
Design, Synthesis and Antiplasmodial Activities of a Library of Fluorine-Based 3-Benzylmenadiones

 , and
, and
Abstract

1. Introduction
2. Results
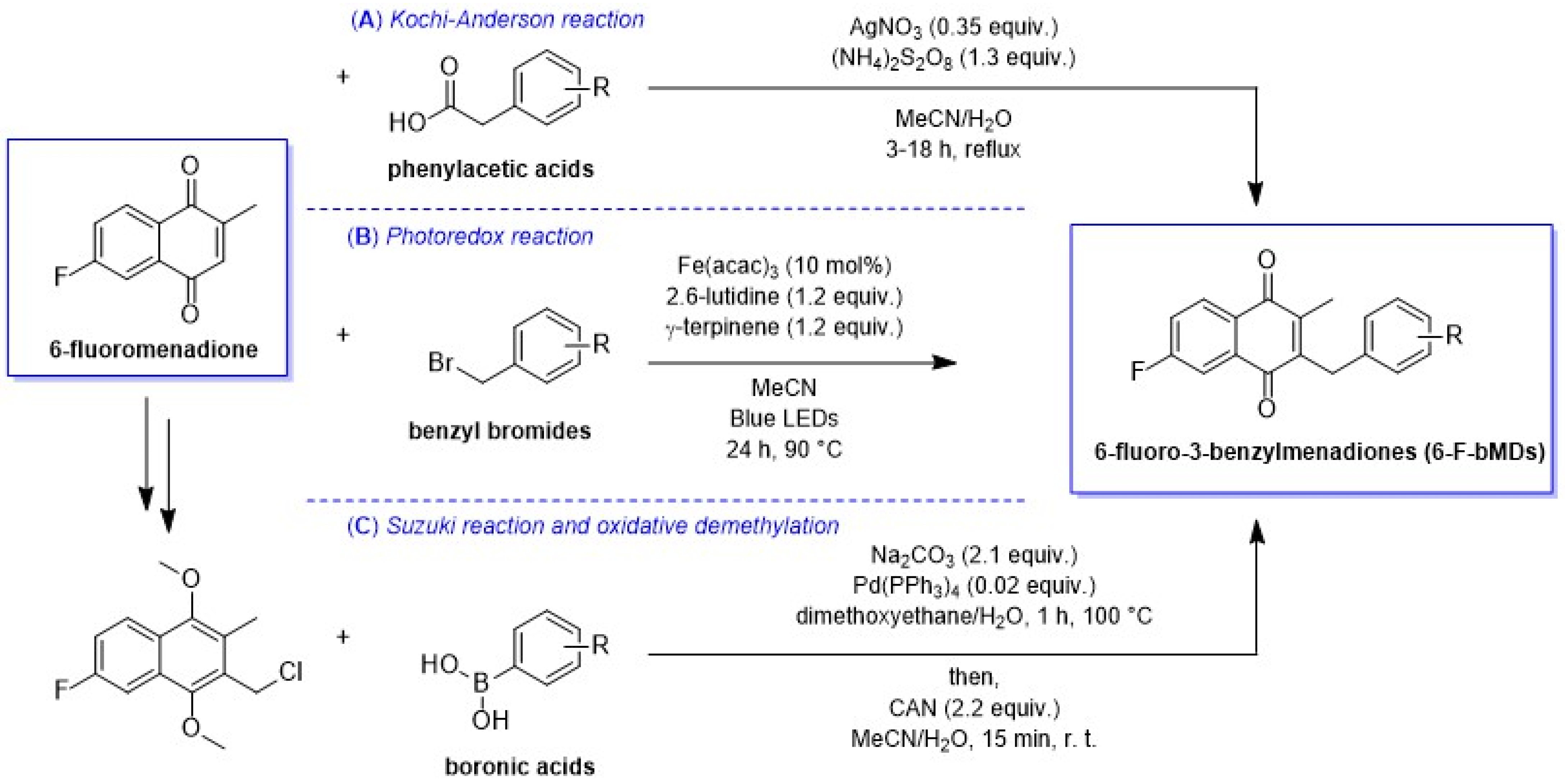
2.1. Synthesis
2.1.1. The Kochi–Anderson Reaction
2.1.2. The Photoredox Reaction
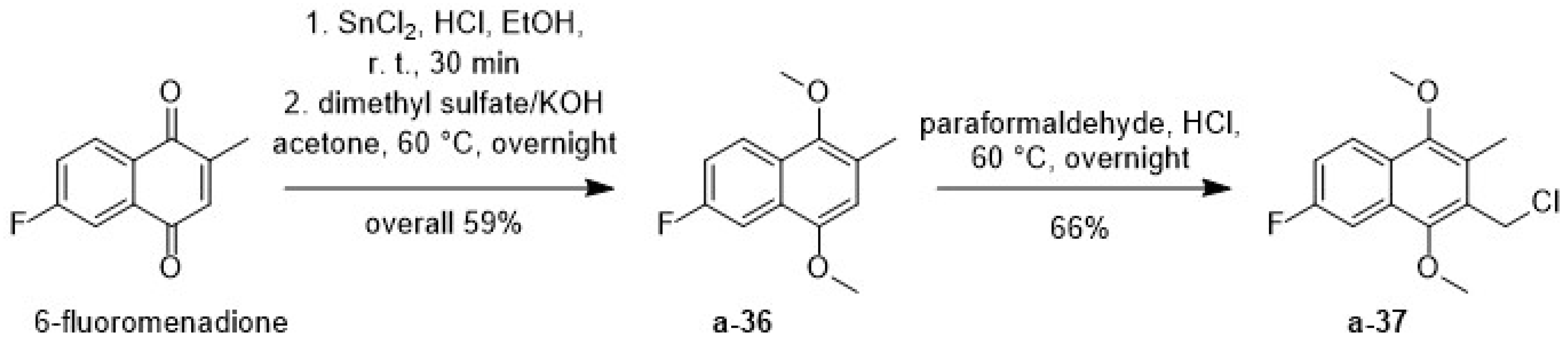
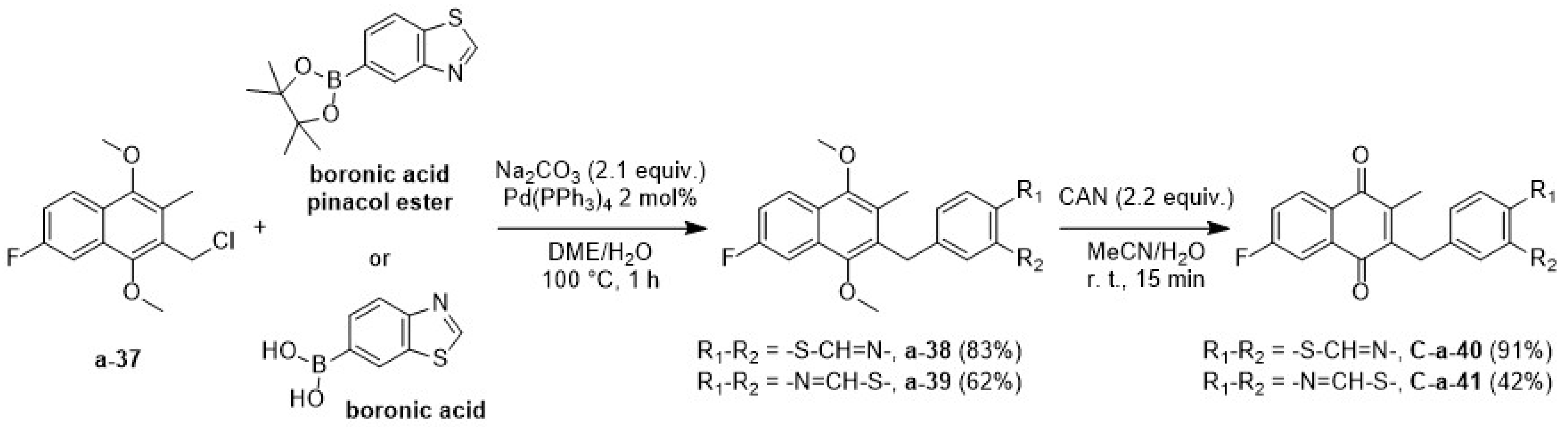
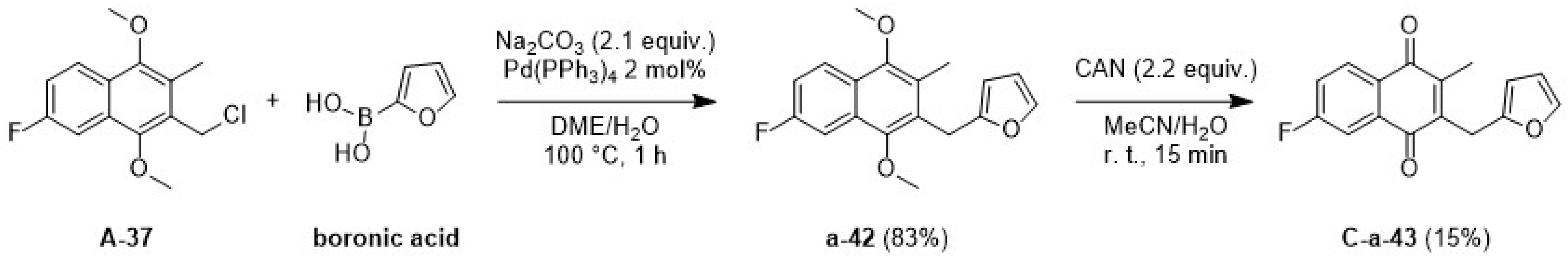
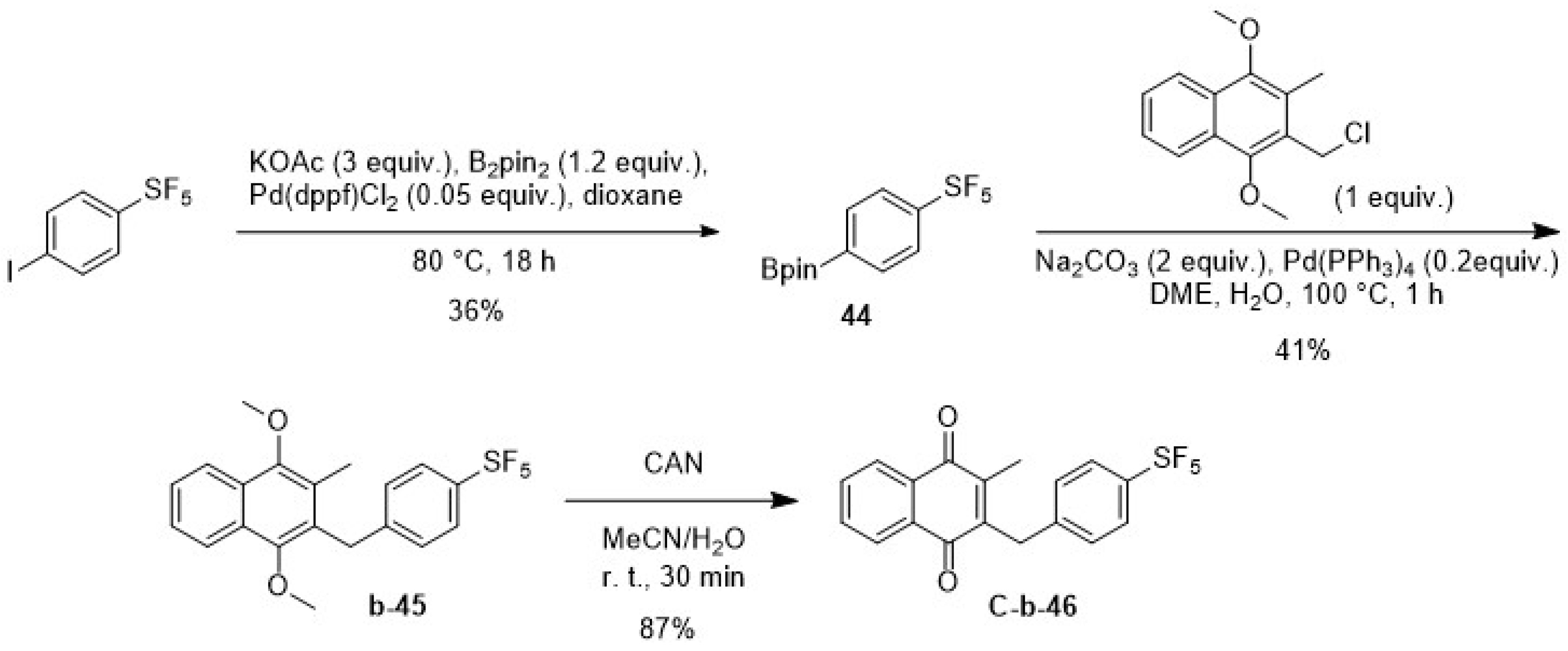
2.1.3. The Synthetic Route via the Suzuki Coupling C
2.2. Physicochemical Properties
2.3. Biological Activities
2.3.1. In Vitro Antiplasmodial Activities
2.3.2. In Vitro Species and Serum Shift Assays
2.3.3. In Vivo Antiplasmodial Activities
3. Discussion and Conclusions
4. Materials and Methods
4.1. General Information
4.2. General Procedure
4.2.1. General Procedure A: Synthesis of 6-R-3-Benzylmenadiones Using the Kochi–Anderson Reaction
4.2.2. General Procedure B: Synthesis of 6-R-3-Benzylmenadiones Using a Photoredox Reaction
4.2.3. General Procedure C: Synthesis of 6-Fluoro-3-benzylmenadiones Derivatives Using the Suzuki Reaction
4.2.4. General Procedure D: Demethylation Reaction
4.3. Pharmacokinetics
4.4. Parasite Culture and Antiplasmodial Drug Assays
4.5. In Vitro Serum Shift Assay
4.6. Cytotoxicity Assays with the Rat L6 Cell Line
4.7. Drug Assay In Vivo in P. berghei-Infected Mice
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. World Malaria Report 2023; Global Report; WHO: Geneva, Switzerland, 2023. [Google Scholar]
- Rosenthal, P.J.; Asua, V.; Bailey, J.A.; Conrad, M.D.; Ishengoma, D.S.; Kamya, M.R.; Rasmussen, C.; Tadesse, F.G.; Uwimana, A.; Fidock, D.A. The emergence of artemisinin partial resistance in Africa: How do we respond? Lancet Infect. Dis. 2024, 24, E591–E600. [Google Scholar] [CrossRef]
- Bégué, J.-P.; Bonnet-Delpon, D. Fluoroartemisinins: Metabolically more stable antimalarial artemisinin derivatives. ChemMedChem 2007, 2, 608–624. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Ehrhardt, K.; Deregnaucourt, C.; Goetz, A.-A.; Tzanova, T.; Pradines, B.; Adjalley, S.H.; Blandin, S.; Bagrel, D.; Lanzer, M.; Davioud-Charvet, E. The redox-cycler plasmodione is a fast-acting antimalarial lead compound with pronounced activity against sexual and early asexual blood-stage parasites. Antimicrob. Agents Chemother. 2016, 60, 5146–5158. [Google Scholar] [CrossRef]
- Bielitza, M.; Belorgey, D.; Ehrhardt, K.; Johann, L.; Lanfranchi, D.A.; Gallo, V.; Schwarzer, E.; Mohring, F.; Jortzik, E.; Williams, D.L.; et al. Antimalarial NADPH-consuming redox-cyclers as superior G6PD deficiency copycats. Antioxid. Redox Signal. 2015, 22, 1337–1351. [Google Scholar] [CrossRef]
- Feng, L.; Lanfranchi, D.A.; Cotos-Munoz, L.; Cesar Rodo, E.; Ehrhardt, K.; Goetz, A.-A.; Zimmerman, H.; Fenaille, F.; Blandin, S.; Davioud-Charvet, E. Synthesis of plasmodione metabolites and 13C-enriched plasmodione as chemical tools for drug metabolism investigation. Org. Biomol. Chem. 2018, 16, 2647–2665. [Google Scholar] [CrossRef]
- Müller, T.; Johann, L.; Jannack, B.; Brückner, M.; Lanfranchi, D.L.; Bauer, H.; Sanchez, C.; Yardley, V.; Deregnaucourt, C.; Schrével, J.; et al. Glutathione Reductase-Catalyzed Cascade of Redox Reactions to Bioactivate Potent Antimalarial 1,4-Naphthoquinones—A New Strategy to Combat Malarial Parasites. J. Am. Chem. Soc. 2011, 133, 11557–11571. [Google Scholar] [CrossRef]
- Donzel, M.; Elhabiri, M.; Davioud-Charvet, E. Bioinspired Photoredox Benzylation of Quinones. J. Org. Chem. 2021, 86, 10055–10066. [Google Scholar] [CrossRef]
- Dupouy, B.; Donzel, M.; Roignant, M.; Charital, S.; Yamaryo-Botté, Y.; Keumoe, R.; Bordat, Y.; Bundschuh, M.; Feckler, A.; Rottmann, M.; et al. 3-Benzylmenadiones and their heteroaromatic analogues target the apicoplast of Apicomplexa parasites: Synthesis and bioimaging studies. ACS Infect. Dis. 2024, 10, 3553–3576. [Google Scholar] [CrossRef]
- Trometer, N.; Roignant, M.; Davioud-Charvet, E. Efficient Multigram-Scale Synthesis of 7-Substituted 3-Methylteral-1-ones and 6-Fluoromenadione. Org. Process Res. Dev. 2022, 26, 1152–1164. [Google Scholar] [CrossRef]
- Rodo, E.C.; Feng, L.; Jida, M.; Ehrhardt, K.; Bielitza, M.; Boilevin, J.; Lanzer, M.; Williams, D.L.; Lanfranchi, D.A.; Davioud-Charvet, E. A Platform of Regioselective Methodologies to Access Polysubstituted 2-Methyl-1,4-naphthoquinone Derivatives: Scope and Limitations. Eur. J. Org. Chem. 2016, 2016, 1982–1993. [Google Scholar] [CrossRef]
- Davioud-Charvet, E.; Müller, T.; Bauer, H.; Schirmer, H. 1,4-Naphthoquinone Derivatives, Their Preparation, Pharmaceutical Compositions, and Use as Antimalarial Agents. WO2009118327 A1, 1 October 2009. [Google Scholar]
- Kohri, M.; Irie, S.; Yamazaki, S.; Kohaku, K.; Taniguchi, T.; Kishikawa, K. Acid-induced Control of Surface Properties Using a Catecholic Silane Coupling Reagent. Chem. Lett. 2019, 48, 551–554. [Google Scholar]
- Newton, J.J.; Brooke, A.J.; Duhamel, B.; Pulfer, J.M.; Britton, R.; Friesen, C.M. Fluorodesulfurization of Thionobenzodioxoles with Silver(I) Fluoride. J. Org. Chem. 2020, 85, 13298–13305. [Google Scholar] [CrossRef]
- Henry, N.; Enguehard-Gueiffier, C.; Thery, I.; Gueiffier, A. One-Pot Dual Substitutions of Bromobenzyl Chloride, 2-Chloromethyl-6-halogenoimidazo[1,2-a]pyridine and -[1,2-b]pyridazine by Suzuki–Miyaura Cross-Coupling Reactions. Eur. J. Org. Chem. 2008, 2008, 4824–4827. [Google Scholar] [CrossRef]
- Trager, W.; Jensen, J.B. Human malaria parasites in continuous culture. Science 1976, 193, 673–675. [Google Scholar] [CrossRef]
- Desjardins, R.E.; Canfield, C.J.; Haynes, J.D.; Chulay, J.D. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob. Agents Chemother. 1979, 16, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Presser, A.; Blaser, G.; Pferschy-Wenzig, E.M.; Kaiser, M.; Mäser, P.; Schuehly, W. Pharmacomodulation of the Redox-Active Lead Plasmodione: Synthesis of Substituted 2-Benzylnaphthoquinone Derivatives, Antiplasmodial Activities, and Physicochemical Properties. Int. J. Mol. Sci. 2025, 26, 2114. [Google Scholar] [CrossRef] [PubMed]
- Boss, C.; Aissaoui, H.; Amaral, N.; Bauer, A.; Bazire, S.; Binkert, C.; Brun, R.; Bürki, C.; Ciana, C.L.; Corminboeuf, O.; et al. Discovery and Characterization of ACT-451840: An Antimalarial Drug with a Novel Mechanism of Action. ChemMedChem 2016, 11, 1995–2014. [Google Scholar] [CrossRef]
- Cesar-Rodo, E.; Dupouy, B.; Häberli, C.; Williams, D.L.; Strub, J.-M.; Mäser, P.; Rottmann, M.; Keiser, J.; Lanfranchi, D.A.; Davioud-Charvet, E. Regioselective synthesis of potential non-quinonoid prodrugs of plasmodione: Drug metabolism studies and antiparasitic properties against two hemoglobin-feeding parasites. Molecules 2024, 29, 5268. [Google Scholar] [CrossRef]
- Vennerstrom, J.L.; Arbe-Barnes, S.; Brun, R.; Charman, S.A.; Chiu, F.C.; Chollet, J.; Dong, Y.; Dorn, A.; Hunziker, D.; Matile, H.; et al. Identification of an antimalarial synthetic trioxolane drug development candidate. Nature 2004, 430, 900–904. [Google Scholar] [CrossRef]
- Ridley, R.G.; Matile, H.; Jaquet, C.; Dorn, A.; Hofheinz, W.; Leupin, W.; Masciadri, R.; Theil, F.P.; Richter, W.F.; Girometta, M.A.; et al. Antimalarial activity of the bisquinoline trans-N1,N2-bis-(7-chloroquinolin-4-yl)cyclohexane-1,2-diamine: Comparison of two stereoisomers and detailed evaluation of the S,S enantiomer, Ro 47-7737. Antimicrob. Agents Chemother. 1997, 41, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Peters, W. Chemotherapy and Drug Resistance in Malaria; Academic Press: Cambridge, MA, USA, 1987; Volume 1. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
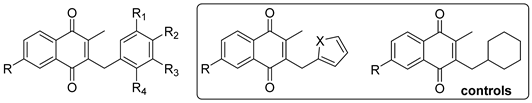
 | ||||||
| Cpnd | R | R1 | R2 | R3 | R4 | Yield (%) |
|---|---|---|---|---|---|---|
| 6-F-PD [12] | F | H | CF3 | H | H | 55 |
| PD [8] | H | H | CF3 | H | H | 80 |
| A-a-1 | F | H | H | CF3 | H | 44 |
| A-a-2 | F | H | H | H | CF3 | 38 |
| A-a-3 | F | H | OCF3 | H | H | 56 |
| A-b-3 [13] | H | H | OCF3 | H | H | 78 |
| A-a-4 | F | H | F | H | H | 77 |
| A-b-4 [13] | H | H | F | H | H | 66 |
| A-a-5 | F | H | H | F | H | 34 |
| A-a-6 | F | H | H | H | F | 45 |
| A-b-6 | H | H | H | H | F | 67 |
| A-a-7 | F | H | CF3 | F | H | 75 |
| A-b-7 | H | H | CF3 | F | H | 82 |
| A-a-8 | F | H | CF3 | Br | H | 64 |
| A-b-8 | H | H | CF3 | Br | H | 84 |
| A-a-9 | F | H | F | CF3 | H | 52 |
| A-b-9 | H | H | F | CF3 | H | 92 |
| A-a-10 | F | H | CN | H | H | 75 |
| A-b-10 [8] | H | H | CN | H | H | 63 |
| A-a-11 | F | H | H | H | CN | 48 |
| A-b-11 | H | H | H | H | CN | 56 |
| A-a-12 | F | H | NO2 | H | H | 72 |
| A-b-12 [8] | H | H | NO2 | H | H | 89 |
| A-a-13 | F | H | I | H | H | 85 |
| A-a-14 | F | H | Cl | H | Cl | 59 |
| A-b-14 | H | H | Cl | H | Cl | 59 |
| A-a-15 | F | H | Cl | H | CF3 | 46 |
| A-b-15 | H | H | Cl | H | CF3 | 53 |
| A-a-16 | F | H | OMe | H | H | 79 |
| A-b-16 [13] | H | H | OMe | H | H | 45 |
| A-a-17 | F | H | H | OMe | H | 59 |
| A-b-17 | H | H | H | OMe | H | 75 |
| A-a-18 | F | H | H | H | OMe | 69 |
| A-b-18 [13] | H | H | H | H | OMe | 82 |
| A-a-19 | F | H | OMe | OMe | H | 52 |
| A-b-19 [13] | H | H | OMe | OMe | H | 94 |
| A-a-20 | F | H | OMe | H | OMe | 71 |
| A-b-20 [13] | H | H | OMe | H | OMe | 37 |
| A-a-21 | F | H | -O-CH2-O- | H | 66 | |
| A-b-21 | H | H | -O-CH2-O- | H | 60 | |
| A-a-22 | F | H | -O-(CH2)2-O- | H | 79 | |
| A-b-22 | H | H | -O-(CH2)2-O- | H | 91 | |
| A-a-23 | F | H | -O-CMe2-O- | H | 83 | |
| A-b-23 | H | H | -O-CMe2-O- | H | 85 | |
| A-a-24 | F | H | -O-CF2-O- | H | 78 | |
| A-b-24 | H | H | -O-CF2-O- | H | 73 | |
| A-a-25 | F | H | OMe | OMe | OMe | 80 |
| A-b-25 [13] | H | H | OMe | OMe | OMe | 76 |
| A-a-26 | F | OMe | H | H | OMe | 63 |
| A-b-26 [13] | H | OMe | H | H | OMe | 80 |
| A-a-27 | F | OMe | OMe | OMe | H | 85 |
| A-b-27 [13] | H | OMe | OMe | OMe | H | 85 |
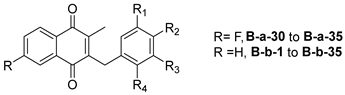 | ||||||
| Cpnd | R | R1 | R2 | R3 | R4 | Yield (%) |
|---|---|---|---|---|---|---|
| 6-F-PD [9] | F | H | CF3 | H | H | 67 |
| B-b-1 [9] | H | H | H | CF3 | H | 76 |
| B-b-2 [9] | H | H | H | H | CF3 | 77 |
| B-b-3 [9] | H | H | OCF3 | H | H | 80 |
| B-b-4 [9] | H | H | F | H | H | 66 |
| B-b-5 | H | H | H | F | H | 60 |
| B-b-26 [9] | H | OMe | H | H | OMe | 78 |
| B-a-30 | F | H | H | CN | H | 43 |
| B-b-30 | H | H | H | CN | H | 66 |
| B-a-31 | F | H | H | H | H | 67 |
| B-a-32 | F | H | NO2 | F | H | 36 |
| B-a-33 | F | H | F | I | H | 36 |
| B-b-33 | H | H | F | I | H | 56 |
| B-a-34 | F | H | Br | H | CF3 | 22 |
| B-b-34 | H | H | Br | H | CF3 | 69 |
| B-a-35 | F | OMe | H | OMe | H | 62 |
| B-b-35 [9] | H | OMe | H | OMe | H | 68 |
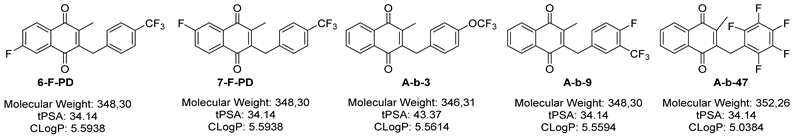
| Cpnd | MW a (g·mol−1) | cLogPa | tPSA a (Å2) | CHI | log D7.4 | n Fluorine | Thermodynamic aq. Solubility in PBS-10% DMSO (µM) |
|---|---|---|---|---|---|---|---|
| PD | 330.31 | 5.416 | 34.14 | 115 | 4.68 | 3 | 0.44 ± 0.05 |
| 6-F-PD | 348.30 | 5.594 | 34.14 | 117 | 4.75 | 4 | 0.06 ± 0.01 |
| 7-F-PD | 348.30 | 5.594 | 34.14 | nd | nd | 4 | 0.59 ± 0.01 |
| A-b-3 | 346.31 | 5.561 | 43.37 | 117 | 4.80 | 3 | 0.16 ± 0.04 |
| A-b-4 | 280.30 | 4.676 | 34.14 | 108 | 4.30 | 1 | 1.33 ± 0.02 |
| A-b-9 | 348.30 | 5.559 | 34.14 | nd | nd | 4 | 0.67 ± 0.01 |
| A-b-10 | 287.32 | 3.966 | 57.93 | 97 | 3.75 | 0 | 0.82 ± 0.01 |
| A-b-12 | 307.30 | 4.276 | 85.95 | 112 | 3.99 | 0 | 0.24 ± 0.01 |
| A-b-16 | 292.33 | 4.452 | 43.37 | 106 | 4.22 | 0 | 1.07 ± 0.02 |
| A-b-26 | 322.36 | 4.541 | 52.6 | 107 | 4.26 | 0 | 0.59 ± 0.01 |
| B-b-35 | 322.36 | 4.541 | 52.6 | 107 | 4.27 | 0 | 0.75 ± 0.14 |
| A-b-47 | 352.26 | 5.038 | 34.14 | 115 | 4.67 | 5 | 0.33 ± 0.01 |
| atovaquone | 366.84 | 6.351 | 54.37 | nd | nd | 0 | 1.55 ± 0.04 |
 | ||||||||
| Cpnd | R | R1 | R2 | R3 | R4 | X | IC50 (µM) | Tox/L6 (µM) |
|---|---|---|---|---|---|---|---|---|
| 6-F-PD [9] | F | H | CF3 | H | H | - | 0.181 | 44.7 |
| PD [10] | H | H | CF3 | H | H | - | 0.043 | 141.7 |
| A-a-1 | F | H | H | CF3 | H | - | 0.082 | 12.5 |
| B-b-1 [9] | H | H | H | CF3 | H | - | 0.062 | 57.1 |
| A-a-2 | F | H | H | H | CF3 | - | 0.138 | 53.4 |
| B-b-2 [9] | H | H | H | H | CF3 | - | 0.026 | 249.8 |
| A-a-3 | F | H | OCF3 | H | H | - | 0.330 | 138.8 |
| A-b-3 | H | H | OCF3 | H | H | - | 0.083 | nd |
| A-a-4 | F | H | F | H | H | - | 0.302 | 174.5 |
| A-b-4 [13] | H | H | F | H | H | - | 0.029 | 51.7 |
| A-a-5 | F | H | H | F | H | - | 0.246 | 6.6 |
| B-b-5 | H | H | H | F | H | - | 0.089 | 71.0 |
| A-a-6 | F | H | H | H | F | - | 0.119 | 7.6 |
| A-b-6 | H | H | H | H | F | - | 0.453 | nd |
| A-a-7 | F | H | CF3 | F | H | - | 0.735 | 85.5 |
| A-b-7 | H | H | CF3 | F | H | - | 2.958 | 49.2 |
| A-a-8 | F | H | CF3 | Br | H | - | 0.167 | nd |
| A-b-8 | H | H | CF3 | Br | H | - | 0.081 | nd |
| A-a-9 | F | H | F | CF3 | H | - | 0.149 | nd |
| A-b-9 | H | H | F | CF3 | H | - | 0.077 | nd |
| A-a-10 | F | H | CN | H | H | - | 0.231 | 95 |
| A-b-10 [8] | H | H | CN | H | H | - | 0.374 | 112.5 |
| A-a-11 | F | H | H | H | CN | - | 0.264 | 327.0 |
| A-b-11 | H | H | H | H | CN | - | 0.312 | 186.2 |
| A-a-12 | F | H | NO2 | H | H | - | 0.177 | 307.0 |
| A-b-12 [8] | H | H | NO2 | H | H | - | 0.201 | 194.5 |
| A-a-13 | F | H | I | H | H | - | 0.198 | 40.9 |
| B-b-13 [9] | H | H | I | H | H | - | 0.115 | 138.6 |
| A-a-14 | F | H | Cl | H | Cl | - | 0.137 | nd |
| A-b-14 | H | H | Cl | H | Cl | - | 0.067 | nd |
| A-a-15 | F | H | Cl | H | CF3 | - | 0.103 | nd |
| A-b-15 | H | H | Cl | H | CF3 | - | 0.080 | nd |
| A-a-16 | F | H | OMe | H | H | - | 1.118 | 195.9 |
| A-a-17 | F | H | H | OMe | H | - | 0.361 | 165.6 |
| A-a-18 | F | H | H | H | OMe | - | 0.572 | 189.3 |
| A-a-19 | F | H | OMe | OMe | H | - | 0.171 | 5.6 |
| A-b-19 [13] | H | H | OMe | OMe | H | - | 0.142 | 5.6 |
| A-a-20 | F | H | OMe | H | OMe | - | 0.316 | 106.6 |
| A-a-21 | F | H | -O-CH2-O- | H | - | 0.070 | 165.6 | |
| A-b-21 | H | H | -O-CH2-O- | H | - | 0.200 | 97.9 | |
| A-a-22 | F | H | -O-(CH2)2-O- | H | - | 0.311 | nd | |
| A-b-22 | H | H | -O-(CH2)2-O- | H | - | 0.280 | nd | |
| A-a-23 | F | H | -O-CMe2-O- | H | - | 0.338 | nd | |
| Cpnd | R | R1 | R2 | R3 | R4 | X | IC50 (µM) | Tox/L6 (µM) |
| A-b-23 | H | H | -O-CMe2-O- | H | - | 0.152 | nd | |
| A-a-24 | F | H | -O-CF2-O- | H | - | 0.206 | nd | |
| A-b-24 | H | H | -O-CF2-O- | H | - | 0.077 | nd | |
| A-a-25 | F | H | OMe | OMe | OMe | - | 0.340 | 215.5 |
| A-a-26 | F | OMe | H | H | OMe | - | 0.291 | 91.7 |
| A-b-26 [9] | H | OMe | H | H | OMe | - | 0.067 | 158.5 |
| A-a-27 | F | OMe | OMe | OMe | H | - | 0.265 | 72.4 |
| A-a-28 | F | - | - | - | - | S | 0.474 | 5.8 |
| A-b-28 | H | - | - | - | - | S | 0.115 | 5.2 |
| B-a-30 | F | H | H | CN | H | - | 0.326 | 33.3 |
| B-b-30 | H | H | H | CN | H | - | 0.190 | 72.6 |
| B-a-31 | F | H | H | H | H | - | 0.300 | 174.5 |
| B-b-31 [9] | H | H | H | H | H | - | 1.695 | 332.8 |
| B-a-32 | F | H | NO2 | F | H | - | 0.705 | 107.4 |
| B-b-32 [9] | H | H | NO2 | F | H | - | 0.598 | 189.2 |
| B-a-33 | F | H | F | I | H | - | 0.376 | nd |
| B-b-33 | H | H | F | I | H | - | 0.136 | nd |
| B-a-34 | F | H | Br | H | CF3 | - | 0.169 | nd |
| B-b-34 | H | H | Br | H | CF3 | - | 0.078 | nd |
| B-a-35 | F | OMe | H | OMe | H | - | 0.711 | 293.0 |
| B-b-35 [9] | H | OMe | H | OMe | H | - | 0.052 | 70.3 |
| C-a-40 | F | H | -S-CH=N- | H | - | 2.062 | nd | |
| C-b-40 | H | H | -S-CH=N- | H | - | 0.235 | 50.29 | |
| C-a-41 | F | H | -N=CH-S- | H | - | 2.527 | nd | |
| C-b-41 | H | H | -N=CH-S- | H | - | 0.370 | 49.81 | |
| C-a-43 | F | - | - | - | - | O | 0.947 | 3.8 |
| A-b-43 [8] | H | - | - | - | - | O | 0.501 | 4.2 |
| C-b-46 | H | H | -SF5 | H | H | - | 0.048 | 35.1 |
| A-b-47 [9] | H | bMD-F5 | - | 0.060 | 161.0 | |||
| A-a-29 | F | Controls | 0.718 | 177.8 | ||||
| A-b-29 | H | 0.913 | 229.9 | |||||
 | |||||
| A. | |||||
| Cpnd | IC50 (nM) | IC50 (nM) | IC50 (nM) | Species Shift | |
| Pf NF54, 72 h | Pf NF54, 24 h | Pb, 24 h | Pf versus Pb | ||
| PD | 59 | 130 | 130 | 1.0 | |
| 6-F-PD | 120 | 104 | 170 | 1.6 | |
| 7-F-PD | 93 | 157 | 138 | 0.9 | |
| A-b-3 | 83 | 223 | 191 | 0.9 | |
| A-b-9 | 62 | 143 | 149 | 1.0 | |
| A-b-47 | 91 | 184 | 265 | 1.4 | |
| AS | 4.8 | 2.1 | 13 | 6.1 | |
| CQ | 11 | 4.9 | 16 | 3.3 | |
| B. | |||||
| Cpnd | IC50 (nM) NF54 | IC50 (nM) NF54 | Serum Shift | ||
| 72 h, 0.5% Albumax | 72 h, 50% Human Serum | ||||
| PD | 57 | 270 | 4.7 | ||
| 6-F-PD | 93 | 80 | 0.9 | ||
| 7-F-PD | 89 | 377 | 4.2 | ||
| A-b-3 | 83 | 473 | 5.7 | ||
| A-b-9 | 59 | 355 | 6.1 | ||
| A-b-47 | 81 | 324 | 4.0 | ||
| AS | 1.7 | 1.0 | 0.6 | ||
| CQ | 5.0 | 6.6 | 1.3 | ||
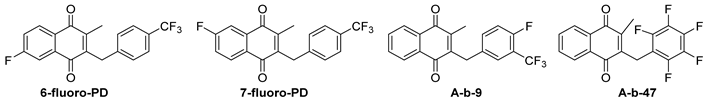 | |||||||||
| Cpnd | Oral Dose (mg/kg) | Parasitized RBC over 100 | Avg. | % of Control | Activity (%) | Avg. Mouse Survival in Days | |||
|---|---|---|---|---|---|---|---|---|---|
| Control | 0 | 59.90 | 51.10 | 71.10 | 68.50 | 58.90 | 4.0 | ||
| PD | 4 × 100 | 61.4 | 64.8 | 57.0 | 60.90 | 103.40 | −3.40 | 4.0 | |
| PD | 4 × 50 | 66.5 | 70.6 | 71.8 | 69.63 | 118.22 | −18.22 | 4.0 | |
| 6-F-PD | 4 × 50 | 28.2 | 35.8 | 27.8 | 30.60 | 51.95 | 48.05 | 7.0 | |
| 7-F-PD 2 | 4 × 50 | 64.7 | 69.0 | 62.2 | 65.30 | 110.87 | −10.87 | 4.0 | |
| A-b-9 | 4 × 50 | 70.3 | 49.1 | 52.4 | 57.27 | 97.23 | 2.77 | 4.0 | |
| A-b-47 3 | 4 × 50 | 65.4 | 62.4 | 72.5 | 66.77 | 113.36 | −13.36 | 4.0 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roignant, M.; Richard, J.; Donzel, M.; Rottmann, M.; Mäser, P.; Davioud-Charvet, E. Design, Synthesis and Antiplasmodial Activities of a Library of Fluorine-Based 3-Benzylmenadiones. Molecules 2025, 30, 2446. https://doi.org/10.3390/molecules30112446
Roignant M, Richard J, Donzel M, Rottmann M, Mäser P, Davioud-Charvet E. Design, Synthesis and Antiplasmodial Activities of a Library of Fluorine-Based 3-Benzylmenadiones. Molecules. 2025; 30(11):2446. https://doi.org/10.3390/molecules30112446
Chicago/Turabian StyleRoignant, Matthieu, Jimmy Richard, Maxime Donzel, Matthias Rottmann, Pascal Mäser, and Elisabeth Davioud-Charvet. 2025. "Design, Synthesis and Antiplasmodial Activities of a Library of Fluorine-Based 3-Benzylmenadiones" Molecules 30, no. 11: 2446. https://doi.org/10.3390/molecules30112446
APA StyleRoignant, M., Richard, J., Donzel, M., Rottmann, M., Mäser, P., & Davioud-Charvet, E. (2025). Design, Synthesis and Antiplasmodial Activities of a Library of Fluorine-Based 3-Benzylmenadiones. Molecules, 30(11), 2446. https://doi.org/10.3390/molecules30112446