

Evaluating Triazole-Substituted Pyrrolopyrimidines as CSF1R Inhibitors

 ,
,  , , ,
, , ,  and
and
Abstract

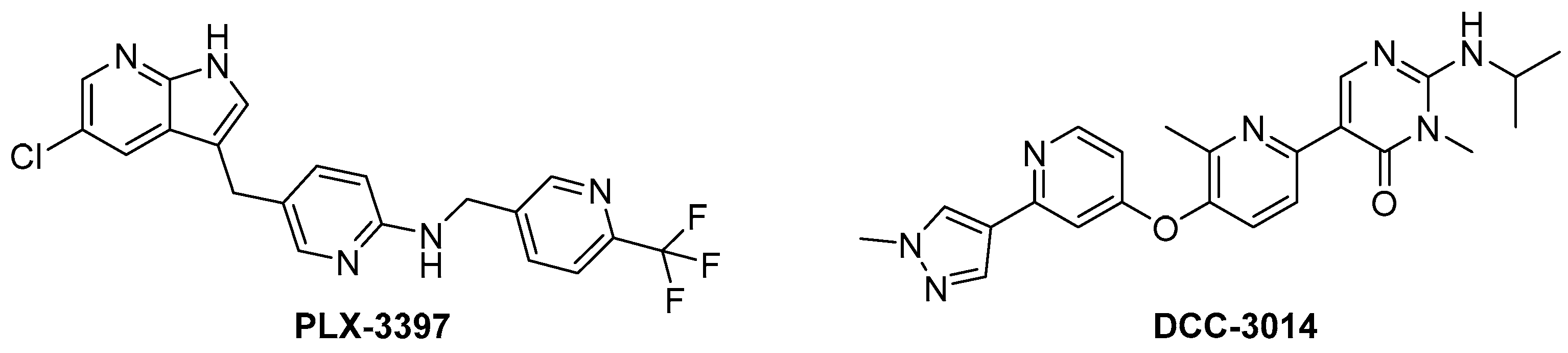
1. Introduction
2. Results and Discussion
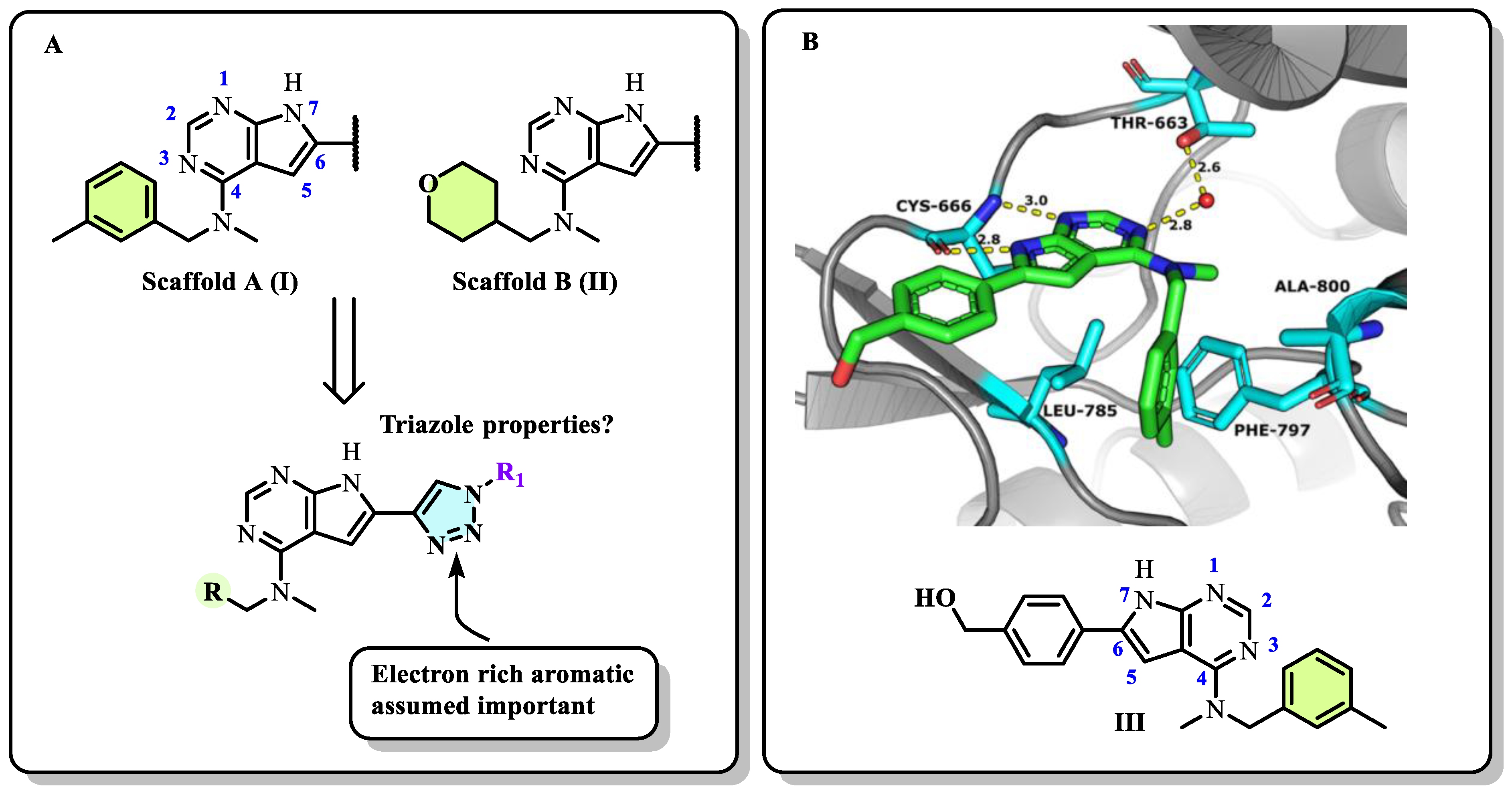
2.1. Designs of the Inhibitors
2.2. Chemistry
2.3. Enzymatic and Cellular Characterisation
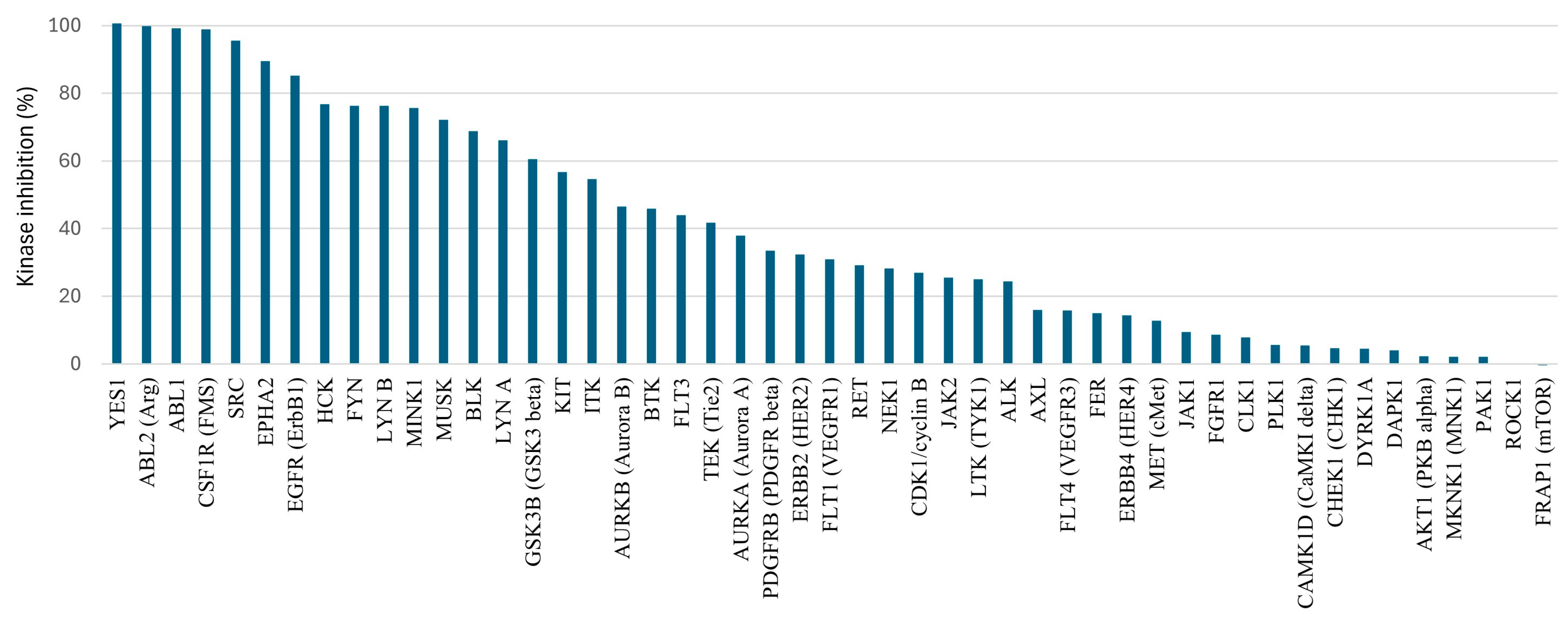
2.4. Initial ADME and Kinase Off-Targets
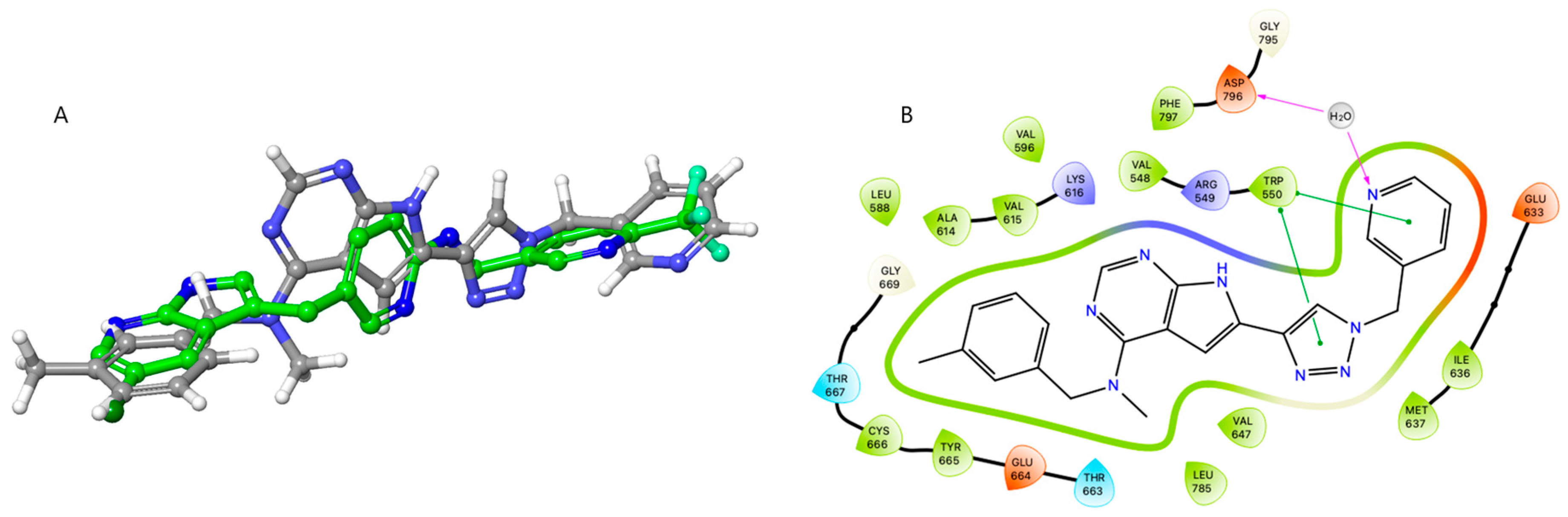
2.5. Molecular Modelling
3. Materials and Methods
3.1. Chemicals and Analysis
3.2. General Procedures
3.2.1. General Procedure A: Synthesis of Triazole Intermediates
3.2.2. General Procedure B: SEM-Deprotection
3.3. Inhibitor Candidates
3.3.1. N-Methyl-N-(3-methylbenzyl)-6-(1-phenyl-1H-1,2,3-triazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (18a)
3.3.2. N-Methyl-6-(1-phenyl-1H-1,2,3-triazol-4-yl)-N-((tetrahydro-2H-pyran-4-yl)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (18b)
3.3.3. 6-(1-(4-Fluorophenyl)-1H-1,2,3-triazol-4-yl)-N-methyl-N-(3-methylbenzyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (19a)
3.3.4. 6-(1-(4-Fluorophenyl)-1H-1,2,3-triazol-4-yl)-N-methyl-N-((tetrahydro-2H-pyran-4-yl)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (19b)
3.3.5. N-Methyl-N-(3-methylbenzyl)-6-(1-(p-tolyl)-1H-1,2,3-triazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (20a)
3.3.6. N-Methyl-N-((tetrahydro-2H-pyran-4-yl)methyl)-6-(1-(p-tolyl)-1H-1,2,3-triazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (20b)
3.3.7. 6-(1-(4-Methoxyphenyl)-1H-1,2,3-triazol-4-yl)-N-methyl-N-(3-methylbenzyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (21a)
3.3.8. 6-(1-(4-Methoxyphenyl)-1H-1,2,3-triazol-4-yl)-N-methyl-N-((tetrahydro-2H-pyran-4-yl)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (21b)
3.3.9. N-Methyl-N-(3-methylbenzyl)-6-(1-(pyridin-3-yl)-1H-1,2,3-triazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (22a)
3.3.10. N-Methyl-6-(1-(pyridin-3-yl)-1H-1,2,3-triazol-4-yl)-N-((tetrahydro-2H-pyran-4-yl)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (22b)
3.3.11. 6-(1-Benzyl-1H-1,2,3-triazol-4-yl)-N-methyl-N-(3-methylbenzyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (23a)
3.3.12. 6-(1-Benzyl-1H-1,2,3-triazol-4-yl)-N-methyl-N-((tetrahydro-2H-pyran-4-yl)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (23b)
3.3.13. 6-(1-(4-Fluorobenzyl)-1H-1,2,3-triazol-4-yl)-N-methyl-N-(3-methylbenzyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (24a)
3.3.14. 6-(1-(4-Fluorobenzyl)-1H-1,2,3-triazol-4-yl)-N-methyl-N-((tetrahydro-2H-pyran-4-yl)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (24b)
3.3.15. N-Methyl-N-(3-methylbenzyl)-6-(1-(4-methylbenzyl)-1H-1,2,3-triazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (25a)
3.3.16. N-Methyl-6-(1-(4-methylbenzyl)-1H-1,2,3-triazol-4-yl)-N-((tetrahydro-2H-pyran-4-yl)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (25b)
3.3.17. 6-(1-(4-Methoxybenzyl)-1H-1,2,3-triazol-4-yl)-N-methyl-N-(3-methylbenzyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (26a)
3.3.18. 6-(1-(4-Methoxybenzyl)-1H-1,2,3-triazol-4-yl)-N-methyl-N-((tetrahydro-2H-pyran-4-yl)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (26b)
3.3.19. N-Methyl-N-(3-methylbenzyl)-6-(1-(pyridin-3-ylmethyl)-1H-1,2,3-triazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (27a)
3.3.20. N-Methyl-6-(1-(pyridin-3-ylmethyl)-1H-1,2,3-triazol-4-yl)-N-((tetrahydro-2H-pyran-4-yl)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (27b)
3.3.21. 3-(4-(4-(Methyl(3-methylbenzyl)amino)-7H-pyrrolo[2,3-d]pyrimidin-6-yl)-1H-1,2,3-triazol-1-yl)propan-1-ol (28a)
3.3.22. 3-(4-(4-(Methyl((tetrahydro-2H-pyran-4-yl)methyl)amino)-7H-pyrrolo[2,3-d]pyrimidin-6-yl)-1H-1,2,3-triazol-1-yl)propan-1-ol (28b)
3.3.23. N-Methyl-N-(3-methylbenzyl)-6-(1-(tetrahydro-2H-pyran-4-yl)-1H-1,2,3-triazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (29a)
3.3.24. N-Methyl-6-(1-(tetrahydro-2H-pyran-4-yl)-1H-1,2,3-triazol-4-yl)-N-((tetrahydro-2H-pyran-4-yl)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (29b)
3.3.25. N-Methyl-N-(3-methylbenzyl)-6-(1-(1-methylpiperidin-4-yl)-1H-1,2,3-triazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (30a)
3.3.26. N-Methyl-6-(1-(1-methylpiperidin-4-yl)-1H-1,2,3-triazol-4-yl)-N-((tetrahydro-2H-pyran-4-yl)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (30b)
3.3.27. N-Methyl-N-(3-methylbenzyl)-6-(1-((tetrahydro-2H-pyran-4-yl)methyl)-1H-1,2,3-triazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (31a)
3.3.28. N-Methyl-N-((tetrahydro-2H-pyran-4-yl)methyl)-6-(1-((tetrahydro-2H-pyran-4-yl)methyl)-1H-1,2,3-triazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (31b)
3.4. Biochemical Assays
3.4.1. KIT Inhibition (KD)
3.4.2. GIST-T1 CTG Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lin, H.; Lee, E.; Hestir, K.; Leo, C.; Huang, M.; Bosch, E.; Halenbeck, R.; Wu, G.; Zhou, A.; Behrens, D.; et al. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science 2008, 320, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Cersosimo, F.; Lonardi, S.; Ulivieri, C.; Martini, P.; Morrione, A.; Vermi, W.; Giordano, A.; Giurisato, E. CSF-1R in Cancer: More than a Myeloid Cell Receptor. Cancers 2024, 16, 282. [Google Scholar] [CrossRef]
- Mun, S.H.; Park, P.S.U.; Park-Min, K.-H. The M-CSF receptor in osteoclasts and beyond. Exp. Mol. Med. 2020, 52, 1239–1254. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Priya, A.; Borade, D.R.; Agrawal-Rajput, R. Macrophage subsets and their role: Co-relation with colony-stimulating factor-1 receptor and clinical relevance. Immunol. Res. 2023, 71, 130–152. [Google Scholar] [CrossRef] [PubMed]
- Mo, H.; Hao, Y.; Lv, Y.; Chen, Z.; Shen, J.; Zhou, S.; Yin, M. Overexpression of macrophage-colony stimulating factor-1 receptor as a prognostic factor for survival in cancer: A systematic review and meta-analysis. Medicine 2021, 100, e25218. [Google Scholar] [CrossRef]
- Riaz, N.; Burugu, S.; Cheng, A.S.; Leung, S.C.Y.; Gao, D.; Nielsen, T.O. Prognostic Significance of CSF-1R Expression in Early Invasive Breast Cancer. Cancers 2021, 13, 5769. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P. The interaction of anticancer therapies with tumor-associated macrophages. J. Exp. Med. 2015, 212, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Xiang, C.; Li, H.; Tang, W. Targeting CSF-1R represents an effective strategy in modulating inflammatory diseases. Pharmacol. Res. 2023, 187, 106566. [Google Scholar] [CrossRef]
- Olmos-Alonso, A.; Gomez-Nicola, D.; Askew, K.; Mancuso, R.; Sri, S.; Schetters, S.T.T.; Perry, V.H.; Vargas-Caballero, M.; Holscher, C. Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain 2016, 139, 891–907. [Google Scholar] [CrossRef]
- Sosna, J.; Philipp, S.; Albay, R., III; Reyes-Ruiz, J.M.; Baglietto-Vargas, D.; LaFerla, F.M.; Glabe, C.G. Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 11. [Google Scholar] [CrossRef] [PubMed]
- Weyer, M.-P.; Strehle, J.; Schäfer, M.K.E.; Tegeder, I. Repurposing of pexidartinib for microglia depletion and renewal. Pharmacol. Ther. 2024, 253, 108565. [Google Scholar] [CrossRef] [PubMed]
- Hume, D.A.; Batoon, L.; Sehgal, A.; Keshvari, S.; Irvine, K.M. CSF1R as a therapeutic target in bone diseases: Obvious but not so simple. Curr. Osteoporos. Rep. 2022, 20, 516–531. [Google Scholar] [CrossRef]
- Alkubaisi, B.O.; Aljobowry, R.; Ali, S.M.; Sultan, S.; Zaraei, S.-O.; Ravi, A.; Al-Tel, T.H.; El-Gamal, M.I. The latest perspectives of small molecules FMS kinase inhibitors. Eur. J. Med. Chem. 2023, 261, 115796. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Wang, S.; Guo, R.; Liu, D. CSF1R inhibitors are emerging immunotherapeutic drugs for cancer treatment. Eur. J. Med. Chem. 2023, 245, 114884. [Google Scholar] [CrossRef]
- Smith, B.D.; Kaufman, M.D.; Wise, S.C.; Ahn, Y.M.; Caldwell, T.M.; Leary, C.B.; Lu, W.-P.; Tan, G.; Vogeti, L.; Vogeti, S.; et al. Vimseltinib: A Precision CSF1R Therapy for Tenosynovial Giant Cell Tumors and Diseases Promoted by Macrophages. Mol. Cancer Ther. 2021, 20, 2098–2109. [Google Scholar] [CrossRef]
- Krauser, J.A.; Jin, Y.; Walles, M.; Pfaar, U.; Sutton, J.; Wiesmann, M.; Graf, D.; Pflimlin-Fritschy, V.; Wolf, T.; Camenisch, G.; et al. Phenotypic and metabolic investigation of a CSF-1R kinase receptor inhibitor (BLZ945) and its pharmacologically active metabolite. Xenobiotica 2015, 45, 107–123. [Google Scholar] [CrossRef]
- Genovese, M.C.; Hsia, E.; Belkowski, S.M.; Chien, C.; Masterson, T.; Thurmond, R.L.; Manthey, C.L.; Yan, X.; Ge, T.; Franks, C.; et al. Results from a Phase IIA Parallel Group Study of JNJ-40346527, an Oral CSF-1R Inhibitor, in Patients with Active Rheumatoid Arthritis despite Disease-modifying Antirheumatic Drug Therapy. J. Rheumatol. 2015, 42, 1752–1760. [Google Scholar] [CrossRef]
- Manthey, C.L.; Moore, B.A.; Chen, Y.; Loza, M.J.; Yao, X.; Liu, H.; Belkowski, S.M.; Raymond-Parks, H.; Dunford, P.J.; Leon, F.; et al. The CSF-1-receptor inhibitor, JNJ-40346527 (PRV-6527), reduced inflammatory macrophage recruitment to the intestinal mucosa and suppressed murine T cell mediated colitis. PLoS ONE 2019, 14, e0223918. [Google Scholar] [CrossRef]
- Aarhus, T.I.; Bjørnstad, F.; Wolowczyk, C.; Larsen, K.U.; Rognstad, L.; Leithaug, T.; Unger, A.; Habenberger, P.; Wolf, A.; Bjørkøy, G.; et al. Synthesis and Development of Highly Selective Pyrrolo[2,3-d]pyrimidine CSF1R Inhibitors Targeting the Autoinhibited Form. J. Med. Chem. 2023, 66, 6959–6980. [Google Scholar] [CrossRef]
- Aarhus, T.I.; Teksum, V.; Unger, A.; Habenberger, P.; Wolf, A.; Eickhoff, J.; Klebl, B.; Wolowczyk, C.; Bjørkøy, G.; Sundby, E.; et al. Negishi Cross-Coupling in the Preparation of Benzyl Substituted Pyrrolo[2,3-d]pyrimidine Based CSF1R Inhibitors. Eur. J. Org. Chem. 2023, 26, e202300052. [Google Scholar] [CrossRef]
- Bjørnstad, F.; Havik, S.; Aarhus, T.I.; Mahdi, I.; Unger, A.; Habenberger, P.; Degenhart, C.; Eickhoff, J.; Klebl, B.M.; Sundby, E.; et al. Pyrrolopyrimidine based CSF1R inhibitors: Attempted departure from Flatland. Eur. J. Med. Chem. 2024, 265, 116053. [Google Scholar] [CrossRef]
- Aarhus, T.I.; Eickhoff, J.; Klebl, B.; Unger, A.; Boros, J.; Choidas, A.; Zischinsky, M.-L.; Wolowczyk, C.; Bjørkøy, G.; Sundby, E.; et al. A highly selective purine-based inhibitor of CSF1R potently inhibits osteoclast differentiation. Eur. J. Med. Chem. 2023, 255, 115344. [Google Scholar] [CrossRef]
- Meanwell, N.A. Improving Drug Candidates by Design: A Focus on Physicochemical Properties As a Means of Improving Compound Disposition and Safety. Chem. Res. Toxicol. 2011, 24, 1420–1456. [Google Scholar] [CrossRef]
- Ritchie, T.J.; Macdonald, S.J.F. Heterocyclic replacements for benzene: Maximising ADME benefits by considering individual ring isomers. Eur. J. Med. Chem. 2016, 124, 1057–1068. [Google Scholar] [CrossRef]
- Subbaiah, M.A.M.; Meanwell, N.A. Bioisosteres of the Phenyl Ring: Recent Strategic Applications in Lead Optimization and Drug Design. J. Med. Chem. 2021, 64, 14046–14128. [Google Scholar] [CrossRef]
- Khan, J.; Rani, A.; Aslam, M.; Maharia, R.S.; Pandey, G.; Nand, B. Exploring triazole-based drugs: Synthesis, application, FDA approvals, and clinical trial updates–A comprehensive review. Tetrahedron 2024, 162, 134122. [Google Scholar] [CrossRef]
- Bozorov, K.; Zhao, J.; Aisa, H.A. 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg. Med. Chem. 2019, 27, 3511–3531. [Google Scholar] [CrossRef] [PubMed]
- Dixit, D.; Verma, P.K.; Marwaha, R.K. A review on ‘triazoles’: Their chemistry, synthesis and pharmacological potentials. J. Iran. Chem. Soc. 2021, 18, 2535–2565. [Google Scholar] [CrossRef]
- Arioli, F.; Borrelli, S.; Colombo, F.; Falchi, F.; Filippi, I.; Crespan, E.; Naldini, A.; Scalia, G.; Silvani, A.; Maga, G.; et al. N-[2-Methyl-5-(triazol-1-yl)phenyl]pyrimidin-2-amine as a Scaffold for the Synthesis of Inhibitors of Bcr-Abl. ChemMedChem 2011, 6, 2009–2018. [Google Scholar] [CrossRef]
- Mortazavi, M.; Eskandari, M.; Moosavi, F.; Damghani, T.; Khoshneviszadeh, M.; Pirhadi, S.; Saso, L.; Edraki, N.; Firuzi, O. Novel quinazoline-1,2,3-triazole hybrids with anticancer and MET kinase targeting properties. Sci. Rep. 2023, 13, 14685. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, M.; Singh Pathania, A.; Mahajan, P.; Verma, P.K.; Chobe, S.S.; Malik, F.A.; Nargotra, A.; Vishwakarma, R.A.; Sawant, S.D. Design and synthesis of 1,4-substituted 1H-1,2,3-triazolo-quinazolin-4(3H)-ones by Huisgen 1,3-dipolar cycloaddition with PI3Kγ isoform selective activity. Bioorg. Med. Chem. Lett. 2018, 28, 1005–1010. [Google Scholar] [CrossRef]
- Safavi, M.; Ashtari, A.; Khalili, F.; Mirfazli, S.S.; Saeedi, M.; Ardestani, S.K.; Rashidi Ranjbar, P.; Barazandeh Tehrani, M.; Larijani, B.; Mahdavi, M. Novel quinazolin-4(3H)-one linked to 1,2,3-triazoles: Synthesis and anticancer activity. Chem. Biol. Drug Des. 2018, 92, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Kettle, J.G.; Anjum, R.; Barry, E.; Bhavsar, D.; Brown, C.; Boyd, S.; Campbell, A.; Goldberg, K.; Grondine, M.; Guichard, S.; et al. Discovery of N-(4-{[5-Fluoro-7-(2-methoxyethoxy)quinazolin-4-yl]amino}phenyl)-2-[4-(propan-2-yl)-1H-1,2,3-triazol-1-yl]acetamide (AZD3229), a Potent Pan-KIT Mutant Inhibitor for the Treatment of Gastrointestinal Stromal Tumors. J. Med. Chem. 2018, 61, 8797–8810. [Google Scholar] [CrossRef]
- Menet, C.J.; Fletcher, S.R.; Van Lommen, G.; Geney, R.; Blanc, J.; Smits, K.; Jouannigot, N.; Deprez, P.; van der Aar, E.M.; Clement-Lacroix, P.; et al. Triazolopyridines as Selective JAK1 Inhibitors: From Hit Identification to GLPG0634. J. Med. Chem. 2014, 57, 9323–9342. [Google Scholar] [CrossRef] [PubMed]
- Kulukian, A.; Lee, P.; Taylor, J.; Rosler, R.; de Vries, P.; Watson, D.; Forero-Torres, A.; Peterson, S. Preclinical Activity of HER2-Selective Tyrosine Kinase Inhibitor Tucatinib as a Single Agent or in Combination with Trastuzumab or Docetaxel in Solid Tumor Models. Mol. Cancer Ther. 2020, 19, 976–987. [Google Scholar] [CrossRef]
- Tap, W.D.; Wainberg, Z.A.; Anthony, S.P.; Ibrahim, P.N.; Zhang, C.; Healey, J.H.; Chmielowski, B.; Staddon, A.P.; Cohn, A.L.; Shapiro, G.I.; et al. Structure-guided blockade of CSF1R kinase in tenosynovial giant-cell tumor. N. Engl. J. Med. 2015, 373, 428–437. [Google Scholar] [CrossRef]
- Pollok, B.A.; Hamman, B.D.; Rodems, S.M.; Makings, L.R. Optical Probes and Assays. U.S. Patent No. 6,410,255, 30 March 2004. [Google Scholar]
- Taguchi, T.; Sonobe, H.; Toyonaga, S.-i.; Yamasaki, I.; Shuin, T.; Takano, A.; Araki, K.; Akimaru, K.; Yuri, K. Conventional and Molecular Cytogenetic Characterization of a New Human Cell Line, GIST-T1, Established from Gastrointestinal Stromal Tumor. Lab. Investig. 2002, 82, 663–665. [Google Scholar] [CrossRef]
- Force, T.; Kolaja, K.L. Cardiotoxicity of kinase inhibitors: The prediction and translation of preclinical models to clinical outcomes. Nat. Rev. Drug Discovery 2011, 10, 111–126. [Google Scholar] [CrossRef]
- Kaspersen, S.J.; Han, J.; Nørsett, K.G.; Rydså, L.; Kjøbli, E.; Bugge, S.; Bjørkøy, G.; Sundby, E.; Hoff, B.H. Identification of new 4-N-substituted 6-aryl-7H-pyrrolo[2,3-d]pyrimidine-4-amines as highly potent EGFR-TK inhibitors with Src-family activity. Eur. J. Pharm. Sci. 2014, 59, 69–82. [Google Scholar] [CrossRef]
- Han, J.; Henriksen, S.; Nørsett, K.G.; Sundby, E.; Hoff, B.H. Balancing potency, metabolic stability and permeability in pyrrolopyrimidine-based EGFR inhibitors. Eur. J. Med. Chem. 2016, 124, 583–607. [Google Scholar] [CrossRef] [PubMed]
- Karaman, M.W.; Herrgard, S.; Treiber, D.K.; Gallant, P.; Atteridge, C.E.; Campbell, B.T.; Chan, K.W.; Ciceri, P.; Davis, M.I.; Edeen, P.T.; et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [Google Scholar] [CrossRef]
- Khatri, A.; Wang, J.; Pendergast, A.M. Multifunctional Abl kinases in health and disease. J. Cell Sci. 2016, 129, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Sun, D.; Tao, J.; Xu, M.; Zhang, X.; Hou, H. Role of YES1 signaling in tumor therapy resistance. Cancer Innov. 2023, 2, 210–218. [Google Scholar] [CrossRef]
- Ortiz, M.A.; Mikhailova, T.; Li, X.; Porter, B.A.; Bah, A.; Kotula, L. Src family kinases, adaptor proteins and the actin cytoskeleton in epithelial-to-mesenchymal transition. Cell Commun. Sig. 2021, 19, 67. [Google Scholar] [CrossRef]
- Garmendia, I.; Redin, E.; Montuenga, L.M.; Calvo, A. YES1: A Novel Therapeutic Target and Biomarker in Cancer. Mol. Cancer Ther. 2022, 21, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.; Shiuan, E.; Brantley-Sieders, D.M. Oncogenic functions and therapeutic targeting of EphA2 in cancer. Oncogene 2021, 40, 2483–2495. [Google Scholar] [CrossRef]
- Schrödinger_Release_2025-1 Glide; Schrödinger, LLC: New York, NY, USA, 2025.
- Wodicka, L.M.; Ciceri, P.; Davis, M.I.; Hunt, J.P.; Floyd, M.; Salerno, S.; Hua, X.H.; Ford, J.M.; Armstrong, R.C.; Zarrinkar, P.P.; et al. Activation state-dependent binding of small molecule kinase inhibitors: Structural insights from biochemistry. Chem. Biol. 2010, 17, 1241–1249. [Google Scholar] [CrossRef]
- Kitagawa, D.; Gouda, M.; Kirii, Y.; Sugiyama, N.; Ishihama, Y.; Fujii, I.; Narumi, Y.; Akita, K.; Yokota, K. Characterization of kinase inhibitors using different phosphorylation states of colony stimulating factor-1 receptor tyrosine kinase. J. Biochem. 2012, 151, 47–55. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
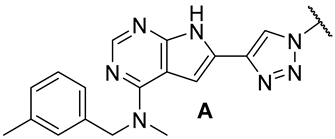 | |||||||
|---|---|---|---|---|---|---|---|
| Group | Comp. | cLogP a | PSA b | Sol. c | Enz. IC50 (nM) d | CSF1R Ba/F3 IC50 (µM) e | IL-3 Ba/F3 IC50 (µM) f |
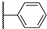 | 18a | 3.08 | 75.5 | 0.3 | 1.9 (0.94) | >10 | >10 |
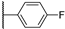 | 19a | 3.18 | 75.5 | 0.4 | 7.7 (0.97) | >10 | >10 |
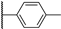 | 20a | 3.43 | 75.5 | 3 | 28.2 (0.98) | >10 | >10 |
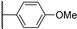 | 21a | 3.01 | 84.8 | 1.3 | 5.9 (0.96) | 6.0 ± 0.0 | >10 |
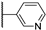 | 22a | 2.08 | 88.4 | 11 | 2.3 (0.95) | 1.2 ± 0.2 | >10 |
 | 23a | 3.43 | 75.5 | 0.3 | 2.0 (0.93) | >10 | >10 |
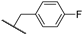 | 24a | 3.44 | 75.5 | 0.2 | 2.0 (0.97) | >10 | >10 |
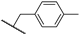 | 25a | 3.68 | 75.5 | 1.1 | 6.3 (0.98) | >10 | >10 |
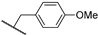 | 26a | 3.27 | 84.8 | 4.6 | 5.8 (0.92) | 2.8 ± 0.3 | >10 |
 | 27a | 2.33 | 88.4 | 3.1 | 1 < (0.98) | 0.3 ± 0.0 | >10 |
 | 28a | 1.76 | 95.8 | 5.5 | 1 < (0.85) | 1.0 ± 0.1 | >10 |
 | 29a | 2.66 | 84.8 | 20 | 1 < (0.99) | >10 | >10 |
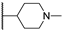 | 30a | 2.77 | 78.8 | 51 | 1 < (0.99) | 1.1 ± 0.1 | >10 |
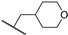 | 31a | 2.41 | 84.8 | 2.5 | 1 < (0.98) | 2.7 ± 1.0 | >10 |
| PLX-3397 | 3.72 | 66.5 | 20 | 21 (0.97) | 0.06 ± 0.1 | >10 | |
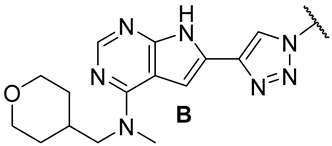 | |||||||
|---|---|---|---|---|---|---|---|
| Group | Comp. | cLogP a | PSA b | Sol. c | Enz. IC50 (nM) d | CSF1R Ba/F3 IC50 (µM) e | IL-3 Ba/F3 IC50 (µM) f |
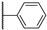 | 18b | 1.91 | 84.8 | 14 | 1.1 (0.99) | >10 | >10 |
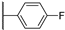 | 19b | 2.01 | 84.8 | 11 | 2.1 (0.97) | >10 | >10 |
 | 20b | 2.25 | 84.8 | 0.7 | 9.1 (0.99) | 3.2 ± 0.9 | >10 |
 | 21b | 1.84 | 94.0 | 7.3 | 3 (0.95) | 0.6 ± 0.2 | >10 |
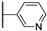 | 22b | 0.91 | 97.6 | 37 | 2.1 (0.97) | 1.6 ± 0.3 | >10 |
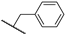 | 23b | 2.16 | 84.8 | 4.9 | 1 < (0.95) | 3.7 ± 1.9 | >10 |
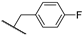 | 24b | 2.26 | 84.8 | 1.7 | 1.5 (0.96) | 4.6 ± 2.7 | >10 |
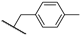 | 25b | 2.50 | 84.8 | 10.6 | 1 < (0.96) | 0.5 ± 0.0 | >10 |
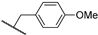 | 26b | 2.09 | 94.0 | 7.2 | 1 < (0.98) | 1.5 ± 0.5 | >10 |
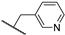 | 27b | 1.16 | 97.6 | 43 | 1.8 (0.98) | 1.7 ± 0.1 | >10 |
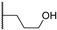 | 28b | 0.59 | 105.0 | 495 | 1.1 (0.93) | >10 | >10 |
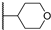 | 29b | 1.48 | 94.0 | 76 | 1 < (0.92) | >10 | >10 |
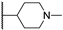 | 30b | 1.59 | 88.0 | 42 | 1 < (0.98) | 2.4 ± 0.5 | >10 |
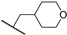 | 31b | 1.24 | 94.0 | 45 | 1 < (0.96) | 1.0 ± 0.2 | >10 |
| PLX-3397 | 3.72 | 66.5 | 20 | 21 (0.97) | 0.06 ± 0.5 | >10 | |
 | ||||||
|---|---|---|---|---|---|---|
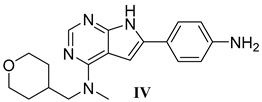
| Assay | 21b | 25b | 27a | III | IV | PLX-3397 |
| CSF1R enz. IC50 (nM) a | 1.2 ± 0.3 | 1.3 ± 0.0 | 0.4 ± 0.2 | 0.5 ± 0.2 | 0.3 ± 1 | 4.7 ± 0.5 |
| KIT enz. KD (nM) b | >1000 | >100 | 793 (0.93) | >3000 | ND | 28 (0.99) |
| KIT cell IC50 (µM) c | >30 | >3 | >30 | ND | ND | 0.03 |
| HLM [µL/min/mg] d | 74.5 | 62.4 | 53.7 | 15.5 | 13.6 | 11.8 |
| MLM [µL/min/mg] e | 288 | 1980 | 295 | 40.7 | 22.0 | 41.4 |
| Papp A > B [10−6 cm/s] f | 10.7 | 10.0 | 1.27 | 0.69 h | 37.9 | 4.45 |
| Papp B > A [10−6 cm/s] f | 13.6 | 25.4 | 3.18 | 0.19 h | 27.3 | 1.49 |
| Ratio Papp B > A/Papp A > B f | 1.3 | 2.6 | 2.5 | 0.28 h | 0.72 | 0.33 |
| PPB (%) g | 76 | 97.5 | >99 | 94.4 | 76.4 | >99 |
| Compound | CSF1R Structure: 8CGC | CSF1R Structure: 4R7H |
|---|---|---|
| Structure III | −11.569 | −9.565 |
| PLX3397 | −7.763 | −12.482 |
| 21b | −9.848 | −7.879 |
| 25b | −9.648 | −8.982 |
| 27a | −10.835 | −10.774 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cherukupalli, S.; Eickhoff, J.; Degenhart, C.; Habenberger, P.; Unger, A.; Hoff, B.H.; Sundby, E. Evaluating Triazole-Substituted Pyrrolopyrimidines as CSF1R Inhibitors. Molecules 2025, 30, 2641. https://doi.org/10.3390/molecules30122641
Cherukupalli S, Eickhoff J, Degenhart C, Habenberger P, Unger A, Hoff BH, Sundby E. Evaluating Triazole-Substituted Pyrrolopyrimidines as CSF1R Inhibitors. Molecules. 2025; 30(12):2641. https://doi.org/10.3390/molecules30122641
Chicago/Turabian StyleCherukupalli, Srinivasulu, Jan Eickhoff, Carsten Degenhart, Peter Habenberger, Anke Unger, Bård Helge Hoff, and Eirik Sundby. 2025. "Evaluating Triazole-Substituted Pyrrolopyrimidines as CSF1R Inhibitors" Molecules 30, no. 12: 2641. https://doi.org/10.3390/molecules30122641
APA StyleCherukupalli, S., Eickhoff, J., Degenhart, C., Habenberger, P., Unger, A., Hoff, B. H., & Sundby, E. (2025). Evaluating Triazole-Substituted Pyrrolopyrimidines as CSF1R Inhibitors. Molecules, 30(12), 2641. https://doi.org/10.3390/molecules30122641