
A Concise Asymmetric Synthesis of Sex Pheromone of Euproctis pseudoconspersa (Strand) and Its Enantiomer

Abstract
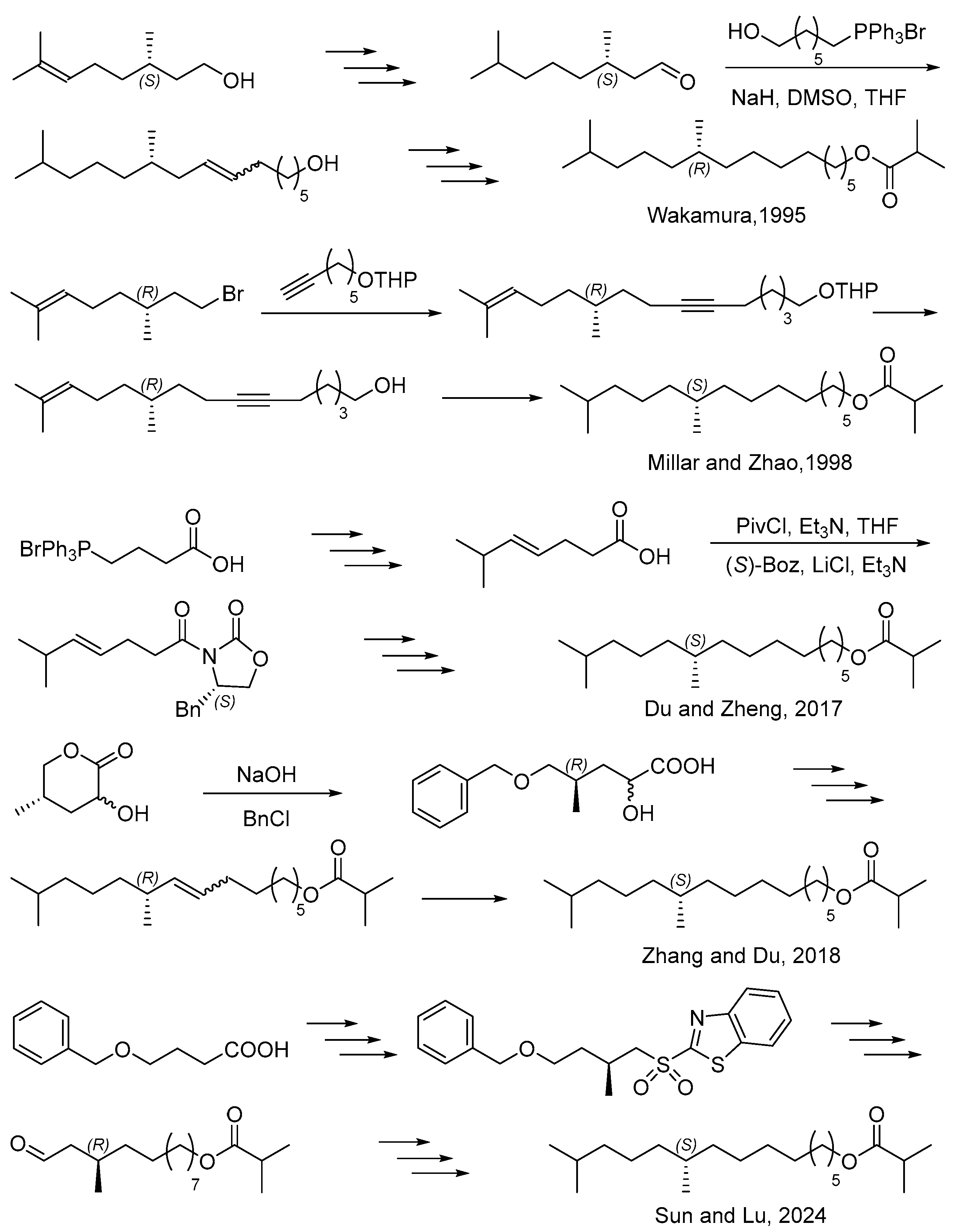
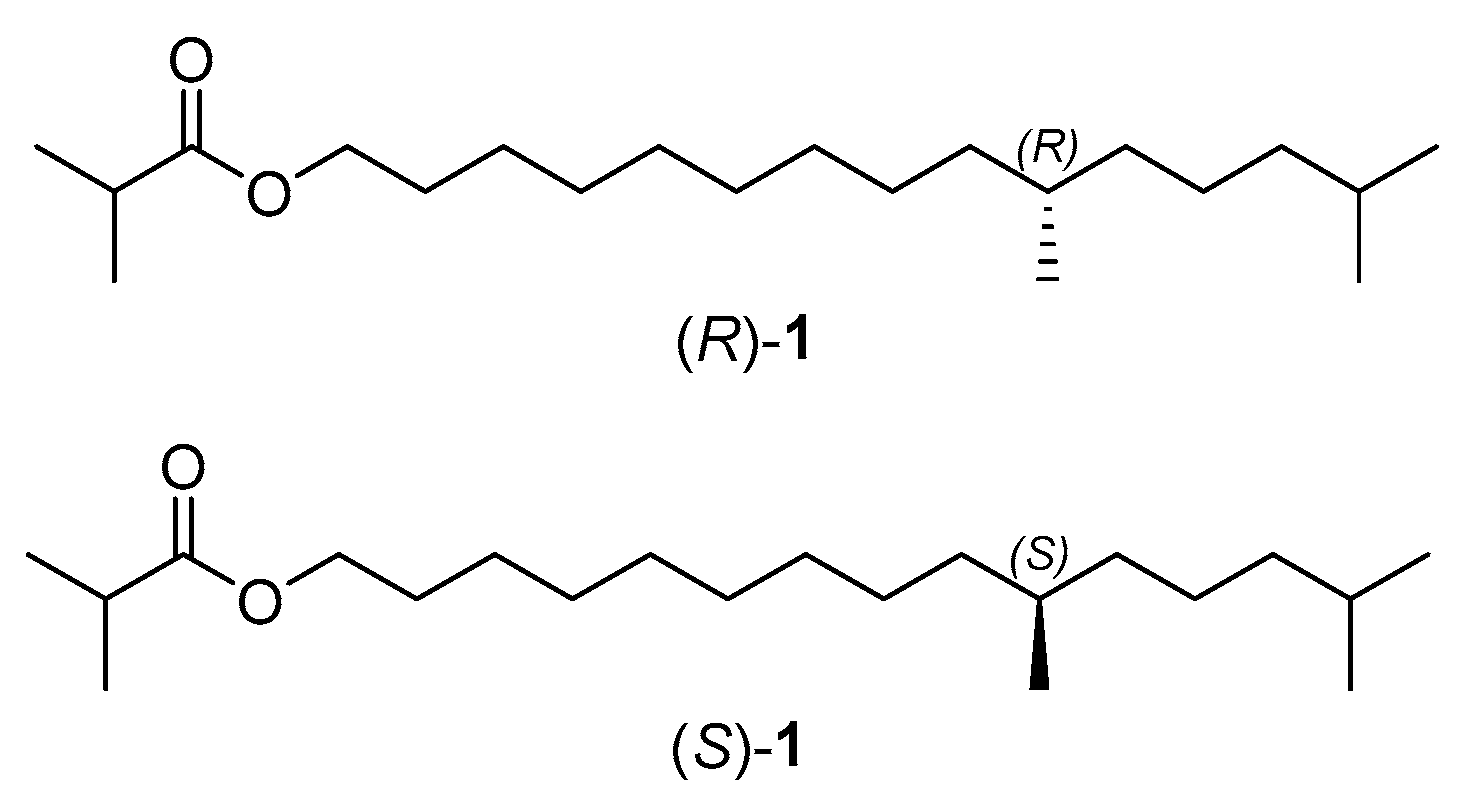
1. Introduction
2. Results and Discussion
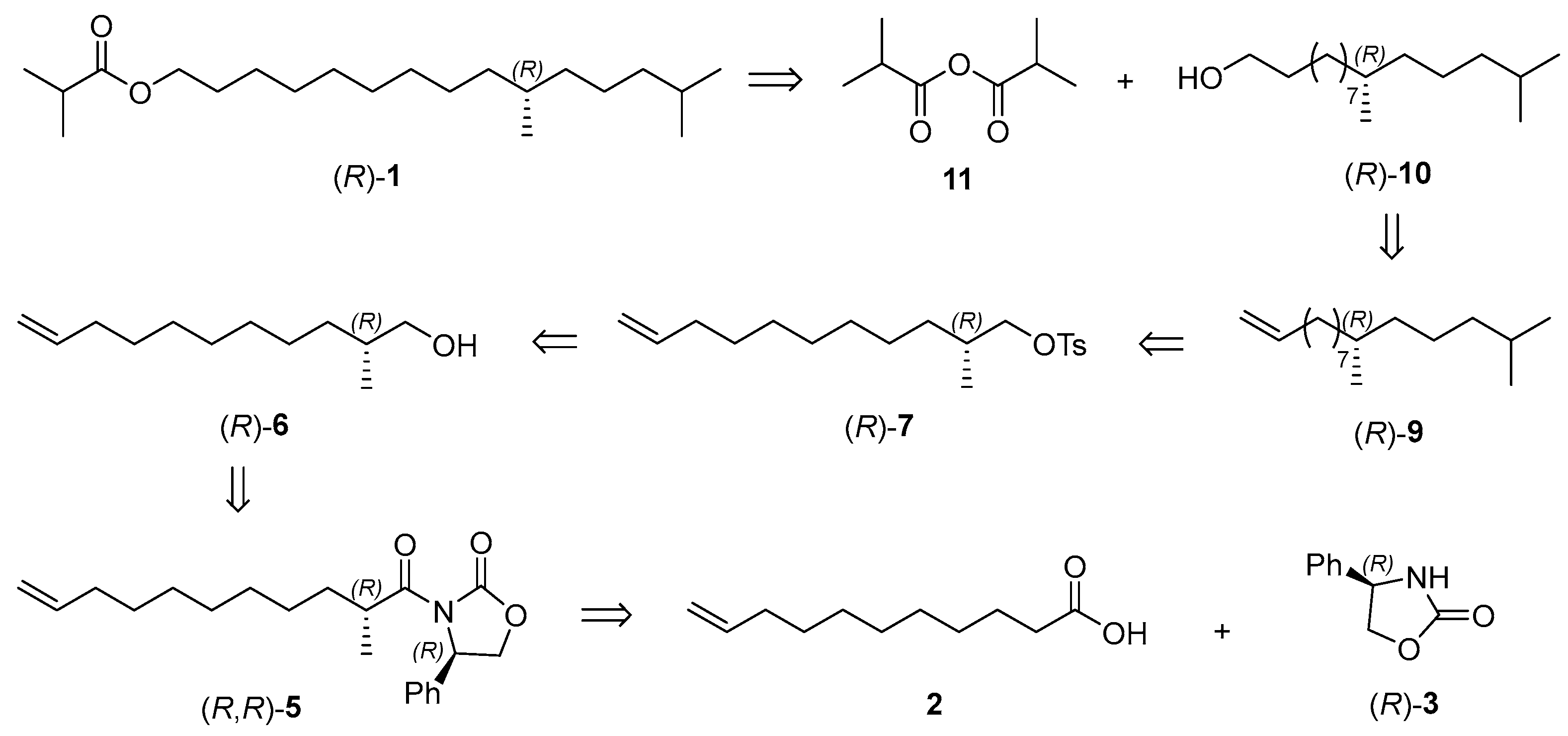
2.1. Retrosynthetic Analysis
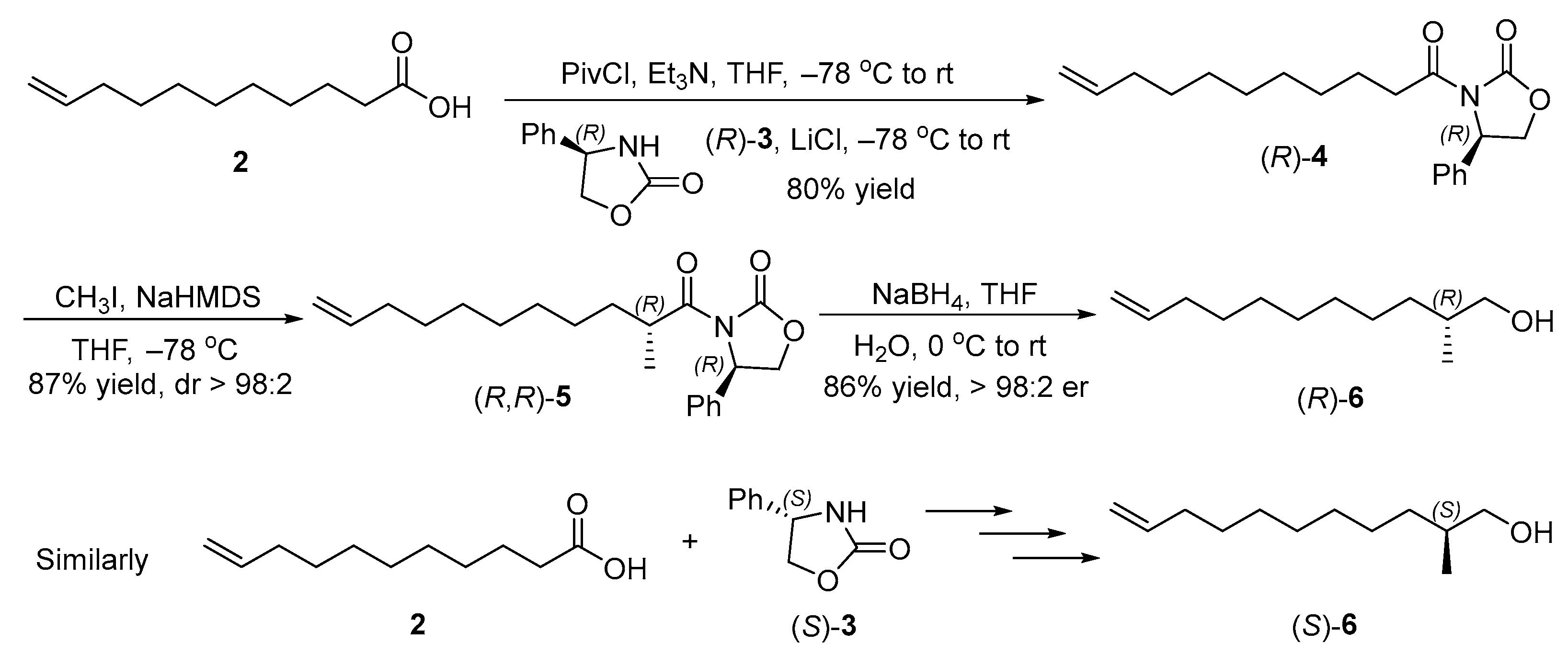
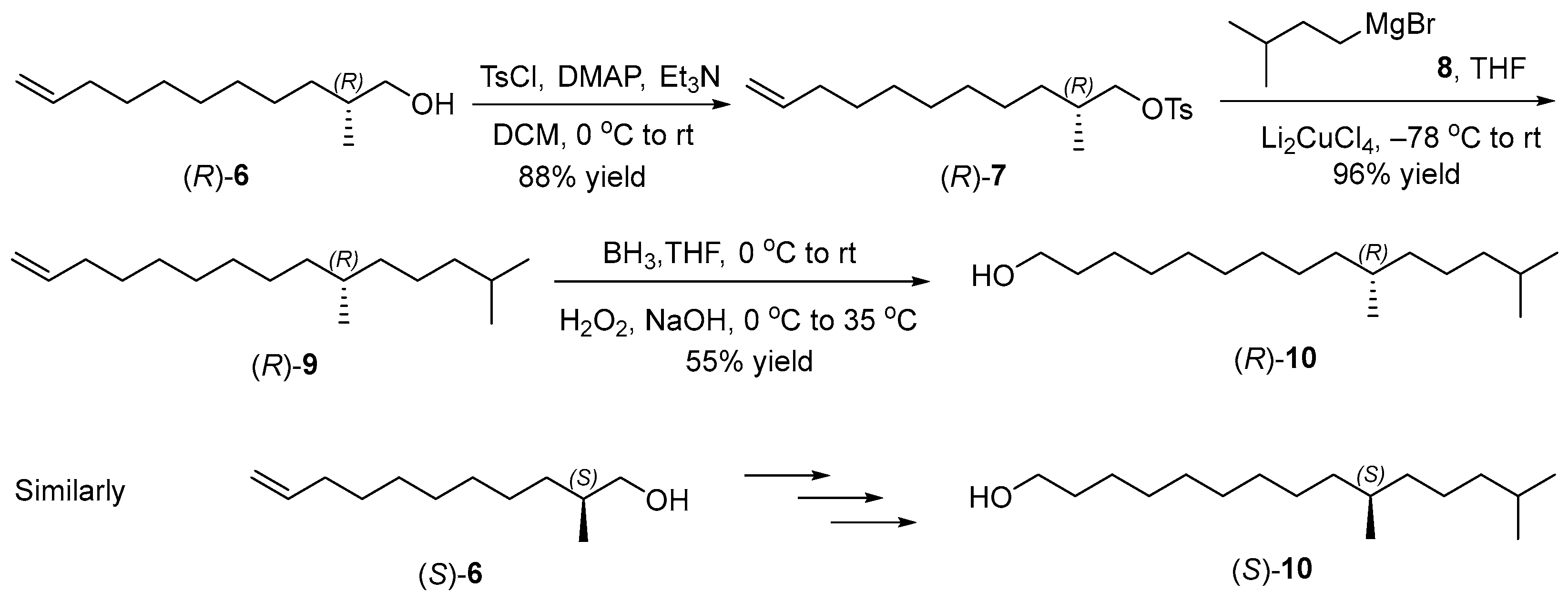
2.2. Synthesis of Chiral Alcohols (R)- and (S)-6
2.3. Synthesis of Chiral Alcohols (R)- and (S)-10
2.4. Synthesis of Sex Pheromone of E. pseudoconspersa and Its Enantiomer
3. Materials and Methods
3.1. General Information
3.2. Synthesis of (R)-4-Phenyl-3-(undec-10-enoyl)oxazolidin-2-one ((R)-4) (New Compound)
- To a 500 mL three-neck flask were added undec-10-enoic acid (2) (5.00 g, 27.13 mmol) Et3N (5.77 g, 56.97 mmol) and THF (150 mL) at room temperature. The solution was cooled to −78 °C, then pivaloyl chloride (3.93 g, 32.55 mmol) was added and stirred for 0.5 h. The reaction solution was allowed to warm to room temperature and stirred for 1 h. (R)-4-Phenyl-2-oxazolidinone ((R)-3) (4.87 g, 29.84 mmol) in THF (40 mL) and LiCl (3.45 g, 81.39 mmol) were then added. The reaction mixture was cooled to −78 °C again and stirred for 1 h. After the reaction mixture was allowed to warm to room temperature and stirred for additional 10 h, it was quenched with saturated NH4Cl aqueous solution (60 mL). Two phases were separated, and the aqueous phase was extracted with EtOAc (60 × 3 mL). The combined organic phases were washed with brine (120 mL), and dried over anhydrous Na2SO4, filtered, and concentrated by a rotary evaporator. The residue was purified by column chromatography on silica gel with an eluent of petroleum ether/EtOAc 10:1 to afford (R)-4-phenyl-3-(pentadec-14-enoyl)oxazolidin-2-one ((R)-4) (7.15 g, 80% yield) as a white solid. [α = −39.042 (c = 3.76, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.39–7.28 (m, 5H), 5.80 (ddt, J = 16.9, 10.3, 6.6 Hz, 1H), 5.42 (dd, J = 8.8, 3.7 Hz, 1H), 4.98 (dd, J = 17.1, 2.0 Hz, 1H), 4.93–4.91 (m, 1H), 4.68 (t, J = 8.8 Hz, 1H), 4.27 (dd, J = 8.9, 3.7 Hz, 1H), 2.97–2.86 (m, 2H), 2.04–2.00 (m, 2H), 1.61–1.58 (m, 2H), 1.36–1.23 (m, 10H). 13C NMR (126 MHz, CDCl3) δ 173.00, 153.87, 139.34, 139.31, 129.29, 128.80, 126.03, 114.24, 70.06, 57.68, 35.66, 33.89, 29.38, 29.15, 29.10, 29.00, 24.25. HRMS (ESI): calculated for C20H28O3N [M + H]+: 330.20637, found: 330.20667.
3.3. Synthesis of (R)-3-((R)-2-Methylundec-10-enoyl)-4-phenyloxazolidin-2-one ((R,R))-5) (New Compound)
- To a 250 mL three-neck flask were added (R)-4-phenyl-3-(pentadec-14-enoyl)oxazolidin-2-one ((R)-4) (2.96 g, 8.98 mmol) and THF (10 mL) at room temperature. The solution was cooled to −78 °C, then NaHMDS (6.75 mL, 2.0 M in THF, 13.50 mmol) was added dropwise and stirred for 1 h. Iodomethane (6.37 g, 44.88 mmol) in THF (50 mL) was added dropwise over 2 h. After the reaction mixture was stirred for 4 h at −78 °C, it was quenched with saturated NH4Cl aqueous solution (40 mL). The two phases were separated, and the aqueous phase was extracted with EtOAc (50 × 3 mL). The combined organic phases were washed with brine (200 mL) and dried over anhydrous Na2SO4, filtered, and concentrated by a rotary evaporator. The residue was purified by column chromatography on silica gel with an eluent of petroleum ether/EtOAc 10:1 to afford (R)-3-((R)-2-methylundec-10-enoyl)-4-phenyloxazolidin-2-one ((R,R)-5) (2.69 g, 87% yield, dr > 98:2, determined by its 13C NMR spectra) as a white solid. m.p. 38–40 °C. [α = −88.197 (c = 3.11, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.38–7.27 (m, 5H), 5.79 (dt, J = 16.7, 8.5 Hz, 1H), 5.43 (dd, J = 8.7, 3.7 Hz, 1H), 4.99 (d, J = 17.1 Hz, 1H), 4.92 (d, J = 10.2 Hz, 1H), 4.67 (t, J = 8.8 Hz, 1H), 4.23 (dd, J = 8.8, 3.7 Hz, 1H), 3.73 (h, J = 6.8 Hz, 1H), 2.05–2.00 (m, 2H), 1.72–1.66 (m, 1H), 1.38–1.27 (m,11H), 1.10 (d, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 176.72, 153.46, 139.43, 139.27, 129.26, 128.70, 125.79, 114.22, 69.82, 57.81, 37.88, 33.86, 33.12, 29.67, 29.41, 29.13, 28.98, 27.35, 17.42. HRMS (ESI): calculated for C21H30O3N [M + H]+: 344.22202, found: 344.22284.
3.4. Synthesis of (R)-2-Methylundec-10-en-1-ol ((R)-6) (CAS 1156505-01-9)
- To a 200 mL Schlenk flask were added (R)-3-((R)-2-methylundec-10-enoyl)-4-phenyloxazolidin-2-one ((R,R)-5) (1.17 g, 3.40 mmol) and THF (25 mL) at room temperature. The solution was cooled to 0 °C, then NaBH4 (0.52 g, 13.74 mmol) in water (2.5 mL) was added dropwise carefully. The reaction mixture was allowed to warm to room temperature, and stirred for 8 h. After being cooled to 0 °C, HCl aqueous solution (1 M) was added dropwise carefully to destroy the excess NaBH4. The two phases were separated, and the aqueous phase was extracted with EtOAc (20 × 3 mL). The combined organic phases were washed with brine (80 mL), and dried over anhydrous Na2SO4, filtered, and concentrated by a rotary evaporator. The residue was purified by column chromatography on silica gel with an eluent of petroleum ether/EtOAc 5:1 to afford (R)-2-methylpentadec-14-en-1-ol ((R)-6) (0.54 g, 86% yield, > 98:2 er, based on the signal/noise ratio of the 19F NMR spectra of its Mosher ester, see the details in Supplementary Materials) as a colorless oil. [α = +8.162 (c = 1.81, CHCl3). Lit. [27] [α = + 8.40 (c = 4.60, CHCl3). 1H NMR (500 MHz, CDCl3) δ 5.81 (ddt, J = 16.9, 10.2, 6.7 Hz, 1H), 4.99 (dq, J = 17.1, 1.8 Hz, 1H), 4.96–4.92 (m, 1H), 3.50 (dd, J = 10.5, 5.8 Hz, 1H), 3.41 (dd, J = 10.5, 6.6 Hz, 1H), 2.06–2.02 (m, 2H), 1.61–1.57 (m, 1H), 1.43–1.25 (m, 12H), 1.11 (dd, J = 12.1, 9.0 Hz, 1H), 0.91 (d, J = 6.8 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 139.35, 114.25, 68.53, 35.89, 33.93, 33.27, 30.01, 29.60, 29.25, 29.06, 27.09, 16.72. HRMS (ESI) m/z calcd. for C12H23O [M − H]+: 183.17434, found183.17367.
3.5. Synthesis of (S)-4-Phenyl-3-(undec-10-enoyl)oxazolidin-2-one((S)-4) (New Compound)
- According to the similar procedure for oxazolidinone imide (R)-4, the acylation of undec-10-enoic acid (2) (6.00 g, 32.56 mmol) with (S)-4-phenyl-2-oxazolidinone ((S)-3) (4.71 g, 28.86 mmol) afforded (S)-4-phenyl-3-(undec-10-enoyl)oxazolidin-2-one ((S)-4) (9.00 g, 84% yield) as a white solid. [α = +44.468 (c = 3.76, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.39–7.28 (m, 5H), 5.80 (ddt, J = 17.0, 10.1, 6.6 Hz, 1H), 5.42 (dd, J = 8.7, 3.7 Hz, 1H), 4.98 (dd, J = 17.1, 2.0 Hz, 1H), 4.92 (dd, J = 10.2, 2.2 Hz, 1H), 4.68 (t, J = 8.8 Hz, 1H), 4.27 (dd, J = 8.9, 3.7 Hz, 1H), 2.97–2.86 (m, 2H), 2.04–2.00 (m, 2H), 1.62–1.56 (m, 2H), 1.36–1.24 (m, 10H). 13C NMR (126 MHz, CDCl3) δ 172.97, 153.85, 139.33, 139.31, 129.28, 128.79, 126.02, 114.24, 70.05, 57.67, 35.65, 33.89, 29.37, 29.14, 29.09, 28.99, 24.23. HRMS (ESI): calculated for C20H27O3NNa [M + Na]+: 352.18831, found: 352.18842.
3.6. Synthesis of (S)-3-((S)-2-Methylundec-10-enoyl)-4-phenyloxazolidin-2-one ((S,S)-5) (New Compound)
- According to the similar procedure for oxazolidinone imide (R,R)-5, the alkylation of (S)-4-phenyl-3-(undec-10-enoyl)oxazolidin-2-one ((S)-4) (4.00 g, 12.14 mmol) with iodomethane (8.62 g, 60.73 mmol) afforded (S)-3-((S)-2-methylundec-10-enoyl)-4-phenyloxazolidin-2-one ((S,S)-5) (3.13 g, 75% yield, dr > 98:2, determined by its 13C NMR spectra) as a white solid. m.p. 37–39 °C. [α = +76.918 (c = 4.07, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.39–7.27 (m, 5H), 5.80 (ddt, J = 16.9, 10.1, 6.6 Hz, 1H), 5.43 (dd, J = 8.8, 3.7 Hz, 1H), 4.99 (dd, J = 8.8, 3.7 Hz, 1H), 4.92 (dd, J = 8.8, 3.7 Hz, 1H), 4.67 (t, J = 8.8 Hz, 1H), 4.24 (ddd, J = 8.9, 3.8, 1.0 Hz, 1H), 3.73 (h, J = 6.8 Hz, 1H), 2.06–2.00 (m, 2H), 1.73–1.66 (m, 1H), 1.38–1.27 (m, 11H), 1.10 (d, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 176.77, 153.49, 139.45, 139.31, 129.30, 128.74, 125.82, 114.24, 69.85, 57.85, 37.92, 33.89, 33.16, 29.70, 29.44, 29.17, 29.01, 27.38, 17.44. HRMS (ESI): calculated for C21H29O3NNa [M + Na]+: 366.20396, found: 366.20413.
3.7. Synthesis of (S)-2-Methylundec-10-en-1-ol ((S)-6) (CAS 2091161-95-2)
- According to the similar procedure for alcohol (R)-6, the reductive cleavage of (S)-3-((S)-2-methylundec-10-enoyl)-4-phenyloxazolidin-2-one ((S,S)-5) (2.01 g, 5.85 mmol) with NaBH4 (0.89 g, 23.40 mmol) afforded (S)-2-methylundec-10-en-1-ol ((S)-6) (0.88 g, 82% yield, >98:2 er, based on the signal/noise ratio of the 19F NMR spectra of its Mosher ester, see the details in Supplementary Materials) as a colorless oil. [α = −7.156 (c = 2.18, CHCl3). Lit. [32] [α = −5.1 (c = 0.85, CHCl3, 82% ee). 1H NMR (500 MHz, CDCl3) δ 5.81 (ddt, J = 16.9, 10.2, 6.7 Hz, 1H), 4.99 (dq, J = 17.2, 1.8 Hz, 1H), 4.93 (ddt, J = 10.2, 2.4, 1.2 Hz, 1H), 3.50 (dd, J = 10.5, 6.2 Hz, 1H), 3.41 (dd, J = 10.5, 6.2 Hz, 1H), 2.06–2.02 (m, 2H), 1.63–1.59 (m, 1H), 1.39–1.24 (m, 12H), 1.13–1.09 (m, 1H), 0.91 (d, J = 6.7 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 139.37, 114.25, 68.55, 35.90, 33.94, 33.27, 30.01, 29.60, 29.26, 29.07, 27.10, 16.72. HRMS (ESI): calculated for C12H23 [M − OH]+: 167.17943, found: 167.18015.
3.8. Synthesis of (R)-2-Methylundec-10-en-1-yl 4-methylbenzenesulfonate ((R)-7) (New Compound)
- To a 200 mL Schlenk flask were added (R)-2-methylpentadec-14-en-1-ol ((R)-6) (2.00 g, 11.40 mmol) and CH2Cl2 (20 mL) at room temperature. The solution was cooled to 0 °C, then Et3N (3.07 g, 30.34 mmol) and DMAP (1.32 g, 10.80 mmol) in CH2Cl2 (20 mL) were added. Subsequently, TsCl (3.93 g, 20.61 mmol) in CH2Cl2 (30 mL) was added dropwise over 30 min. After the reaction mixture was allowed to warm to room temperature and stirred for 8 h, it was quenched with saturated NH4Cl aqueous solution (50 mL). The two phases were separated, and the aqueous phase was extracted with CH2Cl2 (40 × 3 mL). The combined organic phases were washed with brine (150 mL) and dried over anhydrous Na2SO4, filtered, and concentrated by a rotary evaporator. The residue was purified by column chromatography on silica gel with an eluent of petroleum ether/EtOAc 10:1 to afford (R)-2-methylpentadec-14-en-1-yl 4-methylbenzenesulfonate ((R)-7) (3.23 g, 88% yield) as a pale yellow oil. [α = −3.178 (c = 3.15, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.80–7.77 (m, 2H), 7.34 (d, J = 8.1 Hz, 2H), 5.81 (ddt, J = 16.9, 10.2, 6.6 Hz, 1H), 4.99 (dq, J = 17.1, 1.7 Hz, 1H), 4.93 (ddt, J = 10.2, 2.4, 1.3 Hz, 1H), 3.88 (dd, J = 9.3, 5.7 Hz, 1H), 3.80 (dd, J = 9.3, 6.5 Hz, 1H), 2.45 (s, 3H), 2.05–2.00 (m, 2H), 1.79–1.73 (m, 1H), 1.37–1.07 (m, 12H), 0.87 (d, J = 6.8 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 144.71, 139.27, 133.36, 129.90, 128.02, 114.29, 75.26, 33.89, 32.93, 32.78, 29.74, 29.47, 29.18, 29.02, 26.67, 21.75, 16.57. HRMS (ESI): calculated for C19H30O3NaS [M + Na]+: 361.18079, found: 361.18073.
3.9. Synthesis of (R)-10,14-Dimethylpentadec-1-ene ((R)-9) (New Compound)
- To a 250 mL three-neck flask was added (R)-2-methylpentadec-14-en-1-yl 4-methylbenzenesulfonate ((R)-7) (2.95 g, 8.71 mmol) in anhydrous THF (10 mL) at room temperature. After being to 0 °C, isopentyl magnesium bromide (8) (26.80 mL, 1.3 M in THF, 34.84 mmol) was then added dropwise. Subsequently, a solution of Li2CuCl4 (26.10 mL, 0.1 M in THF, 2.61 mmol) was added dropwise at 0 °C. The reaction mixture was cooled to −78 °C and stirred for 1 h, then warmed to −10 °C and stirred for 2 h. After the reaction mixture was allowed to warm to 25 °C and stirred for 8 h, it was quenched with a saturated NH4Cl aqueous solution (80 mL) at 0 °C. The two phases were separated, and the aqueous phase was extracted with EtOAc (100 × 3 mL). The combined organic phases were washed with brine (300 mL) and dried over anhydrous Na2SO4, filtered, and concentrated by a rotary evaporator. The residue was purified by column chromatography on silica gel with an eluent of n-hexane to afford (R)-10,14-dimethylpentadec-1-ene ((R)-9) (1.99 g, 96% yield) as a colorless oil. [α = +0.932 (c = 2.15, CHCl3). 1H NMR (500 MHz, CDCl3) δ 5.82 (ddt, J = 16.9, 10.2, 6.7 Hz, 1H), 4.99 (dq, J = 17.1, 1.8 Hz, 1H), 4.93 (ddt, J = 10.1, 2.3, 1.3 Hz, 1H), 2.06–2.02 (m, 2H), 1.56–1.48 (m, 1H), 1.39–1.22 (m, 19H), 0.86 (d, J = 6.6 Hz, 6H), 0.83 (d, J = 6.6 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 139.44, 114.22, 39.53, 37.48, 37.25, 33.98, 32.92, 30.11, 29.70, 29.32, 29.12, 28.14, 27.22, 27.20, 24.95, 22.87, 22.78, 19.87. HRMS (ESI): calculated for C17H36O [M + H2O]+: 256.27607, found: 256.27584.
3.10. Synthesis of (R)-10,14-Dimethylpentadecan-1-ol ((R)-10) (CAS 164260-11-1)
- To a 50 mL Schlenk tube were added (R)-10,14-dimethylpentadec-1-ene ((R)-9) (1.30 g, 5.45 mmol) and anhydrous THF (10 mL) at room temperature. The resulting solution was cooled to 0 °C, then borane (1.82 mL, 1.0 M in THF, 1.82 mmol) was added dropwise over 20 min. The reaction mixture was warmed to room temperature and stirred for 1 h, and the solution of tris((S)-10,14-dimethylpentadecyl)borane was obtained. Then the solution was cooled to 0 °C, and sodium hydroxide (0.61 mL, 3 N, 1.82 mmol) was added. The resulting solution was cooled to −20 °C, and hydrogen peroxide (0.65 mL, 30%, 6.32 mmol) was added dropwise. After the reaction was warmed to 35 °C and stirred for 1 h, it was quenched with water (10 mL). The two phases were separated, and the aqueous phase was extracted with EtOAc (15 × 3 mL). The combined organic phases were washed with brine (20 mL) and dried over anhydrous Na2SO4, filtered, and concentrated by a rotary evaporator. The residue was purified by column chromatography on silica gel with an eluent of petroleum ether/EtOAc 5:1 to afford (R)-10,14-dimethylpentadecan-1-ol ((R)-10) (0.77 g, 55% yield) as a pale yellow oil. [α = + 0.556 (c = 1.44, CHCl3). 1H NMR (500 MHz, CDCl3) δ 3.64 (t, J = 6.6 Hz, 2H), 1.59–1.49 (m, 3H), 1.35–1.32 (m, 1H), 1.29–1.20 (m, 17H), 1.15–1.11 (m, 2H), 1.09–1.03 (m, 2H), 0.87 (d, J = 6.7 Hz, 6H), 0.84 (d, J = 6.6 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 63.25, 39.52, 37.47, 37.25, 32.97, 32.91, 30.15, 29.79, 29.78, 29.59, 28.13, 27.23, 25.89, 24.95, 22.87, 22.77, 19.86. HRMS (ESI): calculated for C17H37O [M + H]+: 257.28389, found: 257.28122.
3.11. Synthesis of (S)-2-Methylundec-10-en-1-yl 4-methylbenzenesulfonate ((S)-7) (New Compound)
- According to the similar procedure for tosylate (R)-7, the tosylation of (S)-2-methylpentadec-14-en-1-ol ((S)-6) (1.50 g, 8.14 mmol) with TsCl (2.81 g, 14.72 mmol) afforded (S)-2-methylundec-10-en-1-yl 4-methylbenzenesulfonate ((S)-7) (2.42 g, 88% yield) as a pale yellow oil. [α = +3.734 (c = 1.61, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.79 (d, J = 8.3 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 5.81 (ddt, J = 16.9, 10.2, 6.7 Hz, 1H), 4.99 (dq, J = 17.1, 1.7 Hz, 1H), 4.93 (ddt, J = 10.2, 2.3, 1.3 Hz, 1H), 3.88 (dd, J = 9.3, 5.7 Hz, 1H), 3.80 (dd, J = 9.4, 6.5 Hz, 1H), 2.45 (s, 3H), 2.05–2.00 (m, 2H), 1.78–1.74 (m, 1H), 1.37–1.10 (m, 12H), 0.87 (d, J = 6.8 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 144.71, 139.28, 133.36, 129.90, 128.02, 114.29, 75.26, 33.90, 32.94, 32.78, 29.74, 29.48, 29.18, 29.02, 26.67, 21.76, 16.57. HRMS (ESI): calculated for C19H32O4S [M + H2O]+: 356.20158, found: 356.20156.
3.12. Synthesis of (S)-10,14-Dimethylpentadec-1-ene ((S)-9) (New Compound)
- According to the similar procedure for alkene (R)-9, the coupling of (S)-2-methylpentadec-14-en-1-yl 4-methylbenzenesulfonate ((S)-7) (2.02 g, 5.97 mmol) with isopentyl magnesium bromide (8) (18.38 mL, 1.3 M in THF, 23.88 mmol) catalyzed by Li2CuCl4 (18.0 mL, 0.1 M in THF, 1.79 mmol) afforded (S)-10,14-dimethylpentadec-1-ene ((S)-9) (1.28 g, 90% yield) as a colorless oil. [α = −0.631 (c = 1.27, CHCl3). 1H NMR (500 MHz, CDCl3) δ 5.81 (ddt, J = 16.9, 10.2, 6.7 Hz, 1H), 4.99 (dq, J = 17.1, 1.7 Hz, 1H), 4.94–4.91 (m, 1H), 2.06–2.02 (m, 2H), 1.55–1.50 (m, 1H), 1.39–1.15 (m, 19H), 0.87 (d, J = 6.9 Hz, 6H), 0.83 (d, J = 6.6 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 139.41, 114.24, 39.55, 37.50, 37.27, 34.00, 32.94, 30.13, 29.72, 29.34, 29.14, 28.15, 27.24, 24.98, 22.88, 22.79, 19.88. HRMS (ESI): calculated for C17H36O [M + H2O]+: 256.27607, found: 256.27575.
3.13. Synthesis of (S)-10,14-Dimethylpentadecan-1-ol ((S)-10) (CAS 164260-12-2)
- According to the similar procedure for alcohol (R)-10, the hydroboration-oxidation of (S)-10,14-dimethylpentadec-1-ene ((S)-9) (1.26 g, 5.28 mmol) with borane (1.76 mL, 1.0 M in THF, 1.76 mmol) and hydrogen peroxide (0.63 mL, 30%, 6.12 mmol) afforded (S)-10,14-dimethylpentadecan-1-ol ((S)-10) (0.73 g, 54% yield) as a colorless oil. [α = −0.214 (c = 3.74, CHCl3). 1H NMR (500 MHz, CDCl3) δ 3.63 (t, J = 6.7 Hz, 2H), 1.59–1.54 (m, 2H), 1.52–1.48 (m, 1H), 1.45 (br s, 1H), 1.36–1.20 (m, 17H), 1.15–1.11 (m, 2H), 1.09–1.03 (m, 2H), 0.87 (d, J = 6.6 Hz, 6H), 0.84 (d, J = 6.6 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 63.20, 39.51, 37.46, 37.25, 32.96, 32.91, 30.15, 29.79, 29.77, 29.59, 28.12, 27.22, 25.89, 24.94, 22.85, 22.76, 19.85. HRMS (ESI): calculated for C17H35 [M − OH]+: 239.27333, found: 239.27253.
3.14. Synthesis of (R)-10,14-Dimethylpentadecyl Isobutyrate ((R)-1) (164260-03-1)
- To a 20 mL Schlenk tube were added (R)-10,14-dimethylpentadecan-1-ol ((R)-10) (0.40 g, 1.56 mmol) and DMAP (0.19 g, 1.56 mmol) in CH2Cl2 (5 mL) at room temperature. Isobutyric anhydride (11) (0.37 g, 2.34 mmol) was then added dropwise. After the reaction solution was stirred for 9 h at the same temperature, it was diluted with CH2Cl2 (5 mL). The organic solution was washed with HCl aqueous solution (1 N, 20 mL), NaOH aqueous solution (1 N, 20 mL), NaHCO3 solution (20 mL) and brine (20 mL) sequentially. Finaly, the organic solution was dried over anhydrous Na2SO4 and concentrated by a rotary evaporator to afford (R)-10,14-dimethylpentadecyl isobutyrate ((R)-1) (0.47 g, 93% yield) as a pale yellow oil. [α = +1.148 (c = 2.79, CHCl3). Lit. [19] [α = + 0.581 (c = 2.3, CHCl3). 1H NMR (500 MHz, CDCl3) δ 4.06 (t, J = 6.7 Hz, 2H), 2.53 (h, J = 7.0 Hz, 1H), 1.65–1.59 (m, 2H), 1.56–1.48 (m, 1H), 1.36–1.22 (m, 18H), 1.16 (d, J = 7.0 Hz, 6H), 1.14–1.11 (m, 1H), 1.10–1.03 (m, 2H), 0.87 (d, J = 6.6 Hz, 6H), 0.84 (d, J = 6.6 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 177.42, 64.56, 39.52, 37.47, 37.25, 34.20, 32.91, 30.13, 29.75, 29.68, 29.40, 28.80, 28.13, 27.22, 26.05, 24.95, 22.86, 22.77, 19.86, 19.17. HRMS (ESI): calculated for C21H42O2KNa [M + K + Na]+: 388.27141, found: 388.27005.
3.15. Synthesis of (S)-10,14-Dimethylpentadecyl Isobutyrate ((S)-1) (CAS164260-04-2)
- According to the similar procedure for the sex pheromone (R)-1, the esterification of (S)-10,14-dimethylpentadecan-1-ol ((S)-10) (0.30 g, 1.17 mmol) with isobutyric anhydride (11) (0.28 g, 1.77 mmol) afforded (S)-10,14-dimethylpentadecyl isobutyrate ((S)-1) (0.34 g, 90% yield) as a pale yellow oil. [α = −0.563 (c = 1.42, CHCl3). Lit. [18] [α = −0.28 (c = 2.40, CHCl3). 1H NMR (500 MHz, CDCl3) δ 4.06 (t, J = 6.7 Hz, 2H), 2.53 (h, J = 7.0 Hz, 1H), 1.65–1.59 (m, 2H), 1.56–1.48 (m, 1H), 1.37–1.21 (m, 18H), 1.16 (d, J = 7.0 Hz, 6H), 1.14–1.11 (m, 1H), 1.09–1.05 (m, 2H), 0.86 (d, J = 6.6 Hz, 6H), 0.84 (d, J = 6.6 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 177.38, 64.54, 39.51, 37.46, 37.24, 34.19, 32.91, 30.12, 29.75, 29.67, 29.40, 28.80, 28.12, 27.21, 26.05, 24.94, 22.85, 22.76, 19.85, 19.15. HRMS (ESI): calculated for C21H41O2 [M − H]+: 325.31011, found: 325.30765.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hu, J.; Song, H.; Cao, Z.; Chen, Y.; Wang, J.; Zhu, W.; Yao, D.; Han, B. Species and control techniques of main diseases and insect pests of Camellia oleifera in Anhui province. Plant Dis. Pests 2023, 14, 1–4. [Google Scholar]
- Wakamura, S.; Yasuda, T.; Mochizuki, F. Mating behavior of the tea tussock moth, Euproctis pseudoconspersa (Strand) (Lepidoptera: Lymantriidae). Appl. Entomol. Zoolog. 1996, 31, 619–621. [Google Scholar] [CrossRef]
- Zhang, X.Z.; Li, S.S.; Luo, Z.X.; Cai, X.M.; Bian, L.; Xiu, C.L.; Fu, N.X.; Liu, N.Y.; Zhang, Z.Q.; Li, Z.Q. Transcriptome profiling of Euproctis pseudoconspersa reveals candidate olfactory genes for type III sex pheromone detection. Int. J. Mol. Sci. 2025, 26, 1405. [Google Scholar] [CrossRef]
- Tsai, R.-S.; Yang, E.-C.; Wu, C.-Y.; Tseng, H.-K.; Chow, Y.-S. A potent sex attractant of the male tea tussock moth, Euproctis pseudoconspersa (Strand) (Lepidoptera: Lymantriidae) in Taiwan: Field and eag responses. Zool. Stud. 1999, 38, 301–306. [Google Scholar]
- Li, Z.-q.; Yuan, T.-t.; Cui, S.-w.; Zhao, Y.-j.; Shao, Y.-h.; Shang, J.-n.; Luo, Z.-x.; Cai, X.-m.; Bian, L.; Chen, Z.-m. Development of a high-efficiency sex pheromone formula to control Euproctis pseudoconspersa. J. Integr. Agric. 2023, 22, 195–201. [Google Scholar] [CrossRef]
- Wang, X.Q.; Gu, Q.Y.; Zhang, W.; Jiang, H.Y.; Chen, S.C.; Smagghe, G.; Niu, J.Z.; Wang, J.J. Prevalence of a novel bunyavirus in tea tussock moth Euproctis pseudoconspersa (Lepidoptera: Lymantriidae). J. Insect Sci. 2021, 21, 5. [Google Scholar] [CrossRef]
- Zhao, C.H.; Millar, G.J.; Wen, Z.Z.; Wang, S.K.; Wang, X.Y.; Zhu, Y.F. Isolation, identification and synthesis of the female sex pheromone of the tea tussock moth, Euproctis pseudoconspersa (Strand) (Lepidoptera: Lymantriidae). Entomol. Sin. 1996, 3, 58–69. [Google Scholar]
- Huang, D.; Sun, M.; Han, M.; Zhang, Z.; Miao, Y.; Zhang, J.; Yao, Y. Volatile organic compounds (VOCs) regulate the spatial distribution of Lepidoptera insects in an orchard ecosystem. Biol. Control 2020, 149, 104311. [Google Scholar] [CrossRef]
- Roy, S.; Muraleedharan, N. Microbial management of arthropod pests of tea: Current state and prospects. Appl. Microbiol. Biot. 2014, 98, 5375–5386. [Google Scholar] [CrossRef]
- Rizvi, S.A.H.; George, J.; Reddy, G.V.P.; Zeng, X.; Guerrero, A. Latest developments in insect sex pheromone research and its application in agricultural pest management. Insects 2021, 12, 484. [Google Scholar] [CrossRef]
- Souza, J.P.A.; Bandeira, P.T.; Bergmann, J.; Zarbin, P.H.G. Recent advances in the synthesis of insect pheromones: An overview from 2013 to 2022. Nat. Prod. Rep. 2023, 40, 866–889. [Google Scholar] [CrossRef] [PubMed]
- Wakamura, S.; Yasuda, T.; Ichikawa, A.; Fukumoto, T.; Mochizuki, F. Sex attractant pheromone of the tea tussock moth, Euproctis pseudoconspersa (Strand) (Lepidoptera, Lymantriidae)—Identification and field attraction. Appl. Entomol. Zool. 1994, 29, 403–411. [Google Scholar] [CrossRef]
- Ichikawa, A.; Yasuda, T.; Wakamura, S. Absolute configuration of sex pheromone for tea tussock moth, Euproctis pseudoconspersa (Strand) via synthesis of (R)- and (S)-10,14-dimethyl-1-pentadecyl isobutyrates. J. Chem. Ecol. 1995, 21, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.-H.; Millar, J.G.; Pan, K.-H.; Xu, C.-S. Responses of tea tussock moth, Euproctis pseudoconspersa, to its pheromone, (R)-10,14-dimethylpentadecyl isobutyrate, and to the S-enantiomer of its pheromone. J. Chem. Ecol. 1998, 24, 1347–1353. [Google Scholar] [CrossRef]
- Wakamura, S.; Ichikawa, A.; Yasuda, T.; Arakaki, N.; Fukumoto, T. EAG and field responses of the male tea tussock moth, Euproctis pseudoconspersa (Strand) (Lepidoptera: Lymantriidae) to (R)- and (S)-enantiomers and racemic mixture of 10,14-dimethylpentadecyl isobutyrate. Appl. Entomol. Zool. 1996, 31, 623–625. [Google Scholar] [CrossRef]
- Wang, Y.M.; Ge, F.; Liu, X.H.; Feng, F.; Wang, L.J. Evaluation of mass-trapping for control of tea tussock moth Euproctis pseudoconspersa (Strand) (Lepidoptera: Lymantriidae) with synthetic sex pheromone in south China. Int. J. Pest Manag. 2005, 51, 289–295. [Google Scholar]
- Sun, Z.-F.; Zhou, L.-N.; Meng, Y.; Zhang, T.; Du, Z.-T.; Zheng, H. Concise asymmetric synthesis of the sex pheromone of the tea tussock moth. Tetrahedron Asymmetry 2017, 28, 1562–1567. [Google Scholar] [CrossRef]
- Zhang, H.-L.; Sun, Z.-F.; Zhou, L.-N.; Liu, L.; Zhang, T.; Du, Z.-T. Synthesis of the sex pheromone of the tea tussock moth based on a resource chemistry strategy. Molecules 2018, 23, 1347. [Google Scholar] [CrossRef]
- Sun, Z.-F.; Liu, H.; Li, Y.-F.; Duan, Y.-P.; Jin, L.-X.; Ji, X.-H.; Dai, H.-P.; Lu, J.-F. The asymmetric total synthesis of the female-produced sex pheromone of the tea tussock moth. Molecules 2024, 29, 3866. [Google Scholar] [CrossRef]
- Diaz-Munoz, G.; Miranda, I.L.; Sartori, S.K.; de Rezende, D.C.; Alves Nogueira Diaz, M. Use of chiral auxiliaries in the asymmetric synthesis of biologically active compounds: A review. Chirality 2019, 31, 776–812. [Google Scholar] [CrossRef]
- Lin, C.; Zhu, W.; Wu, S.; Bian, Q.; Zhong, J. Asymmetric synthesis and biological activity of contact pheromone of western flower thrips, Frankliniella occidentalis. Int. J. Mol. Sci. 2024, 25, 11699. [Google Scholar] [CrossRef] [PubMed]
- Peddie, V.; Butcher, R.J.; Robinson, W.T.; Wilce, M.C.J.; Traore, D.A.K.; Abell, A.D. Synthesis and conformation of fluorinated β-peptidic compounds. Chem. Eur. J. 2012, 18, 6655–6662. [Google Scholar] [CrossRef]
- Fuwa, H.; Nakajima, M.; Shi, J.; Takeda, Y.; Saito, T.; Sasaki, M. A convergent synthesis of the C1-C16 segment of goniodomin A via palladium-catalyzed organostannane-thioester coupling. Org. Lett. 2011, 13, 1106–1109. [Google Scholar] [CrossRef]
- Wu, J.; Lin, C.; Liu, D.; Bian, Q.; Wang, M.; Zhong, J. Total synthesis of (6R,12R)-6,12-dimethylpentadecan-2-one, the sex pheromone of Diabrotica balteata Leconte. Chirality 2024, 36, e23658. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.A.; Ennis, M.D.; Mathre, D.J. Asymmetric alkylation reactions of chiral imide enolates. A practical approach to the enantioselective synthesis of α-substituted carboxylic acid derivatives. J. Am. Chem. Soc. 1982, 104, 1737–1739. [Google Scholar] [CrossRef]
- Wu, Y.; Shen, X.; Tang, C.-J.; Chen, Z.-L.; Hu, Q.; Shi, W. Synthesis of natural fragrant molecules cis-3-methyl-4-decanolide and aerangis lactone. General enantioselective routes to β,γ-cis-disubstituted γ-lactones and γ,δ-cis-disubstituted δ-lactones. J. Org. Chem. 2002, 67, 3802–3810. [Google Scholar] [CrossRef]
- Kovalenko, V.N.; Mineeva, I.V. Cyclopropane intermediates in the synthesis of chiral alcohols with methyl-branched carbon skeleton. Application in the synthesis of insect pheromones. Russ. J. Org. Chem. 2014, 50, 934–942. [Google Scholar] [CrossRef]
- Yu, S.H.; Liu, J.W.; An, B.Y.; Bian, Q.H.; Min, W.; Zhong, J.C. Asymmetric synthesis of the contact sex pheromone of Neoclytus acuminatus acuminatus (Fabricius). Chin. J. Org. Chem. 2024, 44, 301–308. [Google Scholar] [CrossRef]
- Yang, S.; Che, W.; Wu, H.L.; Zhu, S.F.; Zhou, Q.L. Neutral iridium catalysts with chiral phosphine-carboxy ligands for asymmetric hydrogenation of unsaturated carboxylic acids. Chem. Sci. 2017, 8, 1977–1980. [Google Scholar] [CrossRef]
- Kono, H.; Hooz, J. Ketones and alcohols from organoboranes: Phenyl heptyl ketone, 1-hexanol, and 1-octanol. Org. Syn. 1973, 53, 77–86. [Google Scholar]
- Goncalves, S.M.C.; Silva, G.N.; Pitta, I.d.R.; Rego, M.J.B.d.M.; Gnoato, S.C.B.; Pitta, M.G.d.R. Novel betulin derivatives inhibit IFN-γ and modulates COX-2 expression. Nat. Prod. Res. 2020, 34, 1702–1711. [Google Scholar] [CrossRef] [PubMed]
- Filippini, G.; Silvi, M.; Melchiorre, P. Enantioselective formal α-methylation and α-benzylation of aldehydes by means of photo-organocatalysis. Angew. Chem. Int. Ed. 2017, 56, 4447–4451. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M.; Toriyabe, Y.; Endo, T.; Kobayashi, J. Application of modified Mosher’s method for primary alcohols with a methyl group at C2 position. Chem. Pharm. Bull. 2003, 51, 448–451. [Google Scholar] [CrossRef] [PubMed]
- Dale, J.A.; Mosher, H.S. Nuclear magnetic resonance enantiomer regents. Configurational correlations via nuclear magnetic resonance chemical shifts of diastereomeric mandelate, O-methylmandelate, and α-methoxy-α-trifluoromethylphenylacetate (MTPA) esters. J. Am. Chem. Soc. 1973, 95, 512–519. [Google Scholar] [CrossRef]
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
An, B.; Liu, S.; Wang, J.; Liu, D.; Bian, Q.; Zhong, J. A Concise Asymmetric Synthesis of Sex Pheromone of Euproctis pseudoconspersa (Strand) and Its Enantiomer. Molecules 2025, 30, 2494. https://doi.org/10.3390/molecules30122494
An B, Liu S, Wang J, Liu D, Bian Q, Zhong J. A Concise Asymmetric Synthesis of Sex Pheromone of Euproctis pseudoconspersa (Strand) and Its Enantiomer. Molecules. 2025; 30(12):2494. https://doi.org/10.3390/molecules30122494
Chicago/Turabian StyleAn, Biyu, Shengli Liu, Jianan Wang, Dan Liu, Qinghua Bian, and Jiangchun Zhong. 2025. "A Concise Asymmetric Synthesis of Sex Pheromone of Euproctis pseudoconspersa (Strand) and Its Enantiomer" Molecules 30, no. 12: 2494. https://doi.org/10.3390/molecules30122494
APA StyleAn, B., Liu, S., Wang, J., Liu, D., Bian, Q., & Zhong, J. (2025). A Concise Asymmetric Synthesis of Sex Pheromone of Euproctis pseudoconspersa (Strand) and Its Enantiomer. Molecules, 30(12), 2494. https://doi.org/10.3390/molecules30122494