptors. J. Mammary Gland Biol. Neoplasia 2000, 5, 307–324. [Google Scholar] [CrossRef] [PubMed]
- Pearce, S.T.; Liu, H.; Jordan, V.C. Modulation of estrogen receptor α function and stability by tamoxifen and a critical amino acid (Asp-538) in helix 12. J. Biol. Chem. 2003, 278, 7630–7638. [Google Scholar] [CrossRef] [PubMed]
- Kirk, D.; King, R.J.; Heyes, J.; Peachey, L.; Hirsch, P.J.; Taylor, R.W. Normal human endometrium in cell culture. I. Separation and characterization of epithelial and stromal components in vitro. Vitro 1978, 14, 651–662. [Google Scholar] [CrossRef]
- Fleming, H.; Gurpide, E. Growth characteristics of primary cultures of stromal cells from human endometrium. J. Steroid Biochem. 1982, 16, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, R.; Halvatsiotis, P.; Schimke, J.C.; Nair, K.S. Gene expression profile in skeletal muscle of type 2 diabetes and the effect of insulin treatment. Diabetes 2002, 51, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, R.; Unnikrishnan, J.; Fu, A.; Nygren, J.; Short, K.R.; Schimke, J.; Barazzoni, R.; Nair, K.S. Impact of high-fat diet and antioxidant supplement on mitochondrial functions and gene transcripts in rat muscle. Am. J. Physiol. Endocrinol. Metabol. 2002, 282, E1055–E1061. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, C.-E.; Zhu, X.; Li, J.; Lyle, C.; Dowdy, S.; Podratz, K.C.; Byck, D.; Chen, H.-B.; Jiang, S.-W. Microarray Analysis on Gene Regulation by Estrogen, Progesterone and Tamoxifen in Human Endometrial Stromal Cells. Int. J. Mol. Sci. 2015, 16, 5864-5885. https://doi.org/10.3390/ijms16035864
Ren C-E, Zhu X, Li J, Lyle C, Dowdy S, Podratz KC, Byck D, Chen H-B, Jiang S-W. Microarray Analysis on Gene Regulation by Estrogen, Progesterone and Tamoxifen in Human Endometrial Stromal Cells. International Journal of Molecular Sciences. 2015; 16(3):5864-5885. https://doi.org/10.3390/ijms16035864
Chicago/Turabian StyleRen, Chun-E, Xueqiong Zhu, Jinping Li, Christian Lyle, Sean Dowdy, Karl C. Podratz, David Byck, Hai-Bin Chen, and Shi-Wen Jiang. 2015. "Microarray Analysis on Gene Regulation by Estrogen, Progesterone and Tamoxifen in Human Endometrial Stromal Cells" International Journal of Molecular Sciences 16, no. 3: 5864-5885. https://doi.org/10.3390/ijms16035864
APA StyleRen, C.-E., Zhu, X., Li, J., Lyle, C., Dowdy, S., Podratz, K. C., Byck, D., Chen, H.-B., & Jiang, S.-W. (2015). Microarray Analysis on Gene Regulation by Estrogen, Progesterone and Tamoxifen in Human Endometrial Stromal Cells. International Journal of Molecular Sciences, 16(3), 5864-5885. https://doi.org/10.3390/ijms16035864