
Graphene as Nanocarrier for Gold(I)-Monocarbene Complexes: Strength and Nature of Physisorption



Abstract
1. Introduction
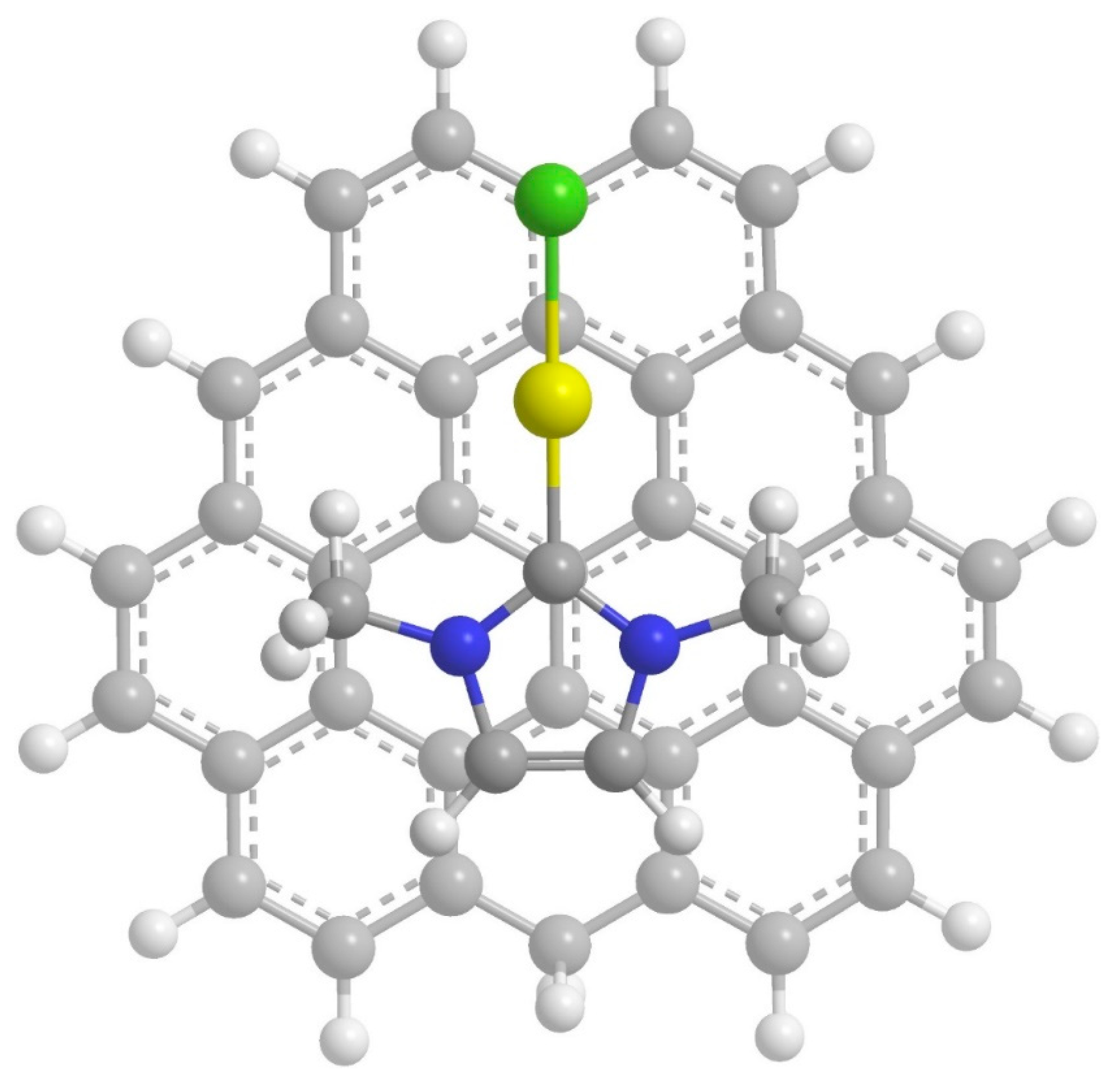
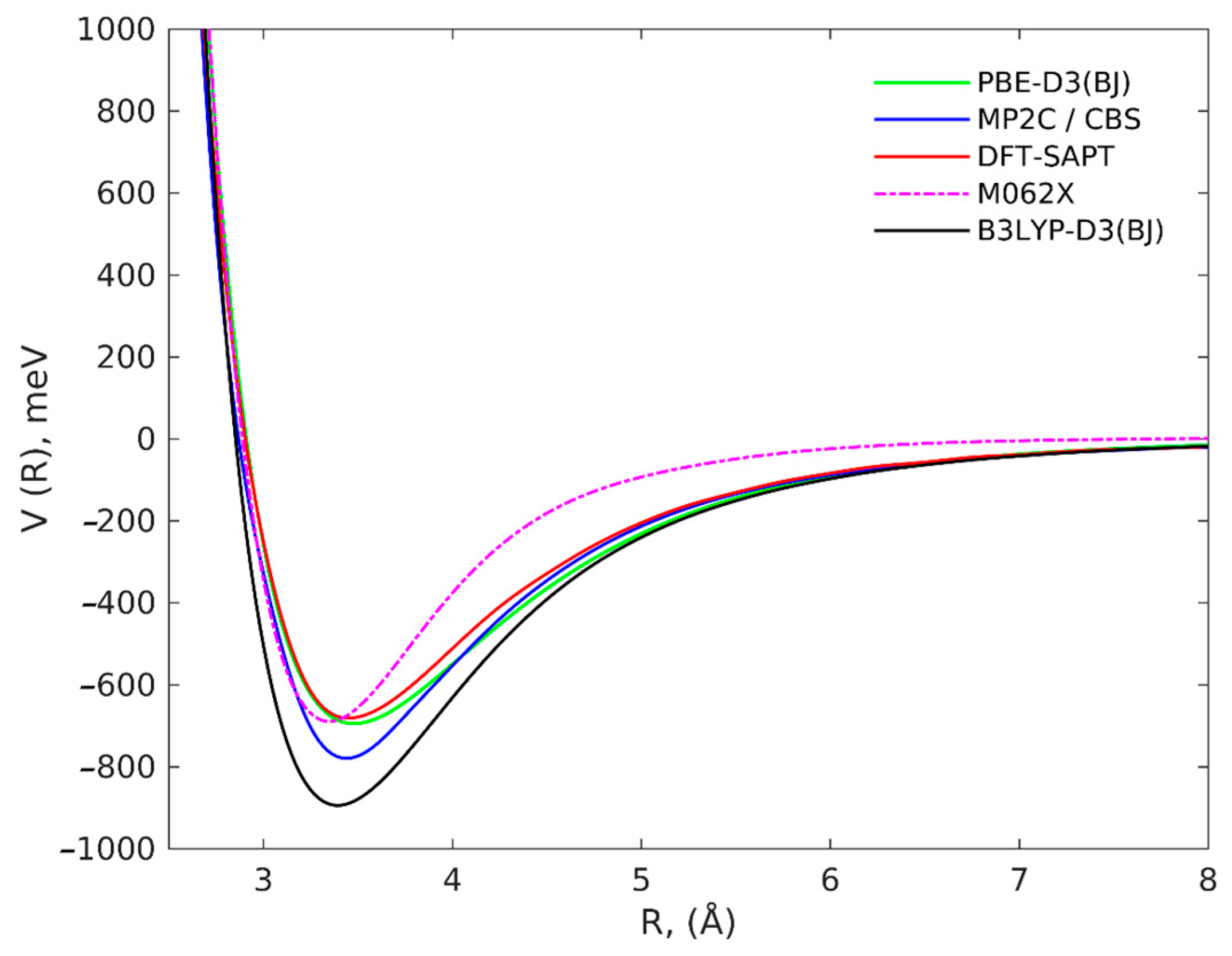
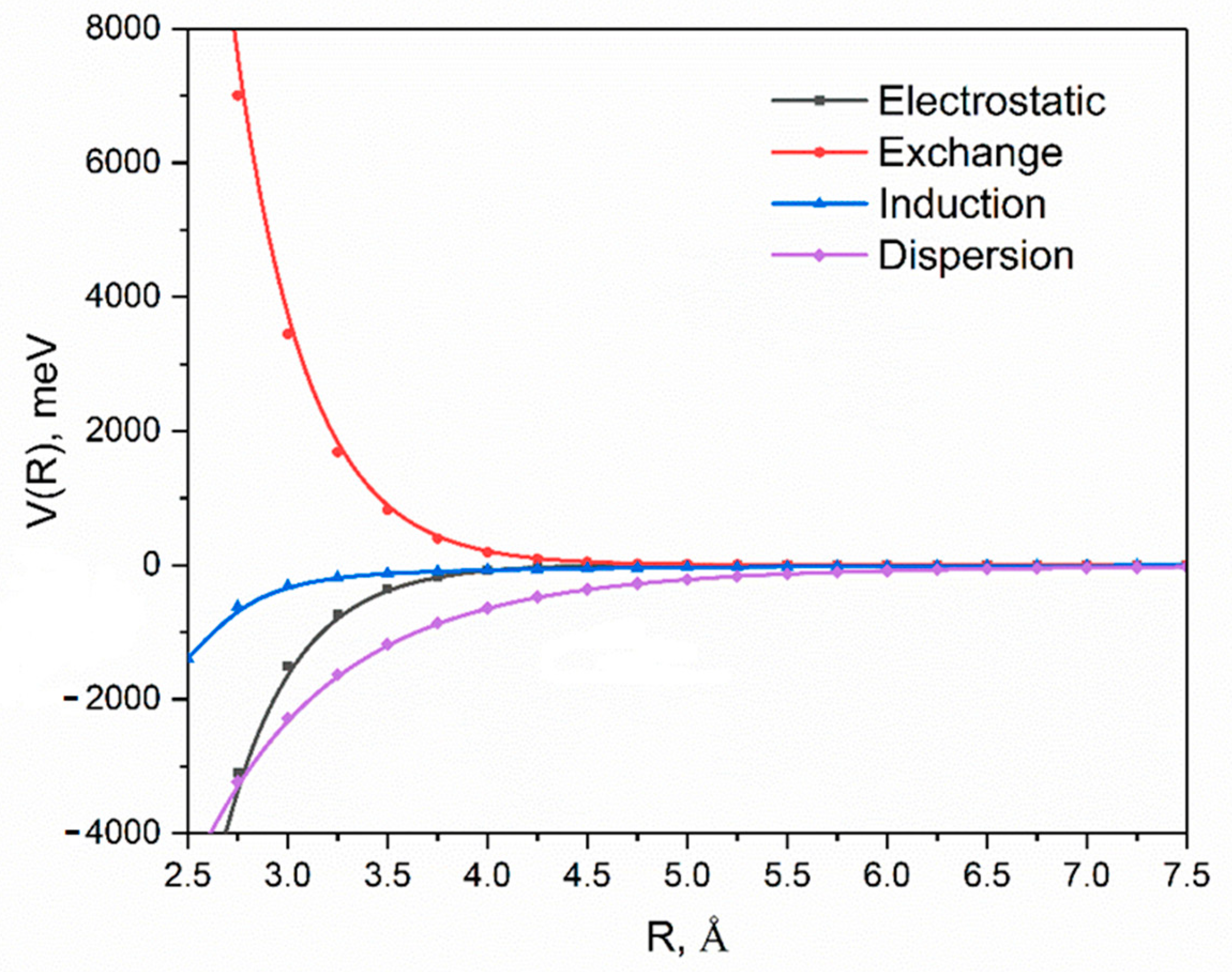
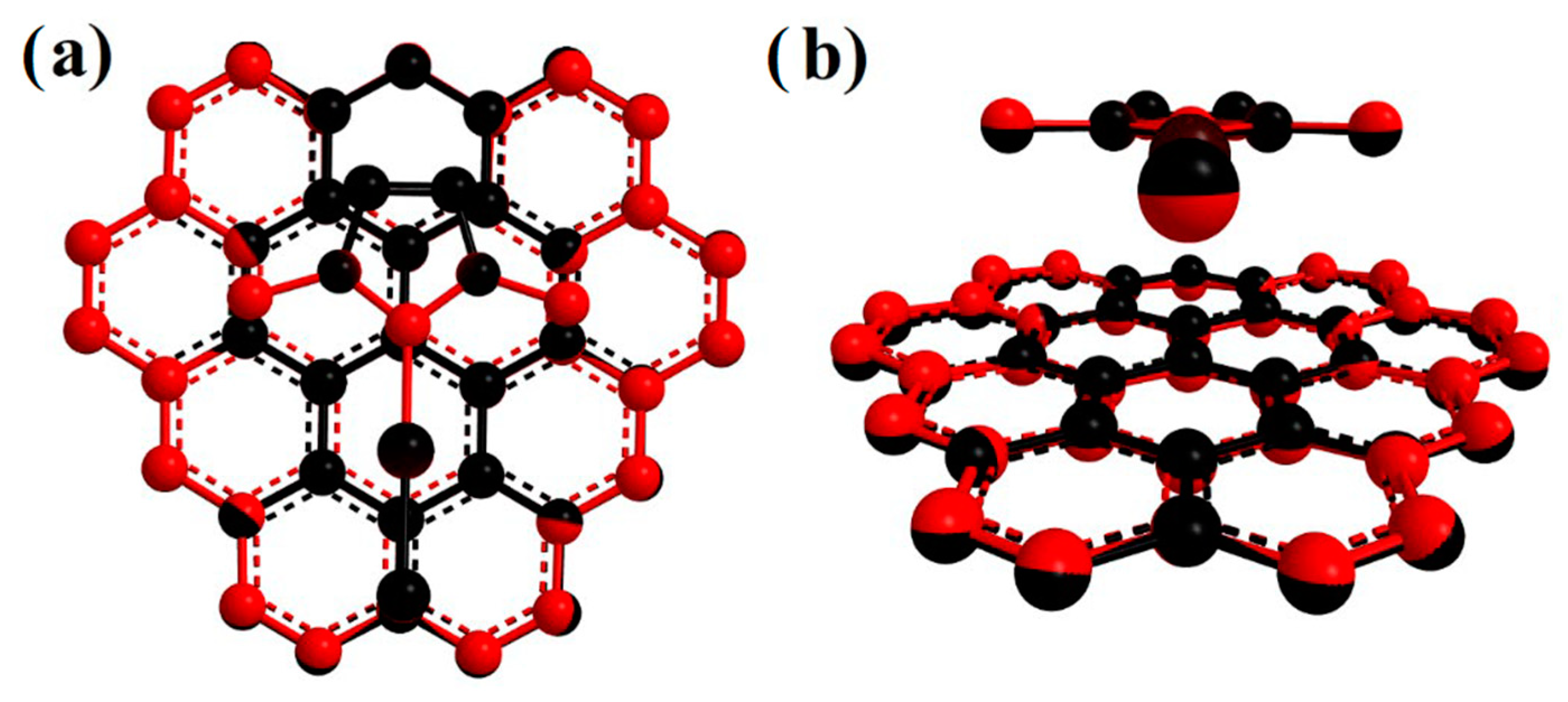
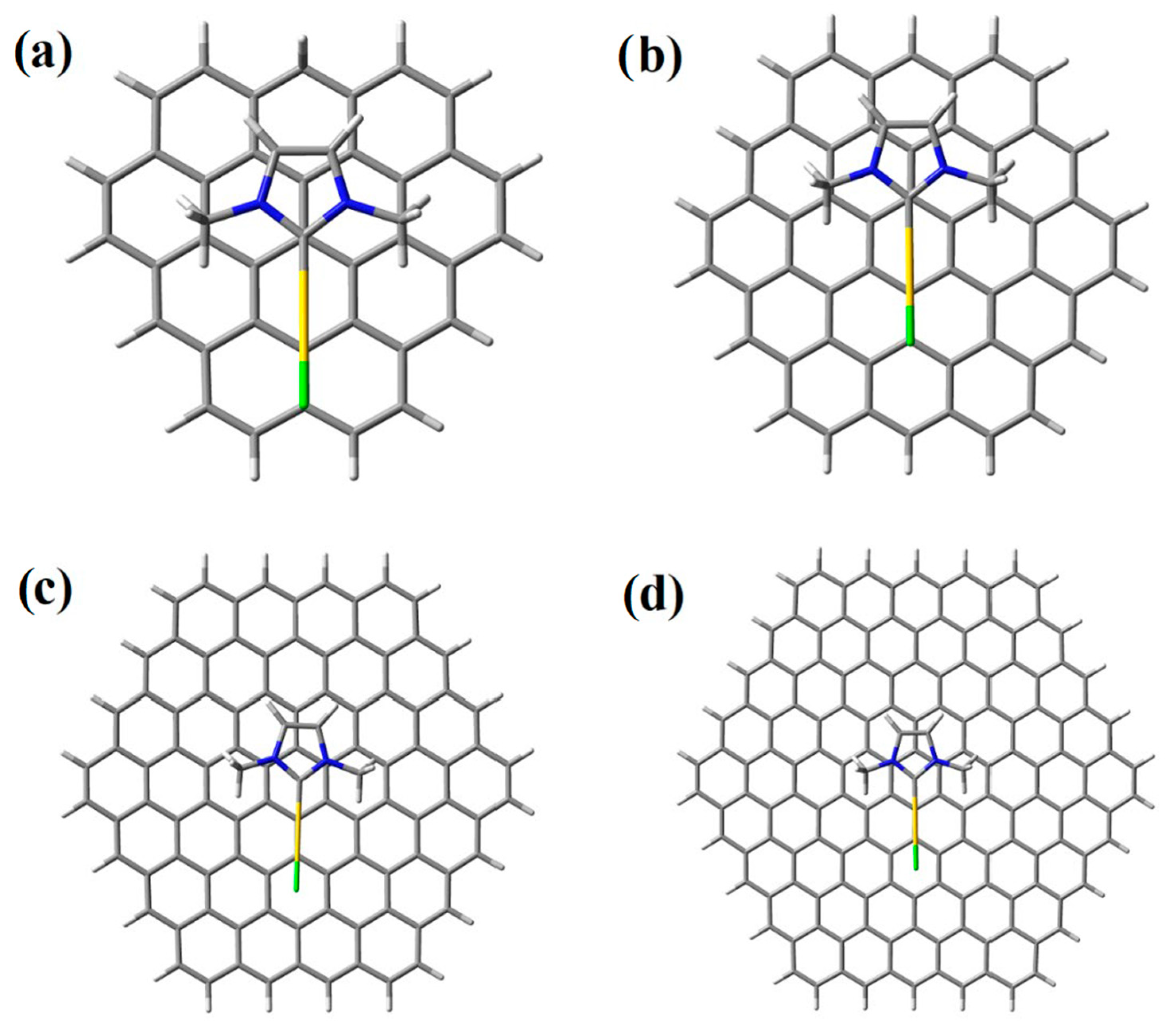
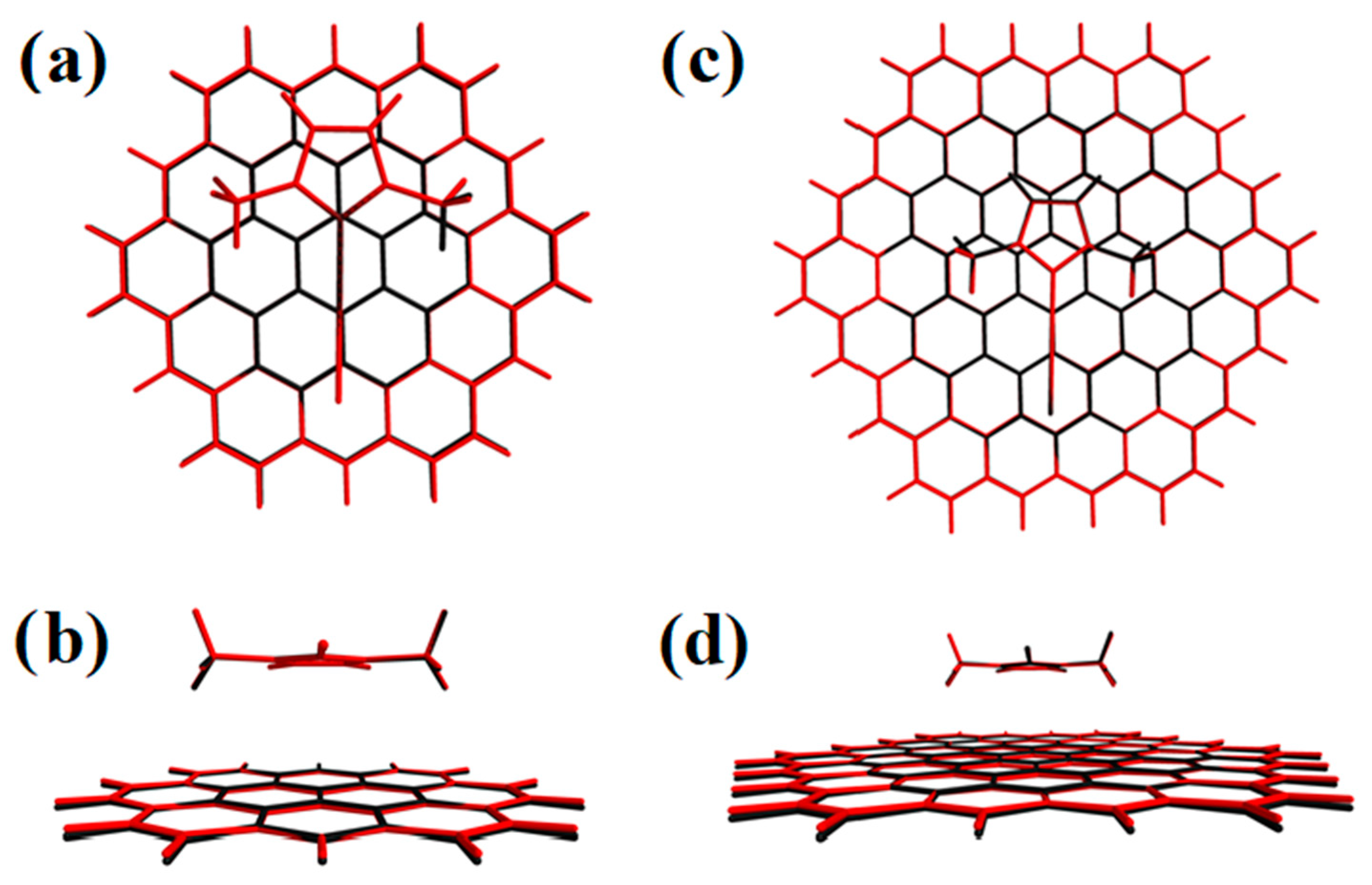
2. Results and Discussion
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Velazquez, H.D.; Verpoort, F. N-Heterocyclic Carbene Transition Metal Complexes for Catalysis in Aqueous Media. Chem. Soc. Rev. 2012, 41, 7032. [Google Scholar] [CrossRef] [PubMed]
- Ott, I. On the Medicinal Chemistry of Gold Complexes as Anticancer Drugs. Coord. Chem. Rev. 2009, 253, 1670–1681. [Google Scholar] [CrossRef]
- Glišić, B.Đ.; Djuran, M.I. Gold Complexes as Antimicrobial Agents: An Overview of Different Biological Activities in Relation to the Oxidation State of the Gold Ion and the Ligand Structure. Dalt. Trans. 2014, 43, 5950–5969. [Google Scholar] [CrossRef]
- Messori, L.; Marcon, G. Gold Complexes in the Treatment of Rheumatoid Arthritis. Met. Ions Biol. Syst. 2004, 41, 279–304. [Google Scholar]
- Sullivan, M.P.; Holtkamp, H.U.; Hartinger, C.G. Antitumor metallodrugs that target proteins. In Metallo-Drugs: Development and Action of Anticancer Agents; De Gruyter: Vienna, Austria, 2018; pp. 351–386. [Google Scholar]
- Yang, Z.; Jiang, G.; Xu, Z.; Zhao, S.; Liu, W. Advances in Alkynyl Gold Complexes for Use as Potential Anticancer Agents. Coord. Chem. Rev. 2020, 423, 213492. [Google Scholar] [CrossRef]
- Messori, L.; Marchetti, L.; Massai, L.; Scaletti, F.; Guerri, A.; Landini, I.; Nobili, S.; Perrone, G.; Mini, E.; Leoni, P.; et al. Chemistry and Biology of Two Novel Gold(I) Carbene Complexes as Prospective Anticancer Agents. Inorg. Chem. 2014, 53, 2396–2403. [Google Scholar] [CrossRef] [PubMed]
- Oberkofler, J.; Aikman, B.; Bonsignore, R.; Pöthig, A.; Platts, J.; Casini, A.; Kühn, F.E. Exploring the Reactivity and Biological Effects of Heteroleptic N-Heterocyclic Carbene Gold(I)-Alkynyl Complexes. Eur. J. Inorg. Chem. 2020, 2020, 1040–1051. [Google Scholar] [CrossRef]
- Arcau, J.; Andermark, V.; Rodrigues, M.; Giannicchi, I.; Pérez-Garcia, L.; Ott, I.; Rodríguez, L. Synthesis and Biological Activity of Gold(I) N-Heterocyclic Carbene Complexes with Long Aliphatic Side Chains. Eur. J. Inorg. Chem. 2014, 2014, 6117–6125. [Google Scholar] [CrossRef]
- Tolbatov, I.; Marrone, A.; Coletti, C.; Re, N. Computational Studies of Au(I) and Au(III) Anticancer MetalLodrugs: A Survey. Molecules 2021, 26, 7600. [Google Scholar] [CrossRef]
- Messori, L.; Scaletti, F.; Massai, L.; Cinellu, M.A.; Gabbiani, C.; Vergara, A.; Merlino, A. The Mode of Action of Anticancer Gold-Based Drugs: A Structural Perspective. Chem. Commun. 2013, 49, 10100. [Google Scholar] [CrossRef]
- Tolbatov, I.; Coletti, C.; Marrone, A.; Re, N. Reactivity of Gold(I) Monocarbene Complexes with Protein Targets: A Theoretical Study. Int. J. Mol. Sci. 2019, 20, 820. [Google Scholar] [CrossRef]
- Hickey, J.L.; Ruhayel, R.A.; Barnard, P.J.; Baker, M.V.; Berners-Price, S.J.; Filipovska, A. Mitochondria-Targeted Chemotherapeutics: The Rational Design of Gold(I) N-Heterocyclic Carbene Complexes That Are Selectively Toxic to Cancer Cells and Target Protein Selenols in Preference to Thiols. J. Am. Chem. Soc. 2008, 130, 12570–12571. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, H.F.; Vieira, M.A.; Sánchez Delgado, G.Y.; Paschoal, D. Ligand Exchange Reaction of Au(I) R-N-Heterocyclic Carbene Complexes with Cysteine. J. Phys. Chem. A 2016, 120, 2250–2259. [Google Scholar] [CrossRef]
- Tolbatov, I.; Coletti, C.; Marrone, A.; Re, N. Insight into the Substitution Mechanism of Antitumor Au(I) N-Heterocyclic Carbene Complexes by Cysteine and Selenocysteine. Inorg. Chem. 2020, 59, 3312–3320. [Google Scholar] [CrossRef] [PubMed]
- Tolbatov, I.; Marzo, T.; Coletti, C.; La Mendola, D.; Storchi, L.; Re, N.; Marrone, A. Reactivity of Antitumor Coinage Metal-Based N-Heterocyclic Carbene Complexes with Cysteine and Selenocysteine Protein Sites. J. Inorg. Biochem. 2021, 223, 111533. [Google Scholar] [CrossRef]
- Schuh, E.; Pflüger, C.; Citta, A.; Folda, A.; Rigobello, M.P.; Bindoli, A.; Casini, A.; Mohr, F. Gold(I) Carbene Complexes Causing Thioredoxin 1 and Thioredoxin 2 Oxidation as Potential Anticancer Agents. J. Med. Chem. 2012, 55, 5518–5528. [Google Scholar] [CrossRef] [PubMed]
- Arambula, J.F.; McCall, R.; Sidoran, K.J.; Magda, D.; Mitchell, N.A.; Bielawski, C.W.; Lynch, V.M.; Sessler, J.L.; Arumugam, K. Targeting Antioxidant Pathways with Ferrocenylated N-Heterocyclic Carbene Supported Gold(I) Complexes in A549 Lung Cancer Cells. Chem. Sci. 2016, 7, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Pratesi, A.; Gabbiani, C.; Michelucci, E.; Ginanneschi, M.; Papini, A.M.; Rubbiani, R.; Ott, I.; Messori, L. Insights on the Mechanism of Thioredoxin Reductase Inhibition by Gold N-Heterocyclic Carbene Compounds Using the Synthetic Linear Selenocysteine Containing C-Terminal Peptide HTrxR(488-499): An ESI-MS Investigation. J. Inorg. Biochem. 2014, 136, 161–169. [Google Scholar] [CrossRef]
- Bertrand, B.; de Almeida, A.; van der Burgt, E.P.M.; Picquet, M.; Citta, A.; Folda, A.; Rigobello, M.P.; Le Gendre, P.; Bodio, E.; Casini, A. New Gold(I) Organometallic Compounds with Biological Activity in Cancer Cells. Eur. J. Inorg. Chem. 2014, 2014, 4532–4536. [Google Scholar] [CrossRef]
- Oehninger, L.; Rubbiani, R.; Ott, I. N-Heterocyclic Carbene Metal Complexes in Medicinal Chemistry. Dalt. Trans. 2013, 42, 3269–3284. [Google Scholar] [CrossRef]
- Duan, C.; Townley, H.E. Nanoparticles as Vectors to Tackle Cancer. Biomolecules 2021, 11, 1729. [Google Scholar] [CrossRef]
- Wang, Y.; Li, J.; Li, X.; Shi, J.; Jiang, Z.; Zhang, C.Y. Graphene-Based Nanomaterials for Cancer Therapy and Anti-Infections. Bioact. Mater. 2022, 14, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Magne, T.M.; de Oliveira Vieira, T.; Alencar, L.M.R.; Junior, F.F.M.; Gemini-Piperni, S.; Carneiro, S.V.; Fechine, L.M.U.D.; Freire, R.M.; Golokhvast, K.; Metrangolo, P.; et al. Graphene and Its Derivatives: Understanding the Main Chemical and Medicinal Chemistry Roles for Biomedical Applications. J. Nanostruct. Chem. 2022, 12, 693–727. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, S.M.; Hashemi, S.A.; Kalashgrani, M.Y.; Omidifar, N.; Bahrani, S.; Vijayakameswara Rao, N.; Babapoor, A.; Gholami, A.; Chiang, W.-H. Bioactive Graphene Quantum Dots Based Polymer Composite for Biomedical Applications. Polymers 2022, 14, 617. [Google Scholar] [CrossRef]
- Liu, J.; Cui, L.; Losic, D. Graphene and Graphene Oxide as New Nanocarriers for Drug Delivery Applications. Acta Biomater. 2013, 9, 9243–9257. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xia, J.; Zhao, Q.; Liu, L.; Zhang, Z. Functional Graphene Oxide as a Nanocarrier for Controlled Loading and Targeted Delivery of Mixed Anticancer Drugs. Small 2010, 6, 537–544. [Google Scholar] [CrossRef]
- Sui, X.; Luo, C.; Wang, C.; Zhang, F.; Zhang, J.; Guo, S. Graphene Quantum Dots Enhance Anticancer Activity of Cisplatin via Increasing Its Cellular and Nuclear Uptake. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 1997–2006. [Google Scholar] [CrossRef]
- Tian, L.; Pei, X.; Zeng, Y.; He, R.; Li, Z.; Wang, J.; Wan, Q.; Li, X. Functionalized Nanoscale Graphene Oxide for High Efficient Drug Delivery of Cisplatin. J. Nanopart. Res. 2014, 16, 2709. [Google Scholar] [CrossRef]
- Augustine, S.; Prabhakar, B.; Shende, P. Adsorption of Cisplatin on Oxidized Graphene Nanoribbons for Improving the Uptake in Non-Small Cell Lung Carcinoma Cell Line A549. Curr. Drug Deliv. 2022, 19, 697–705. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, X.; Liu, Z.; Ma, Y.; Huang, Y.; Chen, Y. High-Efficiency Loading and Controlled Release of Doxorubicin Hydrochloride on Graphene Oxide. J. Phys. Chem. C 2008, 112, 17554–17558. [Google Scholar] [CrossRef]
- Rosli, N.F.; Fojtů, M.; Fisher, A.C.; Pumera, M. Graphene Oxide Nanoplatelets Potentiate Anticancer Effect of Cisplatin in Human Lung Cancer Cells. Langmuir 2019, 35, 3176–3182. [Google Scholar] [CrossRef]
- Cuevas-Flores, M.D.R.; Garcia-Revilla, M.A.; Bartolomei, M. Noncovalent Interactions between Cisplatin and Graphene Prototypes. J. Comput. Chem. 2018, 39, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Flores, M.D.R.; Bartolomei, M.; García-Revilla, M.A.; Coletti, C. Interaction and Reactivity of Cisplatin Physisorbed on Graphene Oxide Nano-Prototypes. Nanomaterials 2020, 10, 1074. [Google Scholar] [CrossRef]
- Abdel-Bary, A.S.; Tolan, D.A.; Nassar, M.Y.; Taketsugu, T.; El-Nahas, A.M. Chitosan, Magnetite, Silicon Dioxide, and Graphene Oxide Nanocomposites: Synthesis, Characterization, Efficiency as Cisplatin Drug Delivery, and DFT Calculations. INt. J. Biol. Macromol. 2020, 154, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.E.; Pitoňák, M.; Jurečka, P.; Hobza, P. Stabilization and Structure Calculations for Noncovalent Interactions in Extended Molecular Systems Based on Wave Function and Density Functional Theories. Chem. Rev. 2010, 110, 5023–5063. [Google Scholar] [CrossRef]
- Bartolomei, M.; Pirani, F.; Marques, J.M.C. Modeling Coronene Nanostructures: Analytical Potential, Stable Configurations and Ab Initio Energies. J. Phys. Chem. C 2017, 121, 14330–14338. [Google Scholar] [CrossRef]
- Řezáč, J.; Hobza, P. Extrapolation and Scaling of the DFT-SAPT Interaction Energies toward the Basis Set Limit. J. Chem. Theory Comput. 2011, 7, 685–689. [Google Scholar] [CrossRef]
- Wang, H.M.J.; Vasam, C.S.; Tsai, T.Y.R.; Chen, S.-H.; Chang, A.H.H.; Lin, I.J.B. Gold(I) N-Heterocyclic Carbene and Carbazolate Complexes. Organometallics 2005, 24, 486–493. [Google Scholar] [CrossRef]
- Meyer, J.C.; Geim, A.K.; Katsnelson, M.I.; Novoselov, K.S.; Booth, T.J.; Roth, S. The Structure of Suspended Graphene Sheets. Nature 2007, 446, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Jansen, G. Symmetry-adapted Perturbation Theory Based on Density Functional Theory for Noncovalent Interactions. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 127–144. [Google Scholar] [CrossRef]
- Heßelmann, A.; Jansen, G.; Schütz, M. Density-Functional Theory-Symmetry-Adapted Intermolecular Perturbation Theory with Density Fitting: A New Efficient Method to Study Intermolecular Interaction Energies. J. Chem. Phys. 2005, 122, 014103. [Google Scholar] [CrossRef]
- Pitoňák, M.; Heßelmann, A. Accurate Intermolecular Interaction Energies from a Combination of MP2 and TDDFT Response Theory. J. Chem. Theory Comput. 2010, 6, 168–178. [Google Scholar] [CrossRef]
- Werner, H.J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M. Molpro: A General-Purpose Quantum Chemistry Program Package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 242–253. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Moszynski, R. Symmetry-Adapted Perturbation Theory for the Calculation of Hartree-Fock Interaction Energies. Mol. Phys. 1996, 88, 741–758. [Google Scholar] [CrossRef]
- Figgen, D.; Peterson, K.A.; Dolg, M.; Stoll, H. Energy-Consistent Pseudopotentials and Correlation Consistent Basis Sets for the 5d Elements Hf–Pt. J. Chem. Phys. 2009, 130, 164108. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron Affinities of the First-Row Atoms Revisited. Systematic Basis Sets and Wave Functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Bartolomei, M.; Pirani, F.; Marques, J.M.C. Low-Energy Structures of Benzene Clusters with a Novel Accurate Potential Surface. J. Comput. Chem. 2015, 36, 2291–2301. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The Calculation of Small Molecular Interactions by the Differences of Separate Total Energies. Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Halkier, A.; Helgaker, T.; Jørgensen, P.; Klopper, W.; Koch, H.; Olsen, J.; Wilson, A.K. Basis-Set Convergence in Correlated Calculations on Ne, N2, and H2O. Chem. Phys. Lett. 1998, 286, 243–252. [Google Scholar] [CrossRef]
- Halkier, A.; Klopper, W.; Helgaker, T.; Jo/rgensen, P. Basis-Set Convergence of the Molecular Electric Dipole Moment. J. Chem. Phys. 1999, 111, 4424–4430. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Function. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-Adjustedab Initio Pseudopotentials for the Second and Third Row Transition Elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Binkley, J.S.; Pople, J.A.; Hehre, W.J. Self-Consistent Molecular Orbital Methods. 21. Small Split-Valence Basis Sets for First-Row Elements. J. Am. Chem. Soc. 1980, 102, 939–947. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. GaussiaN 09, RevisioN B.01; Gaussian: Pittsburgh, PA, USA, 2010; p. 2009. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Energy (meV) | |
|---|---|
| Electrostatic | −429.3 |
| Exchange | 1002.4 |
| Induction | −126.9 |
| Dispersion a | −1314.7 |
| δ(HF) | −46.6 |
| Total | −702.9 |
| Crystal Structure [39] | Monomer | Complex | |||||
|---|---|---|---|---|---|---|---|
| PBE-D3(BJ) | B3LYP-D3(BJ) | M062X | PBE-D3(BJ) | B3LYP-D3(BJ) | M062X | ||
| Au–Cl | 2.2882 | 2.2994 | 2.3145 | 2.3297 | 2.3089 | 2.3250 | 2.3506 |
| Au–C1 | 1.9787 | 1.9864 | 2.0066 | 2.0047 | 1.9879 | 2.0067 | 2.0076 |
| C1–N1 | 1.35 | 1.3665 | 1.3560 | 1.3503 | 1.3657 | 1.3551 | 1.3488 |
| C1–N2 | 1.347 | 1.3665 | 1.3560 | 1.3503 | 1.3657 | 1.3551 | 1.3488 |
| C2–N1 | 1.36 | 1.3853 | 1.3831 | 1.3785 | 1.3853 | 1.3833 | 1.3772 |
| C3–N2 | 1.39 | 1.3853 | 1.3831 | 1.3785 | 1.3853 | 1.3833 | 1.3771 |
| C1-Au-Cl | 178.8 | 179.9 | 180.0 | 179.9 | 179.0 | 178.6 | 179.9 |
| N1-C1-N2 | 105.8 | 104.4 | 104.7 | 104.7 | 104.5 | 104.8 | 104.8 |
| C37H16 Geometry | PBE-D3(BJ)/ 6-311++G(2d,2p) | DFT-SAPT/CBS | MP2C/ CBS | B3LYP-D3(BJ)/ 6-311++G(2d,2p) | M062X/ 6-311++G(2d,2p) |
|---|---|---|---|---|---|
| Rigid | −731.6 | −702.9 | −804.54 | −955.3 | −786.6 |
| Relaxed | −783.2 | −734.7 | −855.71 | −1023.3 | −852.5 |
| R (Å) | BSSE Corrected (meV) | BSSE Uncorrected (meV) | |
|---|---|---|---|
| IMeAuCl-C37H16 (rigid/relaxed) | 3.35/3.37 | −731.6/−783.2 | −831.8/885.1 |
| IMeAuCl-C54H18 (rigid/relaxed) | 3.34/3.37 | −805.1/−829.1 | −918.1/−946.6 |
| IMeAuCl-C96H24 (rigid/relaxed) | 3.31/3.38 | −836.9/−853.8 | −951.8/−976.9 |
| IMeAuCl-C150H30 (rigid) | 3.30 | −861.3 | −988.5 |
| BSSE-Corrected (meV) | BSSE-Uncorrected (meV) | |
|---|---|---|
| IMeAuCl-C37H16 | −734.6 | −834.6 |
| IMeAuCl-C54H18/CP-C54H18 [34] | −805.3/−794.1 | −917.7/−939.4 |
| IMeAuCl-C96H24/CP-C96H24 [34] | −840.0/−818.8 | −960.8/−963.7 |
| IMeAuCl-C150H30 | −862.0 | −982.2 |
| Eint | ΔEads | ΔHads | ΔGads | |
|---|---|---|---|---|
| IMeAuCl-C37H16 | −731.6 (−831.8) | −719.9 (−820.1) | −664.4 (−764.6) | −209.7 (−309.9) |
| IMeAuCl-C54H18 | −805.1 (−918.1) | −795.0 (−908.0) | −739.1 (−852.1) | −276.5 (−389.5) |
| IMeAuCl-C96H24 | −836.9 (−951.8) | −829.5 (−944.4) | −774.6 (−889.5) | −286.8 (−401.7) |
| IMeAuCl-C150H30 | −862.0 (−982.2) | −854.6 (−974.8) a | −799.7 (−919.9) a | −311.8 (−432.0) a |
| CP-C32H14 [33] | −744.7 | (−839.3) | (−785.8) | (−324.7) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orek, C.; Bartolomei, M.; Coletti, C.; Bulut, N. Graphene as Nanocarrier for Gold(I)-Monocarbene Complexes: Strength and Nature of Physisorption. Molecules 2023, 28, 3941. https://doi.org/10.3390/molecules28093941
Orek C, Bartolomei M, Coletti C, Bulut N. Graphene as Nanocarrier for Gold(I)-Monocarbene Complexes: Strength and Nature of Physisorption. Molecules. 2023; 28(9):3941. https://doi.org/10.3390/molecules28093941
Chicago/Turabian StyleOrek, Cahit, Massimiliano Bartolomei, Cecilia Coletti, and Niyazi Bulut. 2023. "Graphene as Nanocarrier for Gold(I)-Monocarbene Complexes: Strength and Nature of Physisorption" Molecules 28, no. 9: 3941. https://doi.org/10.3390/molecules28093941
APA StyleOrek, C., Bartolomei, M., Coletti, C., & Bulut, N. (2023). Graphene as Nanocarrier for Gold(I)-Monocarbene Complexes: Strength and Nature of Physisorption. Molecules, 28(9), 3941. https://doi.org/10.3390/molecules28093941