
Nickel-Catalyzed Three-Component Unsymmetrical Bis-Allylation of Alkynes with Alkenes: A Density Functional Theory Study

 , and
, and
Abstract

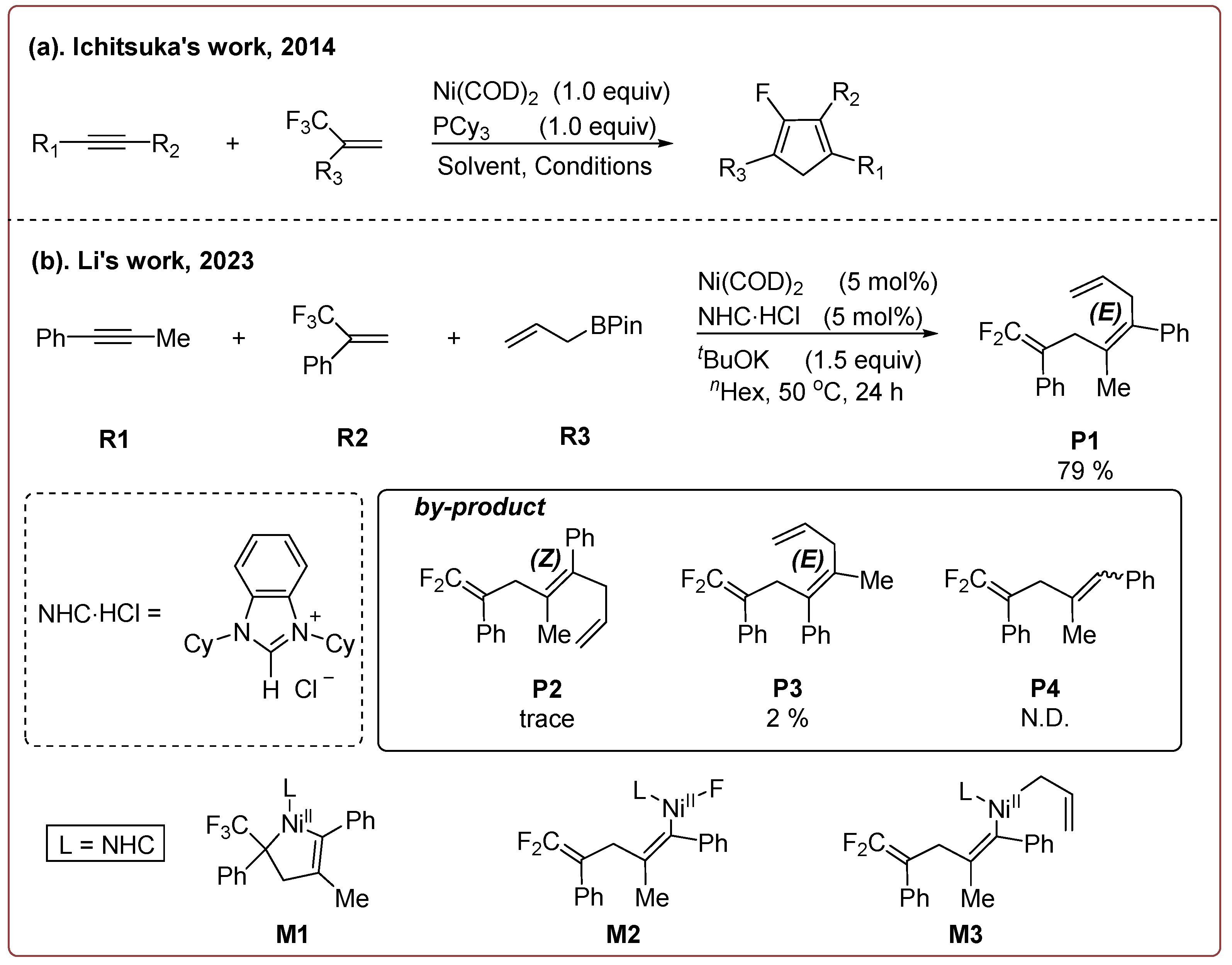
1. Introduction
2. Results and Discussion
2.1. Reaction Mechanism
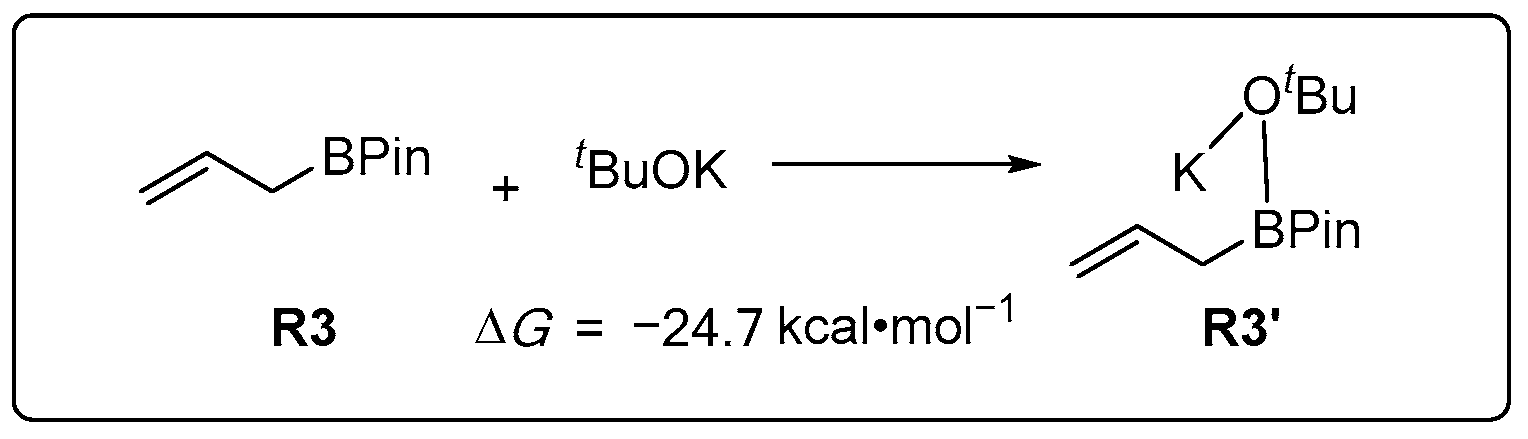
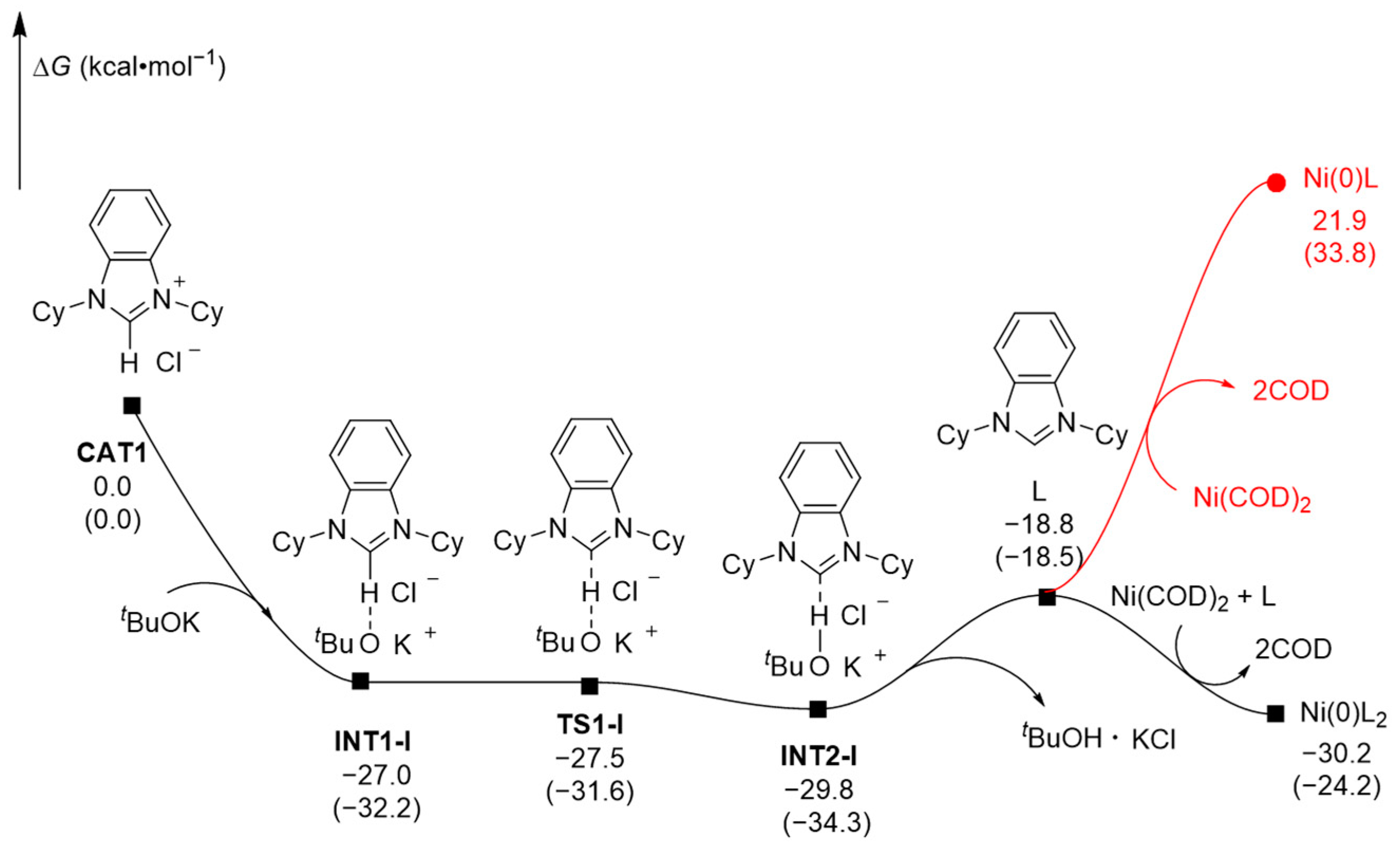
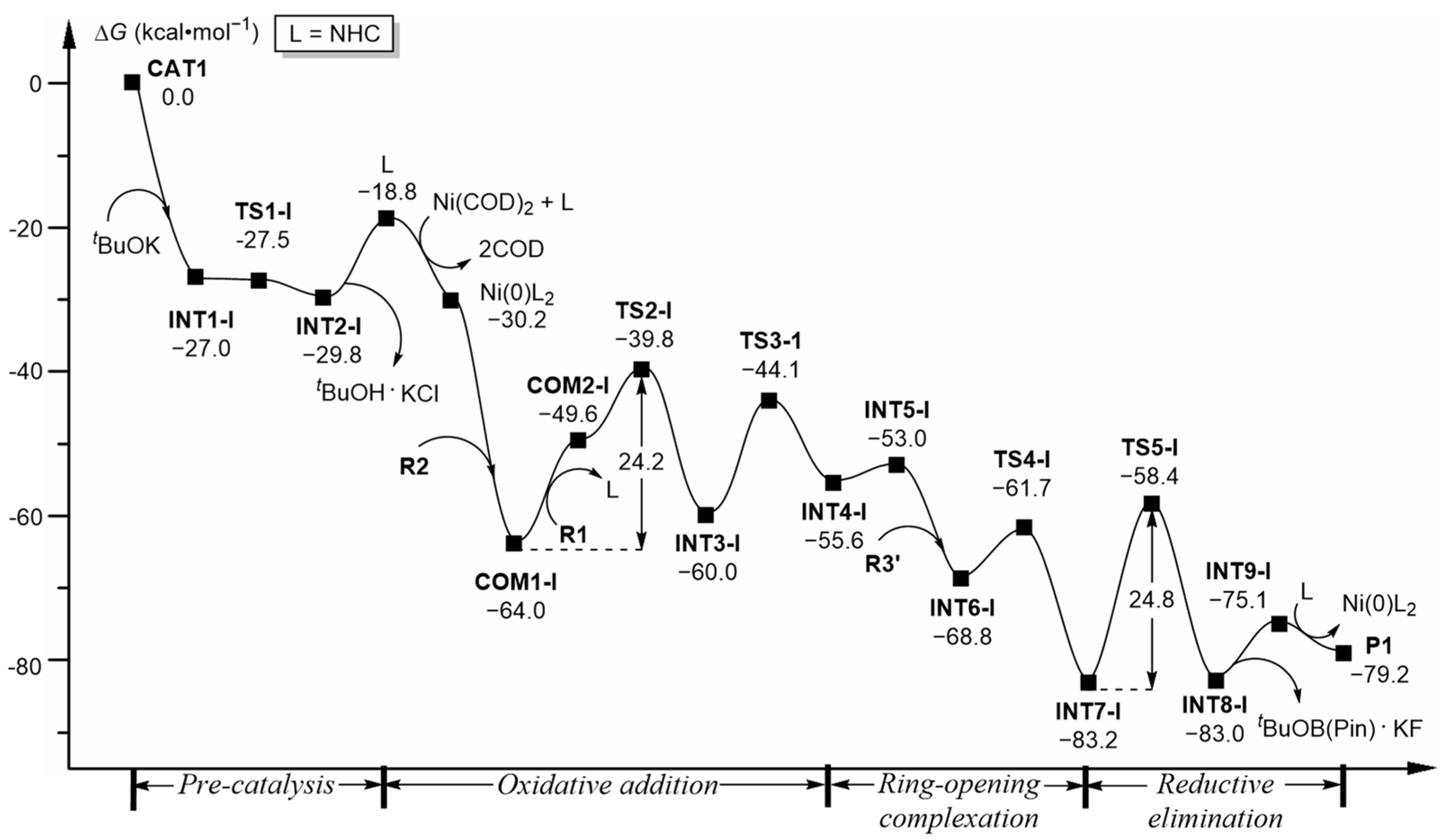
2.1.1. Pre-Catalysis
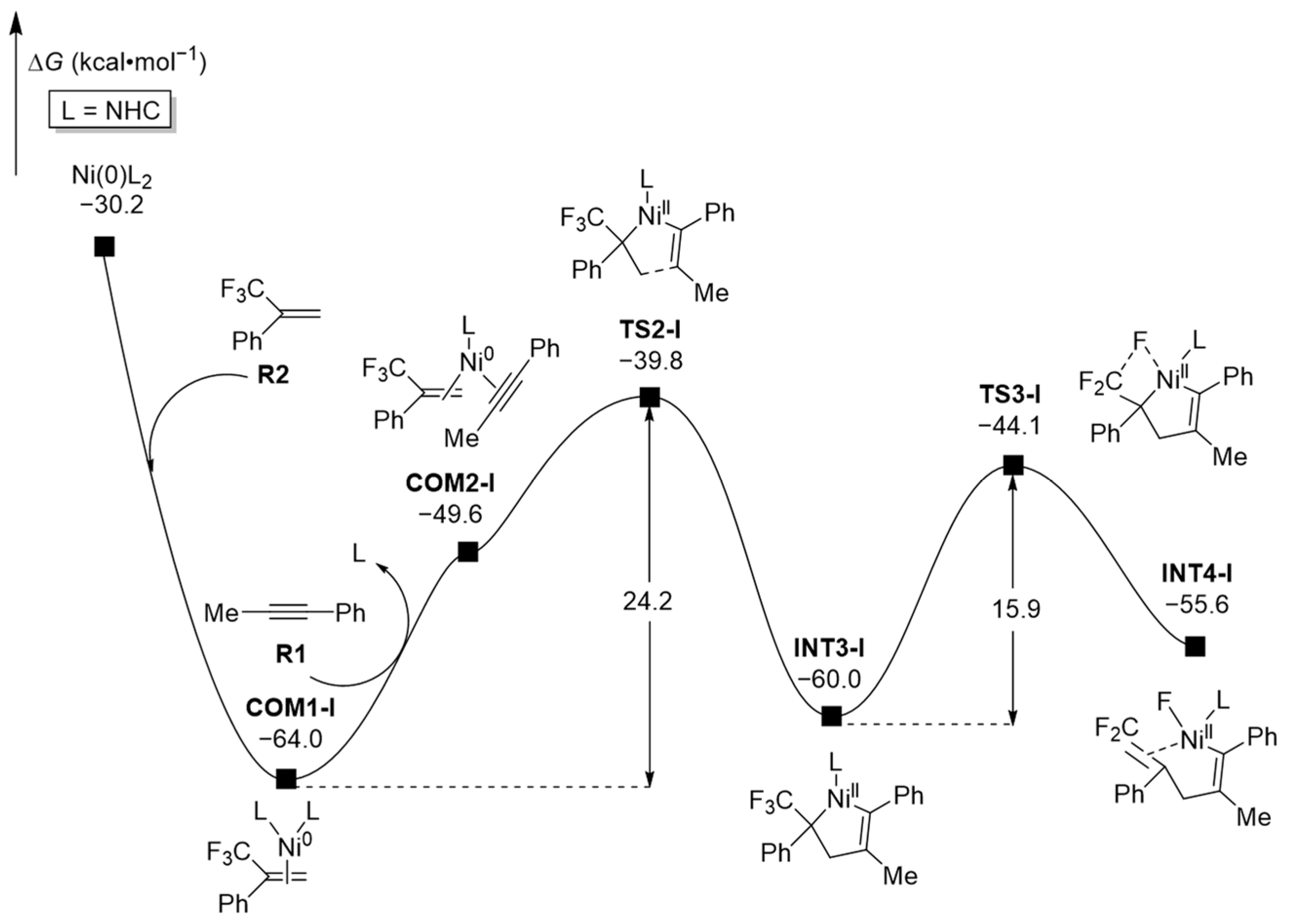
2.1.2. Oxidative Addition (OA)
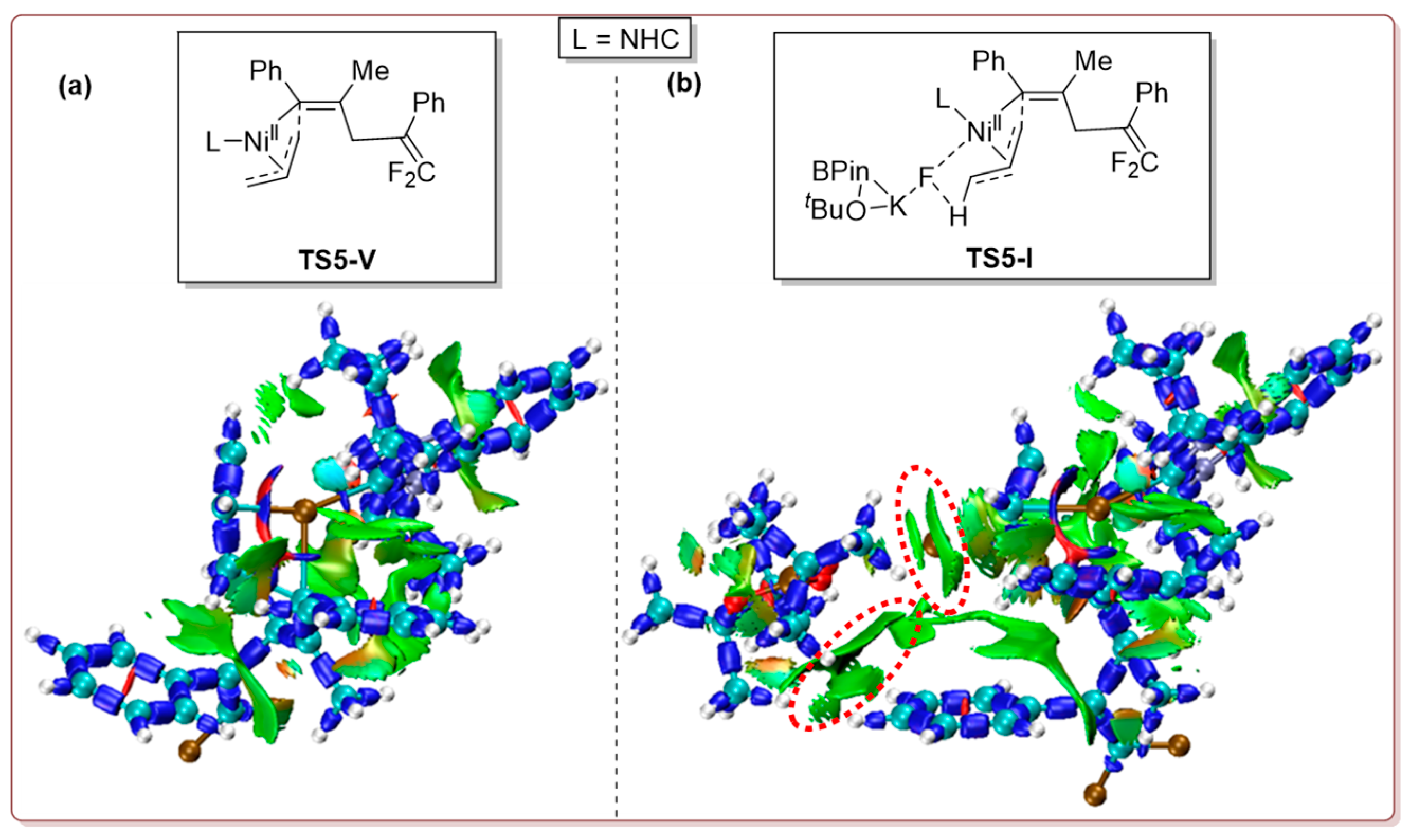
2.1.3. Ring-Opening Complexation
2.1.4. Reductive Elimination (RE)
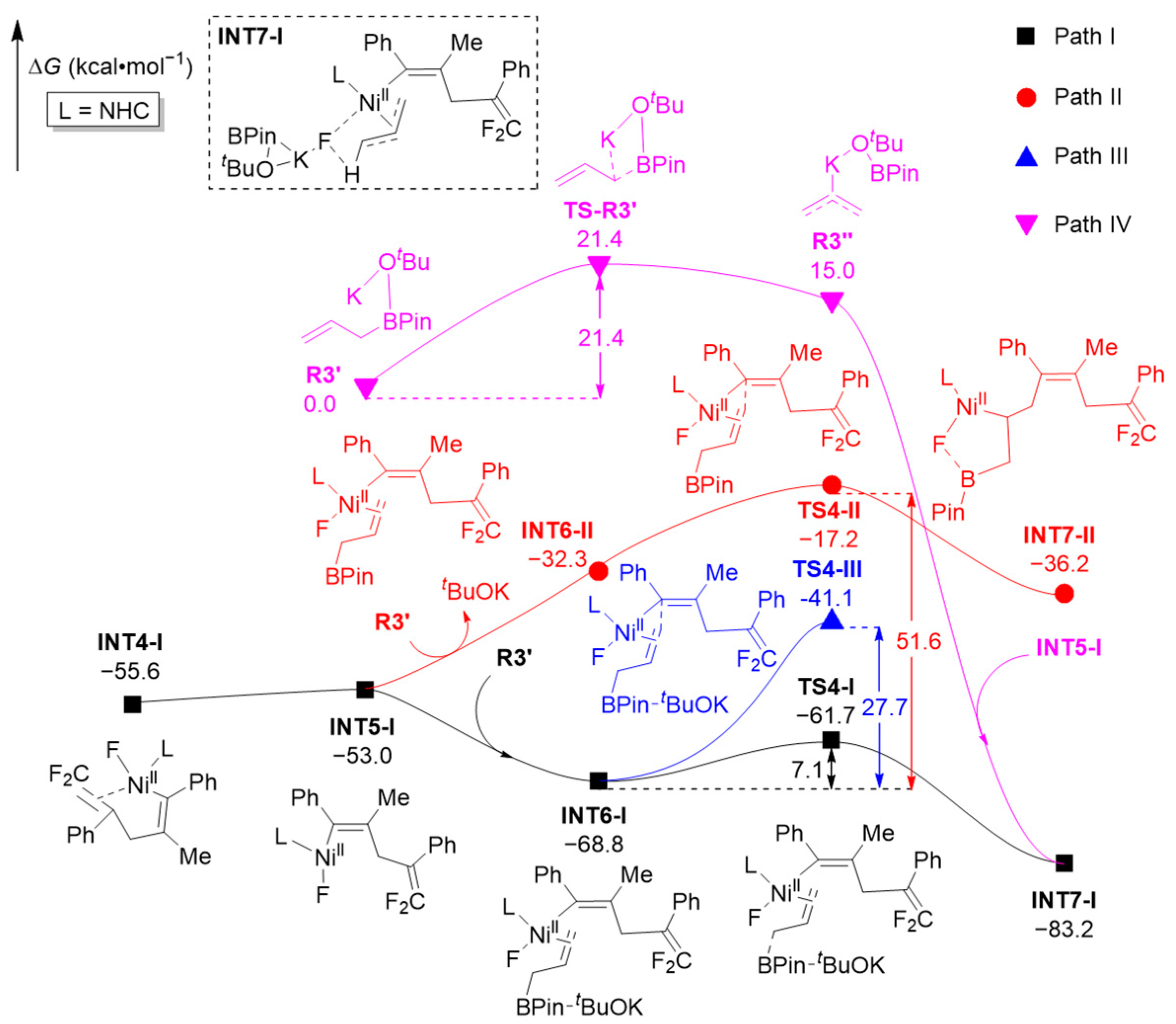
2.2. Selectivities
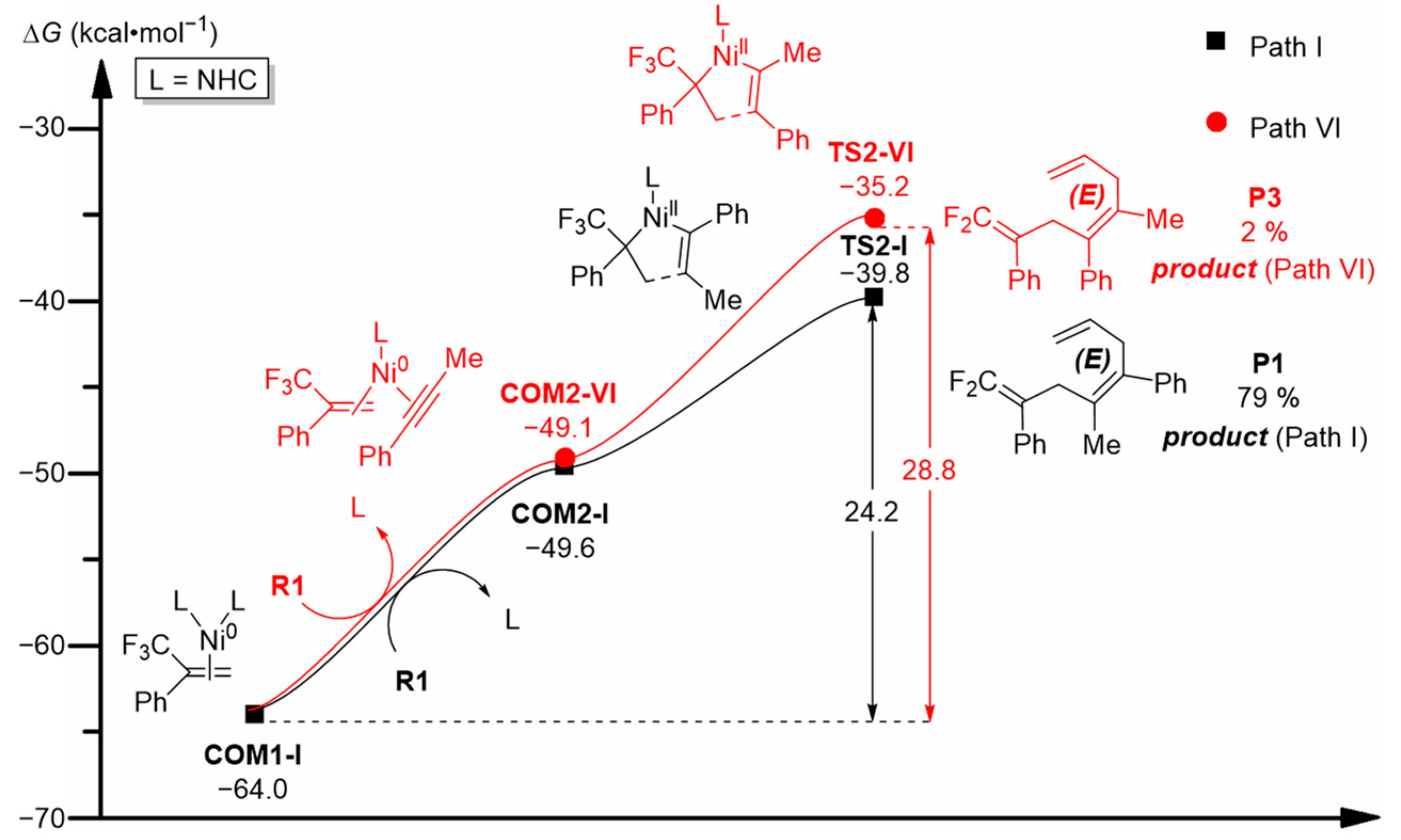
2.2.1. Chemoselectivities
2.2.2. Regioselectivities
2.3. Recycling and Regeneration of Catalyst
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alegre-Requena, J.; Marqués-López, E.; Herrera, R. IntroductIon: Multicomponent Strategies. In Multicomponent Reactions: Concepts and Applications for Design and Synthesis; Herrera, R., Marqués-López, E., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2015; pp. 1–15. [Google Scholar]
- Cioc, R.; Ruijter, E.; Orru, R.V.A. Multicomponent Reactions: Advanced Tools for Sustainable Organic Synthesis. Green Chem. 2014, 16, 2958–2975. [Google Scholar] [CrossRef]
- Sharma, U.; Sharma, N.; Vachhani, D.; Van der Eycken, E.V. Metal-mediated Post-Ugi Transformations for the Construction of Diverse Heterocyclic Scaffolds. Chem. Soc. Rev. 2015, 44, 1836–1860. [Google Scholar] [CrossRef]
- Zhang, J.S.; Liu, L.; Chen, T.Q.; Han, L.B. Recent Advances in Transition Metal-Catalyzed Three-Component Difunctionalization of Alkenes. Chem. Asian J. 2018, 13, 2277–2291. [Google Scholar] [CrossRef] [PubMed]
- Sharma, U.; Ranjan, P.; Van der Eycken, E.V.; You, S.L. Sequential and Direct Multicomponent Reaction (MCR)-Based Dearomatization Strategies. Chem. Soc. Rev. 2020, 49, 8721–8748. [Google Scholar] [CrossRef] [PubMed]
- John, S.; Gulati, S.; Shankaraiah, N. Recent Advances in Multi-Component Reactions and Their Mechanistic Insights: A Triennium Review. Org. Chem. Front. 2021, 8, 4237–4287. [Google Scholar] [CrossRef]
- Tobisu, M.; Chatani, N. Cross-Couplings Using Aryl Ethers via C−O Bond Activation Enabled by Nickel Catalysts. Acc. Chem. Res. 2015, 48, 1717–1726. [Google Scholar] [CrossRef] [PubMed]
- Bhakta, S.; Ghosh, T. Nickel-Catalyzed Cascade Reactions. Eur. J. Org. Chem. 2021, 29, 4201–4215. [Google Scholar] [CrossRef]
- Ichitsuka, T.; Fujita, T.; Ichikawa, J. Nickel-Catalyzed Allylic C(sp3)−F Bond Activation of Trifluoromethyl Groups via β-Fluorine Elimination: Synthesis of Difluoro-1,4-Dienes. ACS Catal. 2015, 5, 5947–5950. [Google Scholar] [CrossRef]
- Pellissier, H. Enantioselective Nickel-Catalyzed Domino and Tandem Processes. Curr. Org. Chem. 2016, 20, 234–265. [Google Scholar] [CrossRef]
- Chen, J.; Zhu, S.L. Nickel-Catalyzed Multicomponent Coupling: Synthesis of α-Chiral Ketones by Reductive Hydrocarbonylation of Alkenes. J. Am. Chem. Soc. 2021, 143, 14089–14096. [Google Scholar] [CrossRef]
- Ding, C.; Ren, Y.Y.; Yu, Y.; Yin, G.Y. Ligand-Modulated Nickel-Catalyzed Regioselective Silylalkylation of Alkenes. Nat. Commun. 2023, 14, 7670–7678. [Google Scholar] [CrossRef]
- Zhang, Y.R.; Wang, H.; Mao, Y.J.; Shi, S.L. Ni-Catalyzed Three-Component Coupling Reaction of Butadiene, Aldimines and Alkenylboronic Acids. Chin. J. Org. Chem. 2022, 42, 1198–1209. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, W.S.; Yang, S.N.; Wang, X.Y.; Liu, Y.; Ji, D.W.; Chen, Q.A. Nickel-Catalyzed Unsymmetrical Bis-Allylation of Alkynes. Angew. Chem. Int. Ed. 2023, 62, e202300036. [Google Scholar] [CrossRef]
- Wang, X.G.; Li, Y.; Liu, H.C.; Zhang, B.S.; Gou, X.Y.; Wang, Q.; Ma, J.W.; Liang, Y.M. Three-Component Ruthenium-Catalyzed Direct Meta-Selective C–H Activation of Arenes: A New Approach to The Alkylarylation of Alkenes. J. Am. Chem. Soc. 2019, 141, 13914–13922. [Google Scholar] [CrossRef]
- Zhang, Z.; Sabat, N.; Frison, G.; Marinetti, A.; Guinchard, X. Enantioselective Au(I)-Catalyzed Multicomponent Annulation via Tethered Counterion-Directed Catalysis. ACS Catal. 2022, 12, 4046–4053. [Google Scholar] [CrossRef]
- Pannilawithana, N.; Son, M.; Hwang, D.; Baik, M.H.; Yi, C. Scope and Mechanistic Studies on the Ruthenium-Catalyzed Multicomponent Deaminative C–H Coupling Reaction of Phenols with Aldehydes and Enamines for the Formation of Xanthene and Dioxacyclic Derivatives. ACS Catal. 2023, 13, 9051–9063. [Google Scholar] [CrossRef]
- Fu, R.; Lu, T.; Chen, F.L. Comparing Methods for Predicting the Reactive Site of Electrophilic Substitution. Acta Phys. Chim. Sin. 2014, 30, 628–639. [Google Scholar]
- Cao, J.J.; Ren, Q.; Chen, F.L.; Lu, T. Comparative Study on the Methods for Predicting the Reactive Site of Nucleophilic Reaction. Sci. China-Chem. 2015, 45, 1281–1290. [Google Scholar] [CrossRef]
- Ichitsuka, T.; Fujita, T.; Arita, T.; Ichikawa, J. Double C-F Bond Activation through β-Fluorine Elimination: Nickel-Mediated [3+2] Cycloaddition of 2-Trifluoromethyl-1-Alkenes with Alkynes. Angew. Chem. Int. Ed. 2014, 53, 7564–7568. [Google Scholar] [CrossRef]
- Bickelhaupt, F.; Houk, K. Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chem. Int. Ed. 2017, 56, 10070–10086. [Google Scholar] [CrossRef]
- Wolters, L.; Bickelhaupt, F. The Activation Strain Model and Molecular Orbital Theory. WIREs. Comput. Mol. Sci. 2015, 5, 324–343. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.W. Interaction Region Indicator: A Simple Real Space Function Clearly Revealing Both Chemical Bonds and Weak Interactions. Chem.-Methods 2021, 1, 231–239. [Google Scholar] [CrossRef]
- Morell, C.; Grand, A.; Toro-Labbé, A. New Dual Descriptor for Chemical Reactivity. J. Phys. Chem. A 2004, 109, 205–212. [Google Scholar] [CrossRef]
- Lessa, M.; Fajardo, J.; Delarmelina, M.; Carneiro, J. A DFT Study on the Mechanism for Polymerization of δ-Valerolactone Initiated by N-heterocyclic Carbene (NHC) Catalysts. Mol. Catal. 2021, 515, 111896–111916. [Google Scholar] [CrossRef]
- Kozuch, S.; Shaik, S. How to Conceptualize Catalytic Cycles? The Energetic Span Model. Acc. Chem. Res. 2011, 44, 101–110. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-Atom Fragments for Describing Molecular Charge Densities. Theor. Chem. Acc. 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Michael, B.; Reimann, C.; Pantazis, D.; Bredow, T.; Neese, F. Geometries of Third-Row Transition-Metal Complexes from Density-Functional Theory. J. Chem. Theory Comput. 2008, 4, 1449–1459. [Google Scholar]
- Steinmetz, M.; Grimme, S. Benchmark Study of the Performance of Density Functional Theory for Bond Activations with (Ni,Pd)-Based Transition-Metal Catalysts. ChemistryOpen 2013, 2, 115–124. [Google Scholar] [CrossRef]
- Dohm, S.; Hansen, A.; Steinmetz, M.; Grimme, S.; Checinski, M. Comprehensive Thermochemical Benchmark Set of Realistic Closed-Shell Metal Organic Reactions. J. Chem. Theory Comput. 2018, 14, 2596–2608. [Google Scholar] [CrossRef]
- Basiuk, V.; Escobar, A.; Molina, H. Basis Set Effects on B3LYP Geometries and Energies: Case Study of Interstellar Reaction HN=CH2 + •C≡N → H2N–C(•)H–C≡N. Int. J. Quantum Chem. 2002, 87, 101–109. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comp. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P. Efficient Diffuse Function-Augmented Basis Sets for Anion Calculations. III. The 3-21+G Basis Set for First-Row Elements, Li-F. J. Comp. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Petersson, G.A.; Bennett, A.; Tensfeldt, T.; Al-Laham, M.A.; Shirley, W.; Mantzaris, J. A Complete Basis Set Model Chemistry. I. The Total Energies of Closed-Shell Atoms and Hydrides of the First-Row Elements. J. Chem. Phys. 1988, 89, 2193–2218. [Google Scholar] [CrossRef]
- Petersson, G.A.; Al-Laham, M.A. A Complete Basis Set Model Chemistry. II. Open-Shell Systems and the Total Energies of the First-Eow Atoms. J. Chem. Phys. 1991, 94, 6081–6090. [Google Scholar] [CrossRef]
- Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. Energyadjusted Ab Initio Pseudopotentials for the First Row Transition Elements. J. Chem. Phys. 1987, 86, 866–872. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian Basis Sets for Molecular Calculations. I. Second Row Atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Selfconsistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A. Selfconsistent Molecular Orbital Methods 25. Supplementary Functions for Gaussian Basis Sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Fukui, K. Formulation of the Reaction Coordinate. J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar] [CrossRef]
- Fukui, K. The Path of Chemical Reactions—The IRC Approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Tao, J.Y.; Mu, W.H.; Chass, G.A.; Tang, T.H.; Fang, D.C. Balancing the Atomic Waistline: Isodensity-Based SCRF Radii for Main-Group Elements and Transition Metals. Int. J. Quantum Chem. 2013, 113, 975–984. [Google Scholar] [CrossRef]
- Fang, D.-C. THERMO Program; Beijing Normal University: Beijing, China, 2013. [Google Scholar]
- Lu, T.; Chen, F.W. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Weinhold, F. NBO 5.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2001. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Reaction Sites | Hirshfeld Charge | Δf | |
|---|---|---|---|---|
| R1 | 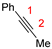 | C1 | −0.07888 | −0.05522 |
| C2 | −0.05603 | −0.04918 | ||
| R2 | 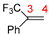 | C3 | −0.02576 | 0.04212 |
| C4 | −0.06044 | 0.05396 | ||
| R3 |  | C5 | −0.10885 | −0.18748 |
| C6 | −0.02911 | −0.13880 | ||
| R3′ | 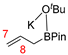 | C7 | −0.12664 | −0.09557 |
| C8 | −0.02076 | −0.01886 | ||
| R3″ |  | C9 | −0.25696 | −0.20117 |
| C10 | −0.08526 | −0.03908 | ||
| C11 | −0.24887 | −0.19177 | ||
| Entry | Substrate A | Substrate B | Compounds | ΔG (kcal·mol−1) |
|---|---|---|---|---|
| 1 | R2 | R1 | 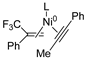 | −49.6 |
| 2 | R2 | R3 | 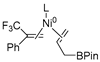 | −22.3 |
| 3 | R1 | R3 | 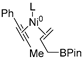 | −18.6 |
| 4 | R2 | R3′ | 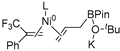 | −47.7 |
| 5 | R1 | R3′ |  | −44.3 |
| 6 | R2 | R3′ | 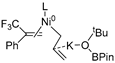 | −36.3 |
| 7 | R1 | R3″ | 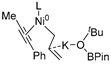 | −26.4 |
| TSs | ΔEdist | ΔEint | ΔΔE | |||
|---|---|---|---|---|---|---|
| Fragment 1 | Fragment 2 | Fragment 3 | Total ΔEdist | |||
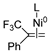 | 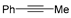 | 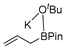 | ||||
| TS5-I | 87.5 | 2.1 | 40.4 | 130.0 | −138.0 | −8.0 |
| TS4-VII | 35.7 | −5.7 | 0.0 | 30.0 | −10.1 | 19.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, T.; Zhang, J.; Liu, G.; Duan, L.; Tian, K.V.; Chass, G.A.; Mu, W. Nickel-Catalyzed Three-Component Unsymmetrical Bis-Allylation of Alkynes with Alkenes: A Density Functional Theory Study. Molecules 2024, 29, 1475. https://doi.org/10.3390/molecules29071475
Yu T, Zhang J, Liu G, Duan L, Tian KV, Chass GA, Mu W. Nickel-Catalyzed Three-Component Unsymmetrical Bis-Allylation of Alkynes with Alkenes: A Density Functional Theory Study. Molecules. 2024; 29(7):1475. https://doi.org/10.3390/molecules29071475
Chicago/Turabian StyleYu, Tao, Jingxuan Zhang, Guo Liu, Liangfei Duan, Kun V. Tian, Gregory A. Chass, and Weihua Mu. 2024. "Nickel-Catalyzed Three-Component Unsymmetrical Bis-Allylation of Alkynes with Alkenes: A Density Functional Theory Study" Molecules 29, no. 7: 1475. https://doi.org/10.3390/molecules29071475
APA StyleYu, T., Zhang, J., Liu, G., Duan, L., Tian, K. V., Chass, G. A., & Mu, W. (2024). Nickel-Catalyzed Three-Component Unsymmetrical Bis-Allylation of Alkynes with Alkenes: A Density Functional Theory Study. Molecules, 29(7), 1475. https://doi.org/10.3390/molecules29071475