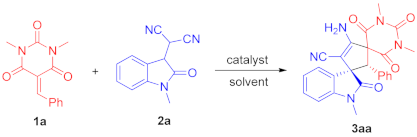
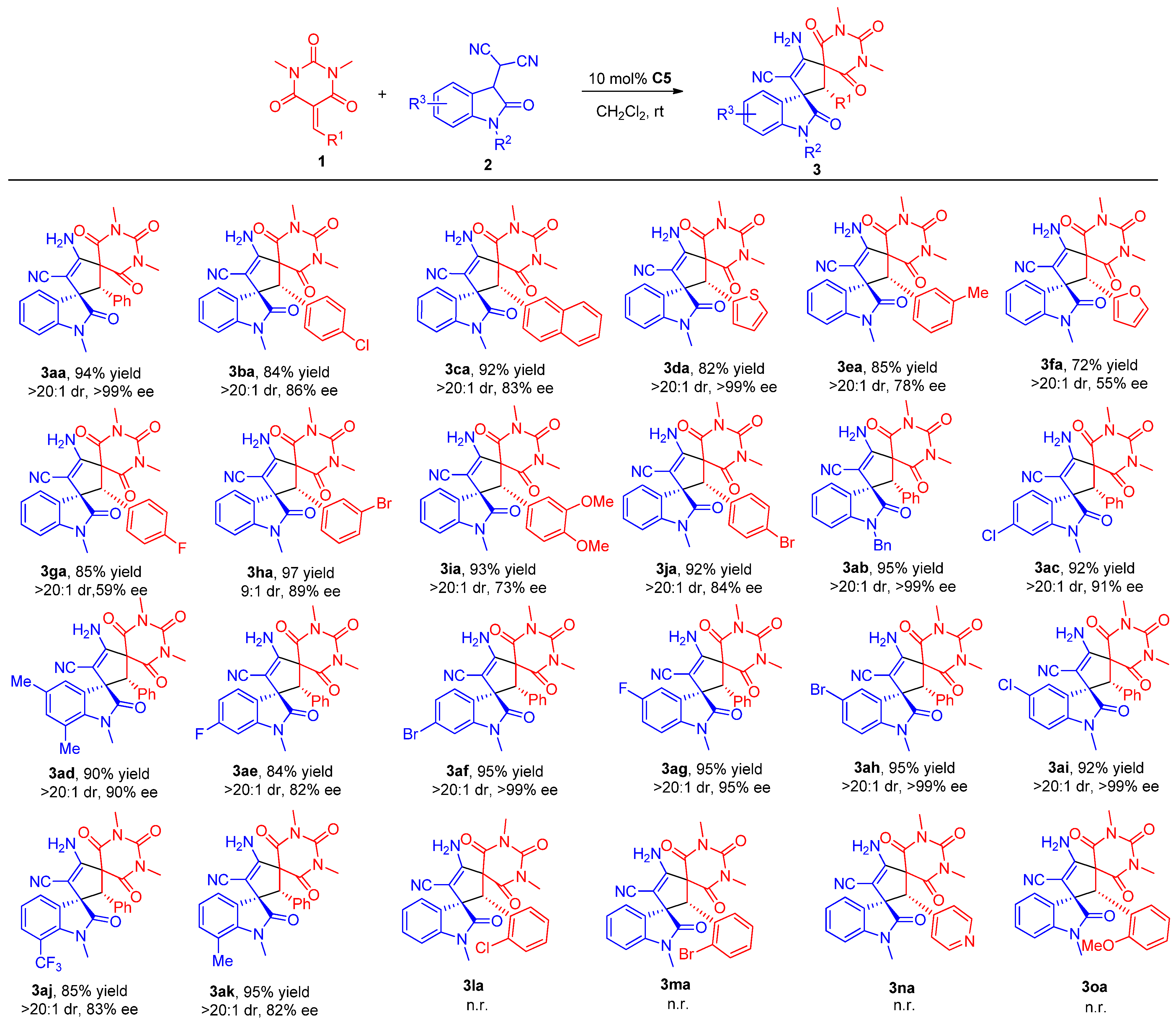
3.4. Procedure for the Asymmetric Michael/Cyclization Reaction
To a dried small vial, barbituric acid 1 (0.24 mmol), oxindolylmalonitrile 2 (0.2 mmol), chiral organocatalyst C5 (5.08 mg, 0.01 mmol, 0.05 equiv), and CH2Cl2 (1.0 mL) were added. After stirring at room temperature under air without gas protection for 8 h, the reaction mixture was concentrated and directly purified by silica gel column chromatography (200–300 mesh) using ethyl acetate/petroleum ether (1:2) as eluent to afford the desired products 3.
(2′
R,3
S)-4′-Amino-1,1″,3″-trimethyl-2,2″,4″,6″-tetraoxo-2′-phenyl-1″,3″,4″,6″-tetrahydro-2″
H-dispiro[indoline-3,1′-cyclopentane-3′,5″-pyrimidin]-4′-ene-5′-carbonitrile (
3aa). According to the general procedure from
1a (58.6 mg, 0.24 mmol) and
2a (42.2 mg, 0.2 mmol) to obtain 85.6 mg (94% yield) compound
3aa as a yellow solid, m.p. 192−195 °C. HPLC (Daicel Chiralpak ADH,
n-hexane/2-propanol = 70:30, flow rate 1.0 mL/min, detection at 254 nm):
tR = 9.2 min (major), >99% ee. [α]
D25 = +9.9 (
c = 0.5, CH
2Cl
2).
1H NMR (700 MHz, DMSO-d
6):
δ 8.05 (dd,
J1 = 7.7 Hz,
J2 = 0.7 Hz, 1H), 7.28 (td,
J1 = 7.7 Hz,
J2 = 1.4 Hz 1H), 7.15–7.18 (m, 4H), 7.06 (t,
J = 8.0 Hz, 2H), 6.86 (d,
J = 7.7 Hz, 1H), 6.76 (d,
J = 7.0 Hz, 2H), 4.24 (s, 1H), 3.07 (s, 3H), 2.99 (s, 3H), 2.87 (s, 3H) ppm.
13C NMR (176 MHz, DMSO-d
6):
δ 176.5, 168.6, 167.8, 158.4, 149.8, 143.0, 131.1, 129.4, 129.3, 129.0, 128.3, 128.0, 127.3, 122.3, 116.3, 108.7, 78.5, 68.1, 64.8, 63.4, 28.5, 28.3, 26.4 ppm. (see
Supplementary Materials) HRMS (ESI):
m/
z calcd. for C
25H
22N
5O
4 [M + H]
+ 456.1666, found 456.1686.
(2′R,3S)-4′-Amino-2′-(4-chlorophenyl)-1,1″,3″-trimethyl-2,2″,4″,6″-tetraoxo-1″,3″,4″,6″-tetrahydro-2″H-dispiro[indoline-3,1′-cyclopentane-3′,5″-pyrimidin]-4′-ene-5′-carbonitrile (3ba). According to the general procedure from 1b (66.7 mg, 0.24 mmol) and 2a (42.2 mg, 0.2 mmol) to obtain 82.2 mg (84% yield) compound 3ba as a yellow solid, m.p. 183–185 °C. HPLC (Daicel Chiralpak IC, n-hexane/2-propanol = 70:30, flow rate 1.0 mL/min, detection at 254 nm): tR = 6.8 min (minor), tR = 9.4 min (major), 86% ee. [α]D25 = +12.2 (c = 0.34, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 8.12 (d, J = 7.2 Hz, 1H), 7.29–7.25 (m, 1H), 7.17 (td, J1 = 7.6 Hz, J2 = 0.8 Hz, 1H), 7.01 (d, J = 8.4 Hz, 2H), 6.78 (d, J = 8.8 Hz, 2H), 6.67 (d, J = 7.6 Hz, 1H), 5.65 (s, 2H), 4.37 (s, 1H), 3.20 (s, 3H), 3.03 (s, 3H), 2.98 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3): δ 176.8, 168.6, 167.1, 158.0, 149.8, 143.9, 135.6, 131.1, 129.8, 129.3, 128.5, 128.2, 127.3, 123.2, 115.3, 108.7, 83.2, 68.6, 64.6, 63.9, 29.3, 28.9, 26.8 ppm. HRMS (ESI): m/z calcd. for C25H21ClN5O4 [M + H]+ 490.1277, found 490.1301.
(2′R,3S)-4′-Amino-1,1″,3″-trimethyl-2′-(naphthalen-2-yl)-2,2″,4″,6″-tetraoxo-1″,3″,4″,6″-tetrahydro-2″H-dispiro[indoline-3,1′-cyclopentane-3′,5″-pyrimidin]-4′-ene-5′-carbonitrile (3ca). According to the general procedure from 1c (70.6 mg, 0.24 mmol) and 2a (42.2 mg, 0.2 mmol) to obtain 93.0 mg (92% yield) compound 3ca as a yellow solid, m.p. 201–203 °C. HPLC (Daicel Chiralpak IC, n-hexane/2-propanol = 70:30, flow rate 1.0 mL/min, detection at 254 nm): tR = 13.0 min (minor), tR = 16.7 min (major), 83% ee. [α]D25 = +16.5 (c = 0.5, CH2Cl2). 1H NMR (400 MHz, DMSO-d6): δ 8.18 (d, J = 7.6 Hz, 1H), 7.85 (d, J = 8.0 Hz, 1H), 7.76 (t, J = 9.4 Hz, 2H), 7.61 (td, J1 = 7.6 Hz, J2 = 1.0 Hz, 1H), 7.50 (t, J = 7.4 Hz, 1H), 7.32–7.22 (m, 4H), 7.12 (t, J = 7.8 Hz, 1H), 7.01 (d, J = 7.2 Hz, 1H), 6.83 (d, J = 7.6 Hz, 1H), 5.49 (s, 1H), 2.88 (s, 3H), 2.81 (s, 3H), 2.60 (s, 3H) ppm. 13C NMR (101 MHz, DMSO-d6): δ 176.5, 168.7, 168.0, 158.5, 149.4, 143.1, 133.0, 131.9, 129.4, 129.3, 129.1, 128.5, 127.5, 127.2, 126.7, 125.9, 123.8, 122.5, 120.7, 116.3, 108.9, 79.1, 68.3, 63.6, 55.9, 28.3, 26.4 ppm. HRMS (ESI): m/z calcd. for C29H24N5O4 [M + H]+ 506.1823, found 506.1818.
(2′R,3S)-4′-Amino-1,1″,3″-trimethyl-2,2″,4″,6″-tetraoxo-2′-(thiophen-2-yl)-1″,3″,4″,6″-tetrahydro-2″H-dispiro[indoline-3,1′-cyclopentane-3′,5″-pyrimidin]-4′-ene-5′-carbonitrile (3da). According to the general procedure from 1d (60.0 mg, 0.24 mmol) and 2a (42.2 mg, 0.2 mmol) to obtain 75.6 mg (82% yield) compound 3da as a brown solid, m.p. 214–215 °C. HPLC (Daicel Chiralpak IA, n-hexane/2-propanol/ethyl acetate = 80:10:10, flow rate 1.0 mL/min, detection at 254 nm): tR = 19.3 min (minor), tR = 26.4 min (major); >99% ee. [α]D25 = +18.5 (c = 0.4, CH2Cl2). 1H NMR (700 MHz, DMSO-d6): δ 8.06 (d, J = 7.7 Hz, 1H), 7.36–7.32 (m, 2H), 7.20–7.16 (m, 3H), 6.94 (d, J = 7.7 Hz, 1H), 6.78 (dd, J1 = 4.9 Hz, J2 = 4.2 Hz, 1H), 6.68 (d, J = 3.5 Hz, 1H), 4.62 (s, 1H), 3.18 (s, 3H), 3.05 (s, 3H), 2.85 (s, 3H) ppm. 13C NMR (176 MHz, DMSO-d6): δ 176.2, 168.3, 167.3, 158.0, 150.0, 143.4, 132.5, 129.8, 129.4, 128.2, 128.0, 127.8, 126.3, 122.4, 116.2, 108.7, 78.5, 68.4, 63.0, 59.7, 28.7, 28.6, 26.5 ppm. HRMS (ESI): m/z calcd. for C23H20N5O4S [M + H]+ 462.1231, found 462.1253.
(2′R,3S)-4′-Amino-1,1″,3″-trimethyl-2,2″,4″,6″-tetraoxo-2′-(m-tolyl)-1″,3″,4″,6″-tetrahydro-2″H-dispiro[indoline-3,1′-cyclopentane-3′,5″-pyrimidin]-4′-ene-5′-carbonitrile (3ea). According to the general procedure from 1e (61.92 mg, 0.24 mmol) and 2a (42.2 mg, 0.2 mmol) to obtain 79.7 mg (85% yield) compound 3ea as a white solid, m.p. 194–196 °C. HPLC (Daicel Chiralpak ADH, n-hexane/2-propanol = 70:30, flow rate 1.0 mL/min, detection at 254 nm): tR = 6.5 min (minor), tR = 11.5 min (major), 78% ee. [α]D25 = +14.8 (c = 0.5, CH2Cl2). 1H NMR (400 MHz, DMSO-d6): δ 8.06 (dd, J1 = 7.6 Hz, J2 = 0.8 Hz, 1H), 7.28 (td, J = 7.6 Hz, J2 = 1.2 Hz, 1H), 7.17 (t, J = 7.2 Hz, 3H), 6.98–6.91 (m, 2H), 6.85 (d, J = 7.6 Hz, 1H), 6.58 (s, 1H), 6.54 (d, J = 7.2 Hz, 1H), 4.20 (s, 1H), 3.08 (s, 3H), 2.98 (s, 3H), 2.86 (s, 3H), 2.03 (s, 3H) ppm. 13C NMR (101 MHz, DMSO-d6): δ 176.5, 168.6, 167.8, 158.4, 149.8, 143.0, 137.2, 131.1, 130.1, 129.5, 129.2, 128.4, 127.8, 127.3, 126.4, 122.1, 116.3, 108.7, 78.5, 68.1, 64.7, 63.4, 28.5, 28.3, 26.4, 20.5. ppm. HRMS (ESI): m/z calcd. for C26H24N5O4 [M + H]+ 470.1823, found 470.1812.
(2′R,3S)-4′-Amino-2′-(furan-2-yl)-1,1″,3″-trimethyl-2,2″,4″,6″-tetraoxo-1″,3″,4″,6″-tetrahydro-2″H-dispiro[indoline-3,1′-cyclopentane-3′,5″-pyrimidin]-4′-ene-5′-carbonitrile (3fa). According to the general procedure from 1f (56.2 mg, 0.24 mmol) and 2a (42.2 mg, 0.2 mmol) to obtain 64.1 mg (72% yield) compound 3fa as a brown solid, m.p. 174–176 °C. HPLC (Daicel Chiralpak IA, n-hexane/2-propanol = 70:30, flow rate 1.0 mL/min, detection at 254 nm): tR = 6.6 min (minor), tR = 8.2 min (major), 55% ee. [α]D25 = −2.0 (c = 0.33, CH2Cl2). 1H NMR (700 MHz, DMSO-d6): δ 7.93 (dd, J = 7.7 Hz, 1H), 7.38 (d, J = 1.4 Hz, 1H), 7.32 (td, J1 = 7.7 Hz, J2 = 0.7 Hz, 1H), 7.17–7.12 (m, 3H), 6.97 (d, J = 7.7 Hz, 1H), 6.15 (dd, J1 = 3.2 Hz, J2 = 1.8 Hz, 1H), 5.74 (d, J = 2.8 Hz, 1H), 4.41 (s, 1H), 3.19 (s, 3H), 3.10 (s, 3H), 2.94 (s, 3H) ppm. 13C NMR (176 MHz, DMSO-d6): δ 176.0, 168.2, 167.2, 157.7, 150.2, 145.9, 144.0, 142.9, 129.3, 128.2, 127.4, 122.3, 116.0, 110.7, 110.4, 108.6, 78.8, 66.9, 61.7, 56.5, 28.7, 28.6, 26.5 ppm. HRMS (ESI): m/z calcd. for C23H20N5O5 [M + H]+ 446.1459, found 446.1472.
(2′R,3S)-4′-Amino-2′-(4-fluorophenyl)-1,1″,3″-trimethyl-2,2″,4″,6″-tetraoxo-1″,3″,4″,6″-tetrahydro-2″H-dispiro[indoline-3,1′-cyclopentane-3′,5″-pyrimidin]-4′-ene-5′-carbonitrile (3ga). According to the general procedure from 1g (62.9 mg, 0.24 mmol) and 2a (42.2 mg, 0.2 mmol) to obtain 80.4 mg (85% yield) compound 3ga as a pink solid, m.p. 178–179 °C. HPLC (Daicel Chiralpak IA, n-hexane/2-propanol/ethyl acetate = 80:15:5, flow rate 1.0 mL/min, detection at 254 nm): tR = 13.4 min (minor), tR = 21.9 min (major); 59% ee. [α]D25 = +9.0 (c = 0.8, CH2Cl2). 1H NMR (700 MHz, DMSO-d6): δ 8.03 (d, J = 7.0 Hz, 1H), 7.30 (t, J = 7.7 Hz, 1H), 7.20–7.16 (m, 3H), 6.94–6.88 (m, 3H), 6.83–6.80 (m, 2H), 4.25 (s, 1H), 3.09 (s, 3H), 3.01 (s, 3H), 2.91 (s, 3H) ppm. 13C NMR (176 MHz, DMSO-d6): δ 176.4, 168.5, 167.7, 162.1 (1JC–F = 246.9 Hz), 158.3, 149.9, 143.0, 131.5 (3JC–F = 8.3 Hz), 129.4, 128.1, 127.3 (4JC–F = 2.6 Hz), 127.2, 122.4, 116.2, 115.0 (2JC–F = 21.5 Hz), 108.8, 78.3, 67.9, 63.7, 63.4, 28.6, 28.4, 26.4 ppm. 19F NMR (659 MHz, DMSO-d6): δ −112.1. HRMS (ESI): m/z calcd. for C25H21FN5O4 [M + H]+ 474.1572, found 474.1587.
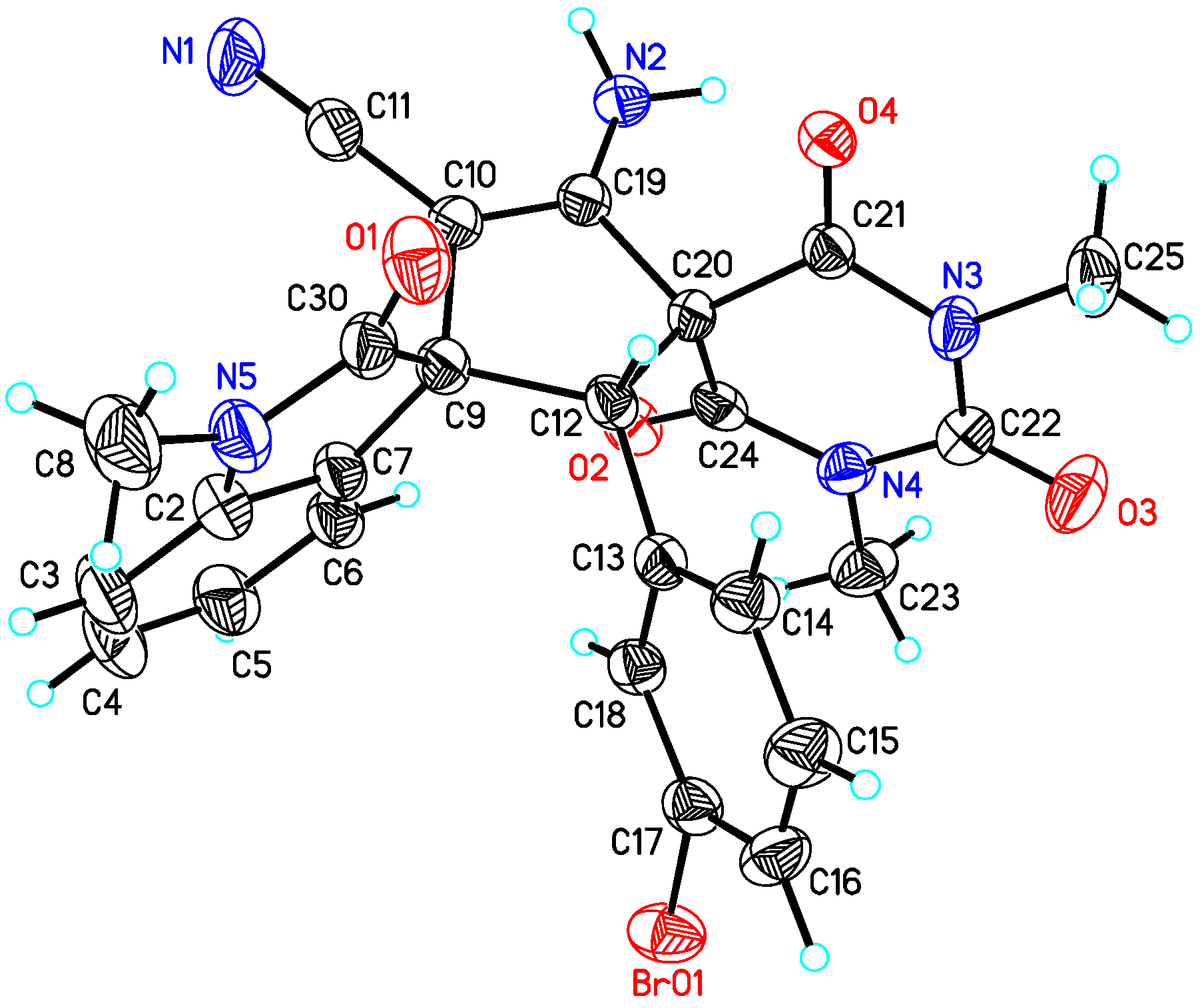
(2′R,3S)-4′-Amino-2′-(3-bromophenyl)-1,1″,3″-trimethyl-2,2″,4″,6″-tetraoxo-1″,3″,4″,6″-tetrahydro-2″H-dispiro[indoline-3,1′-cyclopentane-3′,5″-pyrimidin]-4′-ene-5′-carbonitrile (3ha). According to the general procedure from 1h (77.3 mg, 0.24 mmol) and 2a (42.2 mg, 0.2 mmol) to obtain 103.4 mg (97% yield) compound 3ha as a pink solid, m.p. 194–195 °C. HPLC (Daicel Chiralpak ADH, n-hexane/2-propanol = 75:25, flow rate 1.0 mL/min, detection at 254 nm): tR = 11.8 min (minor), tR = 14.5 min (major); 89% ee. [α]D25 = +14.9 (c = 0.5, CH2Cl2). 1H NMR (700 MHz, DMSO-d6): δ 8.03 (d, J = 7.7 Hz, 1H), 7.39 (d, J = 8.4 Hz, 1H), 7.32 (t, J = 7.7 Hz, 1H), 7.23–7.18 (m, 3H), 7.07 (t, J = 8.0 Hz, 1H), 6.93 (s, 1H), 6.90 (d, J = 7.7 Hz, 1H), 6.85 (d, J = 7.7 Hz, 1H), 4.20 (s, 1H), 3.10 (s, 3H), 3.01 (s, 3H), 2.92 (s, 3H) ppm. 13C NMR (176 MHz, DMSO-d6): δ 176.2, 168.2, 167.7, 158.2, 149.8, 143.0, 133.5, 131.9, 131.6, 130.3, 129.5, 129.1, 127.9, 127.1, 122.3, 121.0, 116.2, 109.0, 78.2, 67.9, 63.6, 63.3, 28.6, 28.3, 26.4 ppm. HRMS (ESI): m/z calcd. for C25H2179BrN5O4 [M + H]+ 534.0771, found 534.0786; calcd. for C25H2181BrN5O4 [M + H]+ 536.0751, found 536.0770.
(2′R,3S)-4′-Amino-2′-(3,4-dimethoxyphenyl)-1,1″,3″-trimethyl-2,2″,4″,6″-tetraoxo-1″,3″,4″,6″-tetrahydro-2″H-dispiro[indoline-3,1′-cyclopentane-3′,5″-pyrimidin]-4′-ene-5′-carbonitrile (3ia). According to the general procedure from 1i (73.0 mg, 0.24 mmol) and 2a (42.2 mg, 0.2 mmol) to obtain 90.2 mg (93% yield) compound 3ia as a yellow solid, m.p. 203–205 °C. HPLC (Daicel Chiralpak ADH, n-hexane/2-propanol = 80:20, flow rate 1.0 mL/min, detection at 254 nm): tR = 19.0 min (minor), tR = 21.3 min (major), 73% ee. [α]D25 = +49.1 (c = 0.7, CH2Cl2). 1H NMR (700 MHz, DMSO-d6): δ 8.10 (d, J = 7.7 Hz, 1H), 7.31 (t, J = 7.7 Hz, 1H), 7.18 (t, J = 7.7 Hz, 3H), 6.90 (d, J = 7.7 Hz, 1H), 6.65 (d, J = 8.4 Hz, 1H), 6.37 (dd, J1 = 8.0 Hz, J2 = 1.4 Hz, 1H), 6.21 (s, 1H), 4.17 (s, 1H), 3.61 (s, 3H), 3.33 (s, 3H), 3.11 (s, 3H), 3.00 (s, 3H), 2.87 (s, 3H) ppm. 13C NMR (176 MHz, DMSO-d6): δ 176.6, 168.7, 167.9, 158.5, 150.0, 148.9, 147.5, 143.2, 129.2, 128.8, 127.2, 123.0, 122.8, 122.1, 116.4, 111.5, 110.7, 108.9, 78.3, 68.2, 64.5, 63.5, 55.1, 55.0, 28.6, 28.4, 26.4 ppm. HRMS (ESI): m/z calcd. for C27H26N5O6 [M + H]+ 516.1878, found 516.1895.
(2′R,3S)-4′-Amino-2′-(4-bromophenyl)-1,1″,3″-trimethyl-2,2″,4″,6″-tetraoxo-1″,3″,4″,6″-tetrahydro-2″H-dispiro[indoline-3,1′-cyclopentane-3′,5″-pyrimidin]-4′-ene-5′-carbonitrile (3ja). According to the general procedure from 1j (77.3 mg, 0.24 mmol) and 2a (42.2 mg, 0.2 mmol) to obtain 98.1 mg (92% yield) compound 3ja as a pink solid, m.p. 217–219 °C. HPLC (Daicel Chiralpak IA, n-hexane/2-propanol = 70:30, flow rate 1.0 mL/min, detection at 254 nm): tR = 7.5 min (minor), tR = 12.4 min (major); 84% ee. [α]D25 = +27.4 (c = 0.9, CH2Cl2). 1H NMR (400 MHz, DMSO-d6): δ 8.01 (dd, J1 = 7.6 Hz, J2 = 0.8 Hz, 1H), 7.32–7.27 (m, 3H), 7.20–7.15 (m, 3H), 6.88 (d, J = 8.0 Hz, 1H), 6.72 (d, J = 8.8 Hz, 2H), 4.22 (s, 1H), 3.09 (s, 3H), 3.01 (s, 3H), 2.93 (s, 3H) ppm. 13C NMR (176 MHz, DMSO-d6): δ 176.4, 168.5, 167.8, 158.4, 149.9, 143.0, 131.5, 131.1, 130.5, 129.5, 128.0, 127.3, 122.6, 122.5, 116.3, 108.9, 78.4, 67.8, 63.9, 63.4, 28.7, 28.5, 26.5 ppm. HRMS (ESI): m/z calcd. for C25H2179BrN5O4 [M + H]+ 534.0771, found 534.0793; calcd. for C25H2181BrN5O4 [M + H]+ 536.0751, found 536.0777.
(2′R,3S)-4′-Amino-1-benzyl-1″,3″-dimethyl-2,2″,4″,6″-tetraoxo-2′-phenyl-1″,3″,4″,6″-tetrahydro-2″H-dispiro[indoline-3,1′-cyclopentane-3′,5″-pyrimidin]-4′-ene-5′-carbonitrile (3ab). According to the general procedure from 1a (58.6 mg, 0.24 mmol) and 2b (57.4 mg, 0.2 mmol) to obtain 100.9 mg (95% yield) compound 3ab as a yellow solid, m.p. 165–167 °C. HPLC (Daicel Chiralpak IC, n-hexane/2-propanol = 70:30, flow rate 1.0 mL/min, detection at 254 nm): tR = 40.3 min (minor), tR = 24.9 min (major), >99% ee. [α]D25 = −11.4 (c = 0.4, CH2Cl2). 1H NMR (400 MHz, CDCl3): δ 8.17 (dd, J1 = 6.4 Hz, J2 = 1.4 Hz, 1H), 7.24–7.20 (m, 1H), 7.17–7.00 (m, 7H), 6.82 (d, J = 7.6 Hz, 2H), 6.61 (d, J = 7.6 Hz, 2H), 6.41 (d, J = 7.2 Hz, 1H), 5.60 (s, 2H), 5.02 (d, J = 16.4 Hz, 1H), 4.49 (d, J = 16.4 Hz, 1H), 4.44 (s, 1H), 3.13 (s, 3H), 2.93 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3): δ 177.2, 168.6, 167.2, 158.2, 149.8, 142.3, 134.4, 130.5, 123.0, 129.55, 129.52, 128.5, 128.30, 128.27, 127.7, 127.2, 126.1, 123.1, 115.5, 109.5, 83.3, 68.8, 66.1, 64.1, 43.8, 29.1, 28.8 ppm. HRMS (ESI): m/z calcd. for C31H26N5O4 [M + H]+ 532.1979, found 532.2000.
(2′R,3S)-4′-Amino-6-chloro-1,1″,3″-trimethyl-2,2″,4″,6″-tetraoxo-2′-phenyl-1″,3″,4″,6″-tetrahydro-2″H-dispiro[indoline-3,1′-cyclopentane-3′,5″-pyrimidin]-4′-ene-5′-carbonitrile (3ac). According to the general procedure from 1a (58.6 mg, 0.24 mmol) and 2c (49.0 mg, 0.2 mmol) to obtain 90.0 mg (92% yield) compound 3ac as a yellow solid, m.p. 172–174 °C. HPLC (Daicel Chiralpak ADH, n-hexane/2-propanol = 70:30, flow rate 1.0 mL/min, detection at 254 nm): tR = 8.0 min (minor), tR = 15.6 min (major), 91% ee. [α]D25 = −30.9 (c = 0.4, CH2Cl2). 1H NMR (700 MHz, DMSO-d6): δ 8.04 (d, J = 8.4 Hz, 1H), 7.25 (dd, J1 = 8.0 Hz, J2 = 2.2 Hz, 3H), 7.19 (t, J = 7.4 Hz, 1H), 7.11 (t, J = 7.7 Hz, 2H), 7.04 (d, J = 1.4 Hz, 1H), 6.75 (d, J = 7.7 Hz, 2H), 4.23 (s, 1H), 3.07 (s, 3H), 3.01 (s, 3H), 2.89 (s, 3H) ppm. 13C NMR (176 MHz, DMSO-d6): δ 176.6, 168.5, 167.9, 158.7, 149.8, 144.5, 133.8, 130.8, 129.3, 129.2, 128.6, 128.2, 127.2, 122.1, 116.1, 109.3, 77.9, 67.9, 64.7, 63.2, 28.6, 28.4, 26.6 ppm. HRMS (ESI): m/z calcd. for C25H21ClN5O4 [M + H]+ 490.1277, found 490.1300.
(2′R,3S)-4′-amino-1,1″,3″,5,7-pentamethyl-2,2″,4″,6″-tetraoxo-2′-phenyl-1″,3″,4″,6″-tetrahydro-2″H-dispiro[indoline-3,1′-cyclopentane-3′,5″-pyrimidin]-4′-ene-5′-carbonitrile (3ad). According to the general procedure from 1a (58.6 mg, 0.24 mmol) and 2d (47.8 mg, 0.2 mmol) to obtain 86.9 mg (90% yield) compound 3ad as a white solid, m.p. 181–182 °C. HPLC (Daicel Chiralpak IC, n-hexane/2-propanol = 70:30, flow rate 1.0 mL/min, detection at 254 nm): tR = 56.3 min (minor), tR = 40.3 min (major), 90% ee. [α]D25 = −40.6 (c = 0.4, CH2Cl2). 1H NMR (700 MHz, DMSO-d6): δ 7.76 (s, 1H), 7.16 (t, J = 7.4 Hz, 1H), 7.13 (s, 2H), 7.07 (t, J = 7.7 Hz, 2H), 6.83 (s, 1H), 6.76 (d, J = 7.7 Hz, 2H), 4.22 (s, 1H), 3.23 (s, 3H), 3.07 (s, 3H), 2.84 (s, 3H), 2.33 (s, 3H), 2.31 (s, 3H) ppm. 13C NMR (176 MHz, DMSO-d6): δ 177.1, 168.7, 167.8, 158.2, 149.8, 138.5, 133.3, 131.3, 130.8, 129.5, 129.1, 128.9, 128.0, 126.0, 119.3, 116.5, 79.2, 68.2, 65.0, 63.0, 29.6, 28.5, 28.4, 20.7, 18.3 ppm. HRMS (ESI): m/z calcd. for C27H26N5O4 [M + H]+ 484.1979, found 484.1992.
(2′R,3S)-4′-Amino-6-fluoro-1,1″,3″-trimethyl-2,2″,4″,6″-tetraoxo-2′-phenyl-1″,3″,4″,6″-tetrahydro-2″H-dispiro[indoline-3,1′-cyclopentane-3′,5″-pyrimidin]-4′-ene-5′-carbonitrile (3ae). According to the general procedure from 1a (58.6 mg, 0.24 mmol) and 2e (45.8 mg, 0.2 mmol) to obtain 79.5 mg (84% yield) compound 3ae as a white solid, m.p. 173–175 °C. HPLC (Daicel Chiralpak IC, n-hexane/2-propanol = 70:30, flow rate 1.0 mL/min, detection at 254 nm): tR = 9.4 min (minor), tR = 11.8 min (major), 82% ee. [α]D25 = −62.8 (c = 1, CH2Cl2). 1H NMR (700 MHz, DMSO-d6): δ 8.04 (dd, J1 = 7.7 Hz, J2 = 5.6 Hz, 1H), 7.23 (s, 2H), 7.18 (t, J = 7.4 Hz, 1H), 7.10 (t, J = 7.4 Hz, 2H), 7.01–6.98 (m, 1H), 6.86 (d, J = 9.1 Hz, 1H), 6.75 (d, J = 8.4 Hz, 2H), 4.22 (s, 1H), 3.07 (s, 3H), 3.00 (s, 3H), 2.88 (s, 3H) ppm. 13C NMR (176 MHz, DMSO-d6): δ 176.9, 168.6, 167.9, 162.8 (d, 1JC–F = 243.8 Hz), 158.5, 149.8, 144.8 (d, 2JC–F = 12.1 Hz), 130.9, 129.4, 129.1, 128.8 (d, 3JC–F = 9.7 Hz), 128.1, 124.0 (d, 4JC–F = 2.5 Hz), 116.2, 108.4 (d, 2JC–F = 22.2 Hz), 97.5 (d, 2JC–F = 27.6 Hz), 78.1, 68.0, 64.7, 63.1, 28.6, 28.4, 26.7 ppm. 19F NMR (659 MHz, DMSO-d6): δ −110.9. HRMS (ESI): m/z calcd. for C25H21FN5O4 [M + H]+ 474.1572, found 474.1602.
(2′R,3S)-4′-Amino-6-bromo-1,1″,3″-trimethyl-2,2″,4″,6″-tetraoxo-2′-phenyl-1″,3″,4″,6″-tetrahydro-2″H-dispiro[indoline-3,1′-cyclopentane-3′,5″-pyrimidin]-4′-ene-5′-carbonitrile (3af). According to the general procedure from 1a (58.6 mg, 0.24 mmol) and 2f (57.8 mg, 0.2 mmol) to obtain 101.3 mg (95% yield) compound 3af as a white solid, m.p. 161–163 °C. HPLC (Daicel Chiralpak IC, n-hexane/2-propanol = 70:30, flow rate 1.0 mL/min, detection at 254 nm): tR = 17.9 min (major); >99% ee. [α]D25 = −42.4 (c = 0.5, CH2Cl2). 1H NMR (700 MHz, DMSO-d6): δ 7.97 (d, J = 7.7 Hz, 1H), 7.39 (dd, J1 = 8.0 Hz, J2 = 1.8 Hz, 1H), 7.25 (s, 2H), 7.19 (t, J = 7.4 Hz, 1H), 7.16 (d, J = 1.4 Hz, 1H), 7.11 (t, J = 7.7 Hz, 2H), 6.75 (d, J = 7.7 Hz, 2H), 4.23 (s, 1H), 3.07 (s, 3H), 3.01 (s, 3H), 2.89 (s, 3H) ppm. 13C NMR (176 MHz, DMSO-d6): δ 176.4, 168.5, 167.8, 158.7, 149.8, 144.6, 130.8, 129.3, 129.2, 129.0, 128.2, 127.6, 125.0, 122.2, 116.1, 112.1, 77.9, 67.9, 64.3, 63.2, 28.6, 28.4, 26.6 ppm. HRMS (ESI): m/z calcd. for C25H2179BrN5O4 [M + H]+ 534.0771, found 534.0785; calcd. for C25H2181BrN5O4 [M + H]+ 536.0751, found 536.0765.
(2′R,3S)-4′-Amino-5-fluoro-1,1″,3″-trimethyl-2,2″,4″,6″-tetraoxo-2′-phenyl-1″,3″,4″,6″-tetrahydro-2″H-dispiro[indoline-3,1′-cyclopentane-3′,5″-pyrimidin]-4′-ene-5′-carbonitrile (3ag). According to the general procedure from 1a (58.6 mg, 0.24 mmol) and 2g (45.8 mg, 0.2 mmol) to obtain 89.9 mg (95% yield) compound 3ag as a white solid, m.p. 201–203 °C. HPLC (Daicel Chiralpak ADH, n-hexane/2-propanol = 70:30, flow rate 1.0 mL/min, detection at 254 nm): tR = 18.7 min (minor), tR = 20.2 min (major); 95% ee. [α]D25 = −58.6 (c = 0.5, CH2Cl2). 1H NMR (700 MHz, DMSO-d6): δ 7.91 (dd, J1 = 8.8 Hz, J2 = 2.4 Hz, 1H), 7.28 (s, 2H), 7.20–7.15 (m, 2H), 7.12 (t, J = 7.7 Hz, 2H), 6.90 (dd, J1 = 8.4 Hz, J2 = 4.2 Hz, 1H), 6.76 (d, J = 7.7 Hz, 2H), 4.26 (s, 1H), 3.07 (s, 3H), 3.00 (s, 3H), 2.91 (s, 3H) ppm. 13C NMR (176 MHz, DMSO-d6): δ 176.2, 168.5, 168.0, 158.8, 158.3 (1JC–F = 237.4 Hz), 149.8, 139.4, 130.8, 130.2 (3JC–F = 8.3 Hz), 129.23, 129.19, 128.3, 116.1, 115.7 (2JC–F = 23.4 Hz), 114.8 (2JC–F = 25.7 Hz), 109.8 (3JC–F = 8.3 Hz), 78.1, 67.9, 64.6, 63.8, 29.9, 28.6, 28.4, 26.6 ppm. 19F NMR (659 MHz, DMSO-d6) δ −120.1. HRMS (ESI): m/z calcd. for C25H21FN5O4 [M + H]+ 474.1572, found 474.1595.
(2′R,3S)-4′-Amino-5-bromo-1,1″,3″-trimethyl-2,2″,4″,6″-tetraoxo-2′-phenyl-1″,3″,4″,6″-tetrahydro-2″H-dispiro[indoline-3,1′-cyclopentane-3′,5″-pyrimidin]-4′-ene-5′-carbonitrile (3ah). According to the general procedure from 1a (58.6 mg, 0.24 mmol) and 2h (57.8 mg, 0.2 mmol) to obtain 101.3 mg (95% yield) compound 3ah as a white solid, m.p. 192–194 °C. HPLC (Daicel Chiralpak IA, n-hexane/2-propanol = 80:20, flow rate 1.0 mL/min, detection at 254 nm): tR = 14.1 min (minor), tR = 11.3 min (major), >99% ee. [α]D25 = −211.6 (c = 0.8, CH2Cl2). 1H NMR (700 MHz, DMSO-d6): δ 8.23 (d, J = 2.1 Hz, 1H), 7.50 (dd, J1 = 8.4 Hz, J2 = 2.1 Hz, 1H), 7.30 (s, 2H), 7.19 (t, J = 7.4 Hz, 1H), 7.12 (t, J = 7.7 Hz, 2H), 6.87 (d, J = 8.4 Hz, 1H), 6.75 (d, J = 7.0 Hz, 2H), 4.24 (s, 1H), 3.07 (s, 3H), 2.99 (s, 3H), 2.90 (s, 3H) ppm. 13C NMR (176 MHz, DMSO-d6): δ 176.0, 168.4, 167.9, 158.8, 149.7, 142.3, 132.0, 130.8, 130.7, 130.0, 129.23, 129.18, 128.3, 116.1, 114.2, 110.8, 77.9, 67.8, 64.6, 63.5, 28.6, 28.4, 26.6 ppm. HRMS (ESI): m/z calcd. for C25H2179BrN5O4 [M + H]+ 534.0771, found 534.0786; calcd. for C25H2181BrN5O4 [M + H]+ 536.0751, found 536.0769.
(2′R,3S)-4′-Amino-5-chloro-1,1″,3″-trimethyl-2,2″,4″,6″-tetraoxo-2′-phenyl-1″,3″,4″,6″-tetrahydro-2″H-dispiro[indoline-3,1′-cyclopentane-3′,5″-pyrimidin]-4′-ene-5′-carbonitrile (3ai). According to the general procedure from 1a (58.6 mg, 0.24 mmol) and 2i (49.0 mg, 0.2 mmol) to obtain 90.0 mg (92% yield) compound 3ai as a white solid, m.p. 201–204 °C. HPLC (Daicel Chiralpak IA, n-hexane/2-propanol = 90:10, flow rate 1.0 mL/min, detection at 254 nm): tR = 45.7 min (minor), tR = 32.5 min (major); >99% ee. [α]D25 = −100.6 (c = 0.5, CH2Cl2). 1H NMR (700 MHz, DMSO-d6): δ 8.11 (d, J = 2.8 Hz, 1H), 7.37 (dd, J1 = 8.4 Hz, J2 = 2.8 Hz, 1H), 7.30 (s, 2H), 7.19 (t, J = 7.7 Hz, 1H), 7.12 (t, J = 7.7 Hz, 2H), 6.92 (d, J = 7.7 Hz, 1H), 6.75 (dd, J1 = 8.4 Hz, J2 = 1.4 Hz, 2H), 4.25 (s, 1H), 3.08 (s, 3H), 3.00 (s, 3H), 2.91 (s, 3H) ppm. 13C NMR (176 MHz, DMSO-d6): δ 176.1, 168.4, 168.0, 158.8, 149.74, 142.0, 130.7, 130.4, 129.23, 129.19, 128.3, 127.2, 126.5, 116.1, 110.4, 77.9, 67.8, 64.6, 63.6, 28.6, 28.4, 26.6 ppm. HRMS (ESI): m/z calcd. for C25H21ClN5O4 [M + H]+ 490.1277, found 490.1304.
(2′R,3S)-4′-Amino-1,1″,3″-trimethyl-2,2″,4″,6″-tetraoxo-2′-phenyl-7-(trifluoromethyl)-1″,3″,4″,6″-tetrahydro-2″H-dispiro[indoline-3,1′-cyclopentane-3′,5″-pyrimidin]-4′-ene-5′-carbonitrile (3aj). According to the general procedure from 1a (58.6 mg, 0.24 mmol) and 2j (55.8 mg, 0.2 mmol) to obtain 88.9 mg (85% yield) compound 3aj as a white solid, m.p. 177–179 °C. HPLC (Daicel Chiralpak IA, n-hexane/2-propanol = 70:30, flow rate 1.0 mL/min, detection at 254 nm): tR = 5.6 min (minor), tR = 7.6 min (major); 83% ee. [α]D25 = −60.8 (c = 0.5, CH2Cl2). 1H NMR (700 MHz, DMSO-d6): δ 8.37 (d, J = 7.0 Hz, 1H), 7.64 (dd, J1 = 8.0 Hz, J2 = 1.0 Hz, 1H), 7.38 (t, J = 8.0 Hz, 3H), 7.17 (t, J = 7.4 Hz, 1H), 7.07 (t, J = 7.7 Hz, 2H), 6.67 (d, J = 7.0 Hz, 2H), 4.22 (s, 1H), 3.14 (s, 3H), 3.08 (s, 3H), 2.89 (s, 3H) ppm. 13C NMR (176 MHz, DMSO-d6): δ 178.0, 168.5, 167.8, 159.3, 149.8, 140.6, 131.4, 131.3, 130.4, 129.2, 128.1, 127.3, 127.2 (q, 3JC–F = 5.5 Hz), 123.2 (q, 1JC–F = 271.2 Hz), 122.2, 116.0, 110.9 (q, 2JC–F = 32.7 Hz), 77.2, 67.8, 65.3, 62.2, 29.0 (q, JC–F = 5.8 Hz), 28.6, 28.4 ppm. 19F NMR (659 MHz, DMSO-d6): δ −52.1. HRMS (ESI): m/z calcd. for C26H21F3N5O4 [M + H]+ 524.1540, found 524.1559.
(2′R,3S)-4′-Amino-1,1″,3″,7-tetramethyl-2,2″,4″,6″-tetraoxo-2′-phenyl-1″,3″,4″,6″-tetrahydro-2″H-dispiro[indoline-3,1′-cyclopentane-3′,5″-pyrimidin]-4′-ene-5′-carbonitrile (3ak). According to the general procedure from 1a (58.6 mg, 0.24 mmol) and 2k (45.0 mg, 0.2 mmol) to obtain 89.1 mg (95% yield) compound 3ak as a white solid, m.p. 182–185 °C. HPLC (Daicel Chiralpak IA, n-hexane/2-propanol = 70:30, flow rate 1.0 mL/min, detection at 254 nm): tR = 7.5 min (minor), tR = 9.3 min (major); 82% ee. [α]D25 = 23.4 (c = 0.4, CH2Cl2). 1H NMR (700 MHz, DMSO-d6): δ 7.92 (d, J = 6.3 Hz, 1H), 7.18–7.14 (m, 3H), 7.08–7.01 (m, 3H), 6.75 (d, J = 7.7 Hz, 2H), 4.23 (s, 1H), 3.27 (s, 3H), 3.07 (s, 3H), 2.84 (s, 3H), 2.38 (s, 3H) ppm. 13C NMR (176 MHz, DMSO-d6): δ 177.2, 168.7, 167.8, 158.3, 149.8, 140.8, 132.9, 131.2, 129.5, 129.0, 128.0, 125.4, 122.1, 119.7, 116.4, 79.0, 68.2, 65.0, 62.9, 29.6, 28.5, 28.3, 18.4 ppm. HRMS (ESI): m/z calcd. for C26H24N5O4 [M + H]+ 470.1823, found 470.1834.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}