
Green Aromatic Epoxidation with an Iron Porphyrin Catalyst for One-Pot Functionalization of Renewable Xylene, Quinoline, and Acridine



Abstract
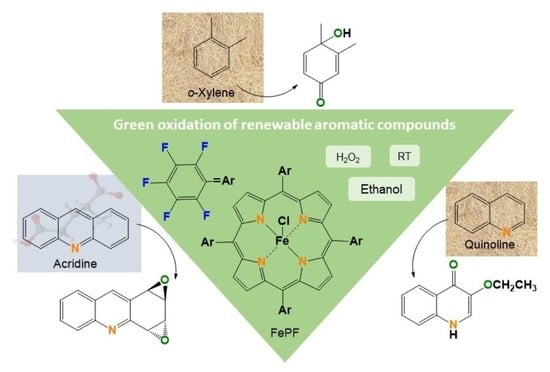
1. Introduction
2. Results and Discussion
2.1. o-Xylene (1)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Entry | [FeP] (mol%) | Time (min) | Conversion (%) c | Selectivity (%) c | TON d | |
| H2O2 (eq.) b | 1a | 1b | ||||
| 1 | 0.3 | 90 3 eq. | 30 | 14 | 86 | 178 |
| 2 | 0.6 | 150 5 eq. | 80 | 14 | 86 | 237 |
2.2. Quinoline (2)
2.3. Acridine (3)
2.4. Catalyst Stability
2.5. Considerations on the Mechanism
3. Materials and Methods
3.1. Materials
3.2. Instrumentation
3.3. Catalytic Oxidation Reactions
3.4. Spectroscopic Data of Products
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, X.; Groves, J.T. Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins. Chem. Rev. 2018, 118, 2491–2553. [Google Scholar] [CrossRef] [PubMed]
- Calvete, M.J.F.; Piñeiro, M.; Dias, L.D.; Pereira, M.M. Hydrogen Peroxide and Metalloporphyrins in Oxidation Catalysis: Old Dogs with Some New Tricks. ChemCatChem 2018, 10, 3615–3635. [Google Scholar] [CrossRef]
- Rebelo, S.L.H.; Moniz, T.; Medforth, C.J.; de Castro, B.; Rangel, M. EPR spin trapping studies of H2O2 activation in metaloporphyrin catalyzed oxygenation reactions: Insights on the biomimetic mechanism. Mol. Catal. 2019, 475, 110500. [Google Scholar] [CrossRef]
- Lipińska, M.E.; Rebelo, S.L.H.; Pereira, M.F.R.; Figueiredo, J.L.; Freire, C. Photoactive Zn(II)Porphyrin–multi-walled carbon nanotubes nanohybrids through covalent β-linkages. Mater. Chem. Phys. 2013, 143, 296–304. [Google Scholar] [CrossRef]
- Shimada, T.; Fujii-Kuriyama, Y. Metabolic activation of polycyclic aromatic hydrocarbons to carcinogens by cytochromes P450 1A1 and 1B1. Cancer Sci. 2004, 95, 1–6. [Google Scholar] [CrossRef]
- Haynes, J.P.; Miller, K.E.; Majestic, B.J. Investigation into Photoinduced Auto-Oxidation of Polycyclic Aromatic Hydrocarbons Resulting in Brown Carbon Production. Environ. Sci. Technol. 2019, 53, 682–691. [Google Scholar] [CrossRef] [PubMed]
- Estrada, A.C.; Simões, M.M.Q.; Santos, I.C.M.S.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S.; Cavaleiro, A.M.V. Oxidation of Polycyclic Aromatic Hydrocarbons with Hydrogen Peroxide in the Presence of Transition Metal Mono-Substituted Keggin-Type Polyoxometalates. ChemCatChem 2011, 3, 771–779. [Google Scholar] [CrossRef]
- Sorokin, A.; Meunier, B. Oxidation of Polycyclic Aromatic Hydrocarbons Catalyzed by Iron Tetrasulfophthalocyanine FePcS: Inverse Isotope Effects and Oxygen Labeling Studies. Eur. J. Inorg. Chem. 1998, 1998, 1269–1281. [Google Scholar] [CrossRef]
- Giri, N.G.; Chauhan, S.M.S. Oxidation of polycyclic aromatic hydrocarbons with hydrogen peroxide catalyzed by Iron(III)porphyrins. Catal. Commun. 2009, 10, 383–387. [Google Scholar] [CrossRef]
- Rebelo, S.L.H.; Simões, M.M.Q.; Neves, M.G.P.M.S.; Silva, A.M.S.; Cavaleiro, J.A.S. An efficient approach for aromatic epoxidation using hydrogen peroxide and Mn(iii) porphyrins. Chem. Commun. 2004, 5, 608–609. [Google Scholar] [CrossRef]
- Costa, P.; Linhares, M.; Rebelo, S.L.H.; Neves, M.G.P.M.S.; Freire, C. Direct access to polycyclic peripheral diepoxy-meso-quinone derivatives from acene catalytic oxidation. RSC Adv. 2013, 3, 5350–5353. [Google Scholar] [CrossRef]
- Linhares, M.; Rebelo, S.L.; Biernacki, K.; Magalhães, A.L.; Freire, C. Biomimetic one-pot route to acridine epoxides. J. Org. Chem. 2015, 80, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Rebelo, S.L.H.; Pires, S.M.G.; Simões, M.M.Q.; de Castro, B.; Neves, M.G.P.M.S.; Medforth, C.J. Biomimetic Oxidation of Benzofurans with Hydrogen Peroxide Catalyzed by Mn(III) Porphyrins. Catalysts 2020, 10, 62. [Google Scholar] [CrossRef]
- Rebelo, S.L.H.; Simões, M.M.Q.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Oxidation of alkylaromatics with hydrogen peroxide catalysed by manganese(III) porphyrins in the presence of ammonium acetate. J. Mol. Catal. A Chem. 2003, 201, 9–22. [Google Scholar] [CrossRef]
- Dutta, S.; Bhat, N.S.; Anchan, H.N. Nanocatalysis for Renewable Aromatics. In Heterogeneous Nanocatalysis for Energy and Environmental Sustainability; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2022; pp. 61–90. [Google Scholar]
- Wollensack, L.; Budzinski, K.; Backmann, J. Defossilization of pharmaceutical manufacturing. Curr. Opin. Green Sustain. Chem. 2022, 33, 100586. [Google Scholar] [CrossRef]
- Niziolek, A.M.; Onel, O.; Guzman, Y.A.; Floudas, C.A. Biomass-Based Production of Benzene, Toluene, and Xylenes via Methanol: Process Synthesis and Deterministic Global Optimization. Energy Fuels 2016, 30, 4970–4998. [Google Scholar] [CrossRef]
- Wang, S.; Li, Z.; Yi, W.; Fu, P.; Zhang, A.; Bai, X. Renewable aromatic hydrocarbons production from catalytic pyrolysis of lignin with Al-SBA-15 and HZSM-5: Synergistic effect and coke behaviour. Renew. Energy 2021, 163, 1673–1681. [Google Scholar] [CrossRef]
- Genuino, H.C.; Muizebelt, I.; Heeres, A.; Schenk, N.J.; Winkelman, J.G.M.; Heeres, H.J. An improved catalytic pyrolysis concept for renewable aromatics from biomass involving a recycling strategy for co-produced polycyclic aromatic hydrocarbons. Green Chem. 2019, 21, 3802–3806. [Google Scholar] [CrossRef]
- Gancedo, J.; Faba, L.; Ordóñez, S. From Biomass to Green Aromatics: Direct Upgrading of Furfural–Ethanol Mixtures. ACS Sustain. Chem. Eng. 2022, 10, 7752–7758. [Google Scholar] [CrossRef]
- Zeng, Y.; Wang, Y.; Liu, Y.; Dai, L.; Wu, Q.; Xia, M.; Zhang, S.; Ke, L.; Zou, R.; Ruan, R. Microwave catalytic co-pyrolysis of waste cooking oil and low-density polyethylene to produce monocyclic aromatic hydrocarbons: Effect of different catalysts and pyrolysis parameters. Sci. Total Environ. 2022, 809, 152182. [Google Scholar] [CrossRef]
- Wellmann, A.; Grazia, L.; Bermejo-Deval, R.; Sanchez-Sanchez, M.; Lercher, J.A. Effect of promoters on o-xylene oxidation pathways reveals nature of selective sites on TiO2 supported vanadia. J. Catal. 2022, 408, 330–338. [Google Scholar] [CrossRef]
- Mei, J.; Shen, Y.; Wang, Q.; Shen, Y.; Li, W.; Zhao, J.; Chen, J.; Zhang, S. Roles of Oxygen Species in Low-Temperature Catalytic o-Xylene Oxidation on MOF-Derived Bouquetlike CeO2. ACS Appl. Mater. Interfaces 2022, 14, 35694–35703. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.-P.; Deng, W.-H.; Wu, Y.; Liao, R.-Z. QM/MM Calculations Suggest Concerted O–O Bond Cleavage and Substrate Oxidation by Nonheme Diiron Toluene/o-Xylene Monooxygenase. Chem. Asian J. 2022, 17, e202200490. [Google Scholar] [CrossRef]
- Batista, V.F.; Pinto, D.C.G.A.; Silva, A.M.S. Synthesis of Quinolines: A Green Perspective. ACS Sustain. Chem. Eng. 2016, 4, 4064–4078. [Google Scholar] [CrossRef]
- Nasseri, M.A.; Zakerinasab, B.; Kamayestani, S. Proficient Procedure for Preparation of Quinoline Derivatives Catalyzed by NbCl5 in Glycerol as Green Solvent. J. Appl. Chem. 2015, 2015, 743094. [Google Scholar] [CrossRef]
- Ortiz-Cervantes, C.; Flores-Alamo, M.; García, J.J. Synthesis of pyrrolidones and quinolines from the known biomass feedstock levulinic acid and amines. Tetrahedron Lett. 2016, 57, 766–771. [Google Scholar] [CrossRef]
- Jiao, Z.; Zhang, X.; Gong, H.; He, D.; Yin, H.; Liu, Y.; Gao, X. CuO-doped Ce for catalytic wet peroxide oxidation degradation of quinoline wastewater under wide pH conditions. J. Ind. Eng. Chem. 2022, 105, 49–57. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, L.; Mou, X.; Chen, Y. Enzymatic approaches to site-selective oxidation of quinoline and derivatives. Org. Biomol. Chem. 2022, 20, 2580–2600. [Google Scholar] [CrossRef]
- Sharma, R.K.; Chan, W.G.; Hajaligol, M.R. Product compositions from pyrolysis of some aliphatic α-amino acids. J. Anal. Appl. Pyrolysis 2006, 75, 69–81. [Google Scholar] [CrossRef]
- Rebelo, S.L.; Silva, A.M.; Medforth, C.J.; Freire, C. Iron(III) Fluorinated Porphyrins: Greener Chemistry from Synthesis to Oxidative Catalysis Reactions. Molecules 2016, 21, 481. [Google Scholar] [CrossRef]
- Rebelo, S.L.H.; Pereira, M.M.; Simões, M.M.Q.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Mechanistic studies on metalloporphyrin epoxidation reactions with hydrogen peroxide: Evidence for two active oxidative species. J. Catal. 2005, 234, 76–87. [Google Scholar] [CrossRef]
- Rebelo, S.L.H.; Pires, S.M.G.; Simões, M.M.Q.; Medforth, C.J.; Cavaleiro, J.A.S.; Neves, M.G.P.M.S. A Green and Versatile Route to Highly Functionalized Benzofuran Derivatives Using Biomimetic Oxygenation. ChemistrySelect 2018, 3, 1392–1403. [Google Scholar] [CrossRef]
- Dalton, T.; Faber, T.; Glorius, F. C–H Activation: Toward Sustainability and Applications. ACS Cent. Sci. 2021, 7, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Kozuch, S.; Martin, J.M.L. “Turning Over” Definitions in Catalytic Cycles. ACS Catal. 2012, 2, 2787–2794. [Google Scholar] [CrossRef]
- Kurganova, E.A.; Frolov, A.S.; Koshel’, G.N.; Nesterova, T.N.; Shakun, V.A.; Mazurin, O.A. A Hydroperoxide Method for 3,4-Xylenol Synthesis. Pet. Chem. 2018, 58, 451–456. [Google Scholar] [CrossRef]
- Zalibera, Ľ.; Milata, V.; Ilavský, D. 1H and 13C NMR spectra of 3-substituted 4-quinolones. Magn. Reson. Chem. 1998, 36, 681–684. [Google Scholar] [CrossRef]
- Bergman, J.J.; Chandler, W.D. A Study of the Barriers to Rotation in Some Highly Substituted Diphenyl Ethers. Can. J. Chem. 1972, 50, 353–363. [Google Scholar] [CrossRef]
- Eddahmi, M.; Moura, N.M.M.; Bouissane, L.; Gamouh, A.; Faustino, M.A.F.; Cavaleiro, J.A.S.; Paz, F.A.A.; Mendes, R.F.; Lodeiro, C.; Santos, S.M.; et al. New nitroindazolylacetonitriles: Efficient synthetic access via vicarious nucleophilic substitution and tautomeric switching mediated by anions. New J. Chem. 2019, 43, 14355–14367. [Google Scholar] [CrossRef]
- Martins, M.B.M.S.; Corrêa, G.A.; Moniz, T.; Medforth, C.J.; de Castro, B.; Rebelo, S.L.H. Nanostructured binuclear Fe(III) and Mn(III) porphyrin materials: Tuning the mimics of catalase and peroxidase activity. J. Catal. 2023, 419, 125–136. [Google Scholar] [CrossRef]
- Rocha, M.; Rebelo, S.L.H.; Freire, C. Enantioselective arene epoxidation under mild conditions by Jacobsen catalyst: The role of protic solvent and co-catalyst in the activation of hydrogen peroxide. Appl. Catal. A Gen. 2013, 460–461, 116–123. [Google Scholar] [CrossRef]









 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | [FeP] (mol%) | Conversion (%) | Selectivity (Yield) (%) | TON h | |||
| 2a | 2b d | 2c d | Other | ||||
| 1 | 0.9 | 57 e,f | 100 (57) g | - | - | - | 61 |
| 2 | 1.3 | 53 f,g | - | 46 (24) g | 22 (12) g | 27 (14) g | - |
| 3 | 1.9 | 70 e | - | - | - | - | 38 |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | [FeP] (mol%) | Additive | Conversion (%) c | Selectivity (%) c | TON d | |
| 3a | 3b | |||||
| 1 | 0.6 | None | 94 | 10 | 90 | 285 |
| 2 | 0.6 | HNO3 (pH 4.6) | 89 | 16 | 84 | 261 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corrêa, G.A.; Rebelo, S.L.H.; de Castro, B. Green Aromatic Epoxidation with an Iron Porphyrin Catalyst for One-Pot Functionalization of Renewable Xylene, Quinoline, and Acridine. Molecules 2023, 28, 3940. https://doi.org/10.3390/molecules28093940
Corrêa GA, Rebelo SLH, de Castro B. Green Aromatic Epoxidation with an Iron Porphyrin Catalyst for One-Pot Functionalization of Renewable Xylene, Quinoline, and Acridine. Molecules. 2023; 28(9):3940. https://doi.org/10.3390/molecules28093940
Chicago/Turabian StyleCorrêa, Gabriela A., Susana L. H. Rebelo, and Baltazar de Castro. 2023. "Green Aromatic Epoxidation with an Iron Porphyrin Catalyst for One-Pot Functionalization of Renewable Xylene, Quinoline, and Acridine" Molecules 28, no. 9: 3940. https://doi.org/10.3390/molecules28093940
APA StyleCorrêa, G. A., Rebelo, S. L. H., & de Castro, B. (2023). Green Aromatic Epoxidation with an Iron Porphyrin Catalyst for One-Pot Functionalization of Renewable Xylene, Quinoline, and Acridine. Molecules, 28(9), 3940. https://doi.org/10.3390/molecules28093940