

Screening of Anti-Inflammatory Activity and Metabolomics Analysis of Endophytic Fungal Extracts; Identification and Characterization of Perylenequinones and Terpenoids from the Interesting Active Alternaria Endophyte

 , , , , and
, , , , and 
Abstract
:1. Introduction
2. Results
2.1. Cytotoxicity and Anti-Inflammatory Activity of the Five Fungal Endophytic Extracts
2.1.1. Cytotoxicity of the Five Fungal Endophytic Extracts on Differentiated THP-1 Cells
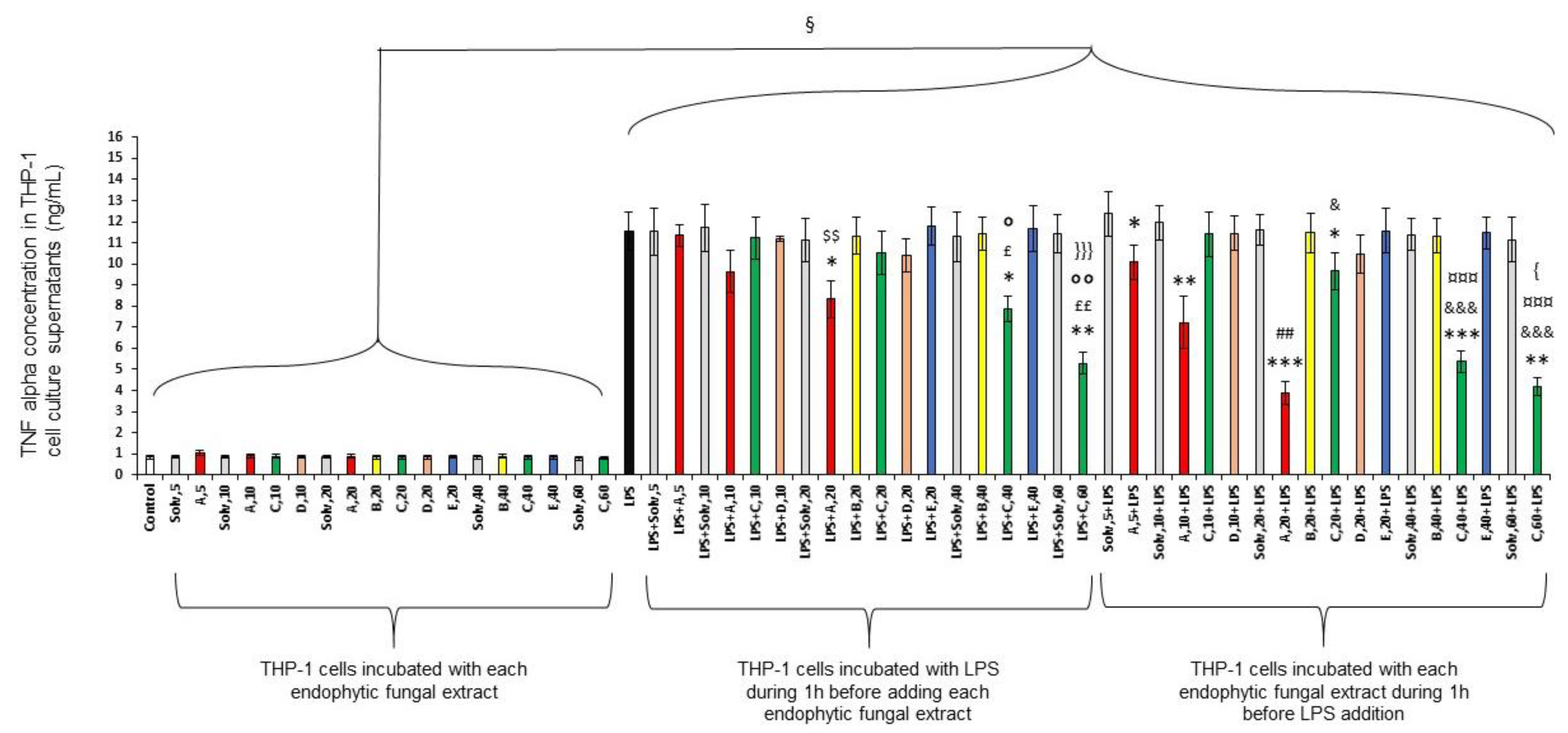
2.1.2. Anti-Inflammatory Activity of the Five Fungal Endophytic Extracts on Differentiated THP-1 Cells
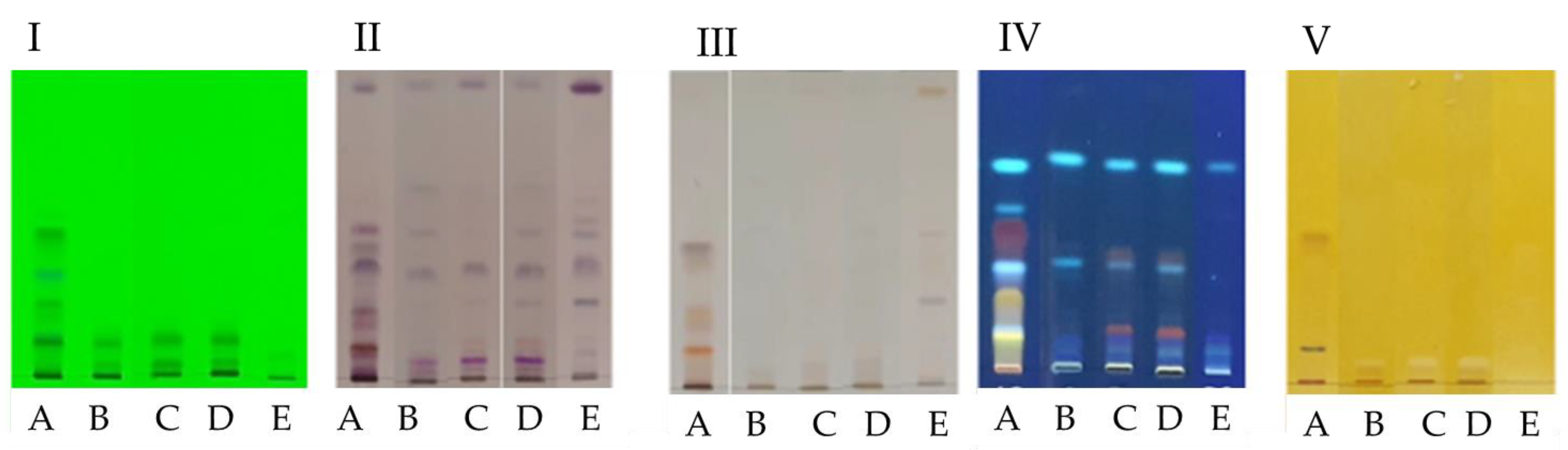
2.2. High Performance Thin Layer Chromatography (HPTLC)
2.3. Gas Chromatography Coupled to Mass Spectrometer (GC-MS)
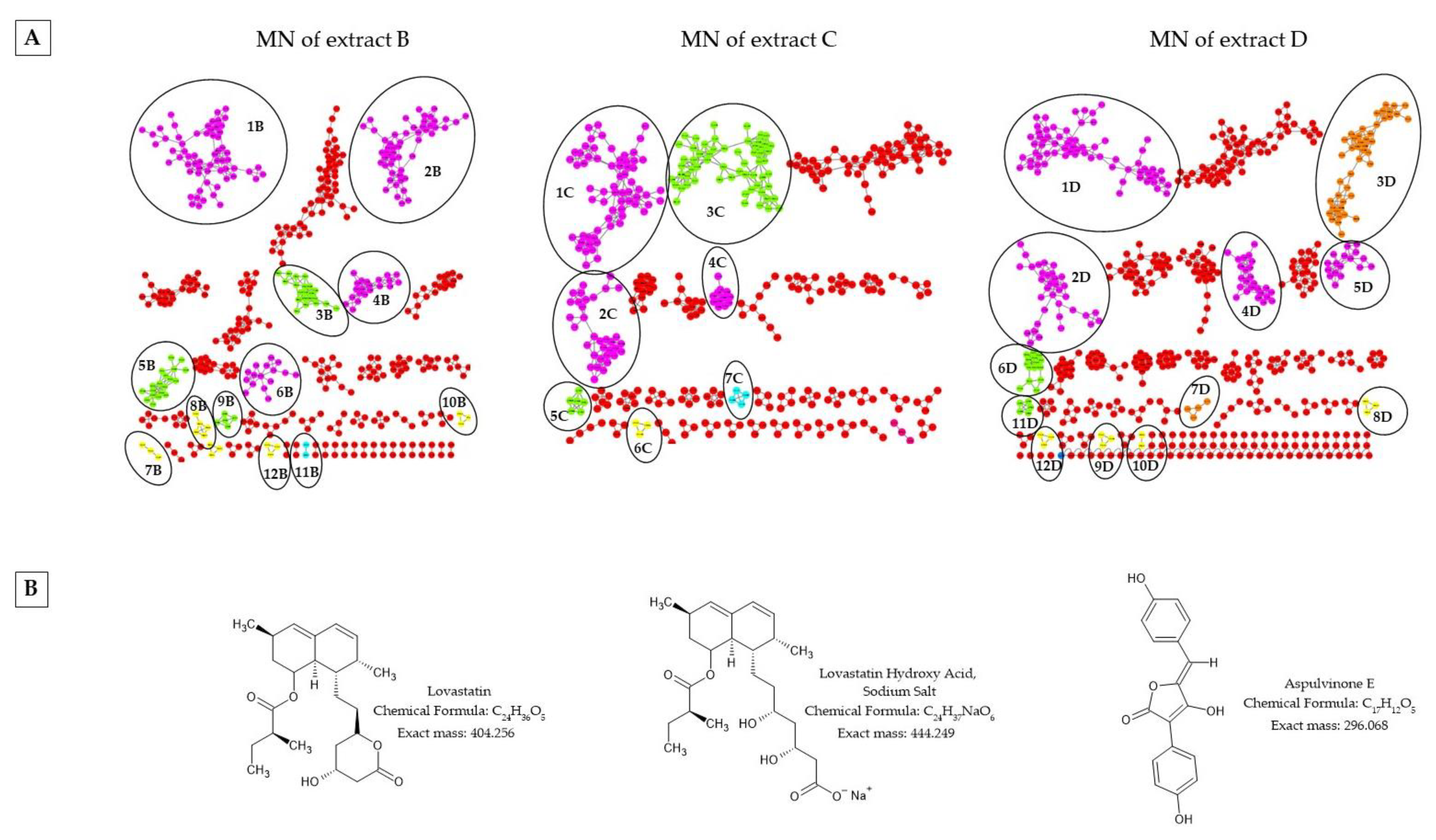
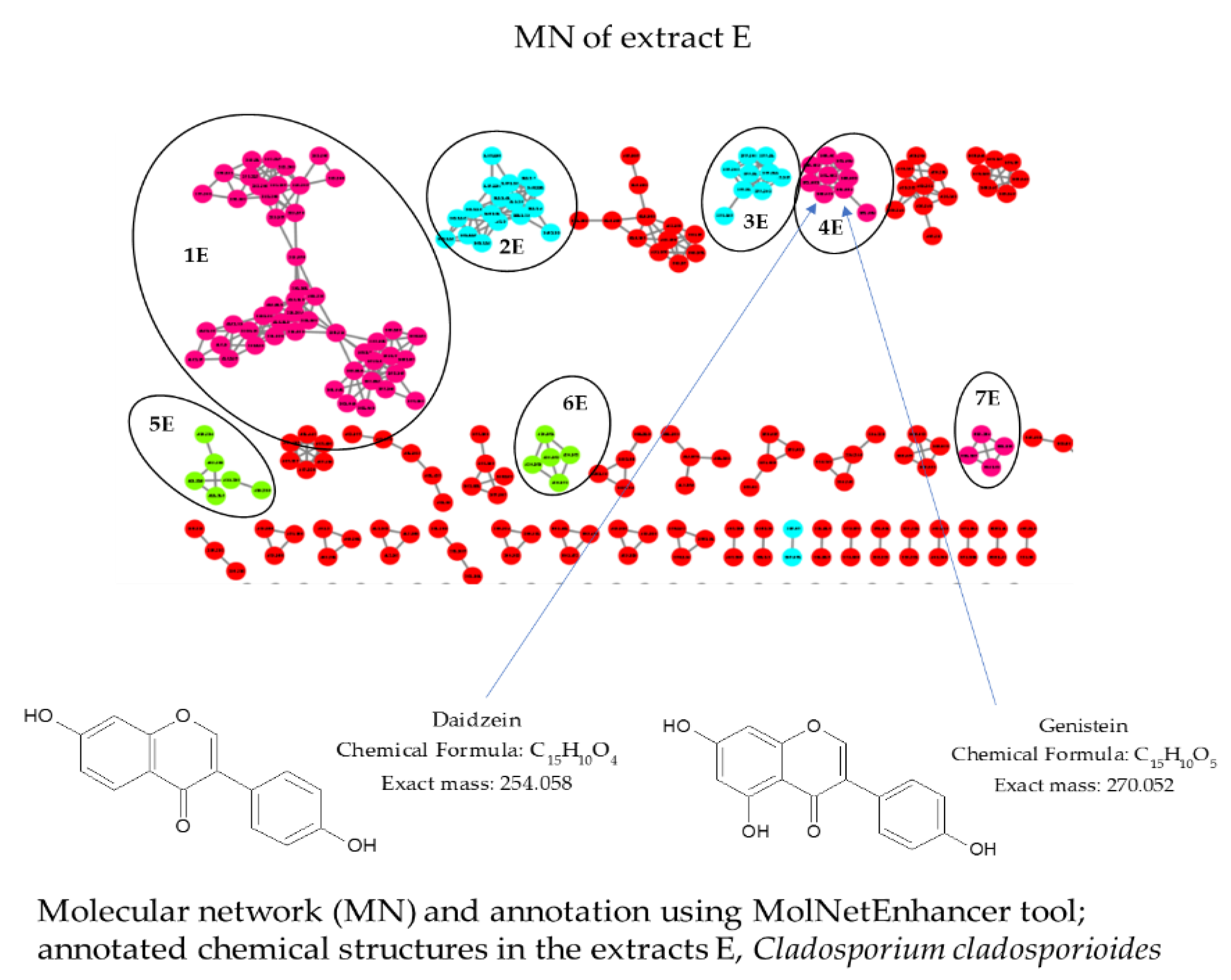
2.4. Liquid Chromatography Coupled with Mass Spectrometry in Tandem (LC/MS-MS) and Molecular Network (MN) of the Fungal Endophytic Crude Extracts
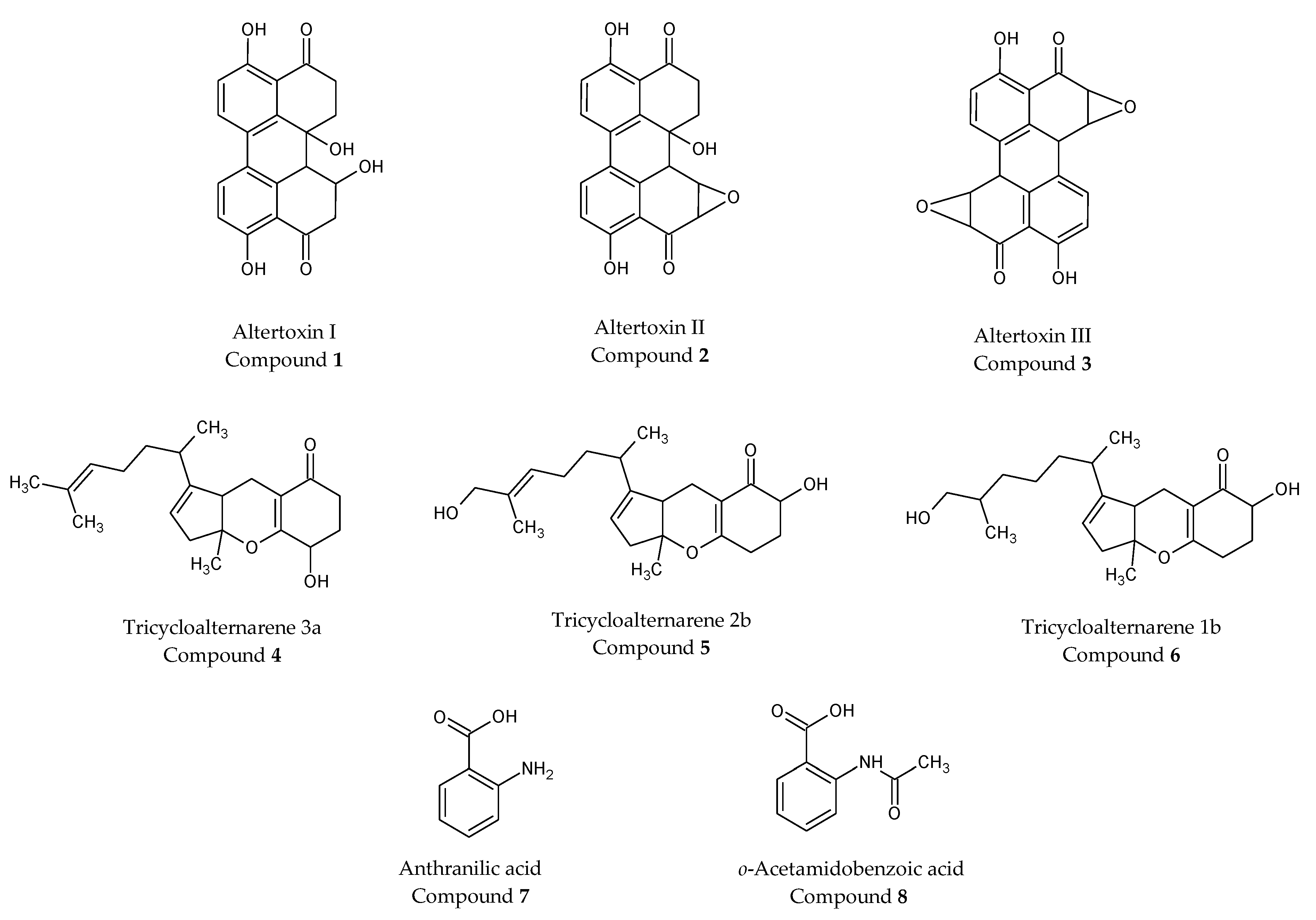
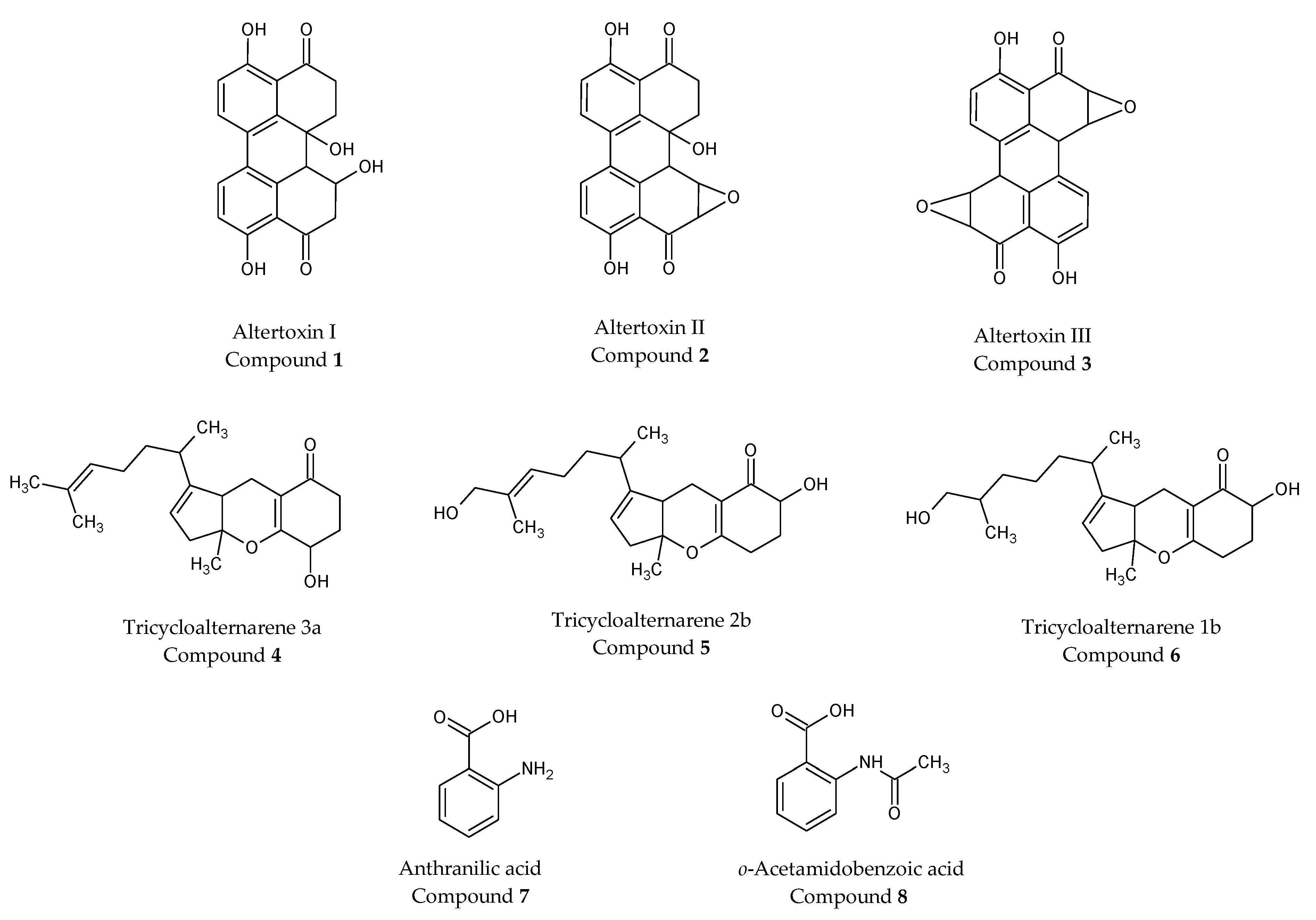
2.5. Purification of Extract a from Alternaria alternata and Characterization of Isolated Compounds
3. Discussion
4. Materials and Methods
4.1. Fungal Endophyte Strains
4.2. Culture of Endophytic Fungi
4.3. Preparation of Fungal Endophytic Extracts
4.4. Cytotoxicity and Anti-Inflammatory Activity
4.4.1. Cell Culture and Treatments of THP-1 Cells
4.4.2. THP-1 Cell Viability Measurement
4.4.3. TNF-α Quantification from THP-1 Cell Culture Supernatants
4.5. Data Analysis
4.6. High Performance Thin Layer Chromatography (HPTLC): Equipment and Method
4.7. Gas Chromatography Coupled to Mass Spectrometer (GC-MS): Equipment and Method
4.8. Liquid Chromatography Coupled to Mass Spectrometer (LC-MS): Equipment, Method and Molecular Networking
4.9. Isolation of Pure Compounds from Extract A, from Alternaria alternata Endophyte
4.9.1. General Experimental Procedures
4.9.2. Isolation of Pure Compounds from Strain A
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Rafei, A.; Elliott, M.R.; Jones, R.E.; Riosmena, F.; Cunningham, S.A.; Mehta, N.K. Obesity Incidence in U.S. Children and Young Adults: A Pooled Analysis. Am. J. Prev. Med. 2022, 63, 51–59. [Google Scholar] [CrossRef]
- Kuenzig, M.E.; Fung, S.G.; Marderfeld, L.; Mak, J.W.Y.; Kaplan, G.G.; Ng, S.C.; Wilson, D.C.; Cameron, F.; Henderson, P.; Kotze, P.G.; et al. Twenty-First Century Trends in the Global Epidemiology of Pediatric-Onset Inflammatory Bowel Disease: Systematic Review. Gastroenterology 2022, 162, 1147–1159.e4. [Google Scholar] [CrossRef] [PubMed]
- Wernecke, C.; Braun, H.J.; Dragoo, J.L. The Effect of Intra-Articular Corticosteroids on Articular Cartilage: A Systematic Review. Orthop. J. Sports Med. 2015, 3, 2325967115581163. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.R.; Olmstead, R.; Carroll, J.E. Sleep Disturbance, Sleep Duration, and Inflammation: A Systematic Review and Meta-Analysis of Cohort Studies and Experimental Sleep Deprivation. Biol. Psychiatry 2016, 80, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Scheijen, B. Molecular Mechanisms Contributing to Glucocorticoid Resistance in Lymphoid Malignancies. Cancer Drug Resist. 2019, 2, 647–664. [Google Scholar] [CrossRef]
- De Iudicibus, S.; Franca, R.; Martelossi, S.; Ventura, A.; Decorti, G. Molecular Mechanism of Glucocorticoid Resistance in Inflammatory Bowel Disease. World J. Gastroenterol. 2011, 17, 1095–1108. [Google Scholar] [CrossRef]
- Olivas-Aguirre, M.; Torres-López, L.; Pottosin, I.; Dobrovinskaya, O. Overcoming Glucocorticoid Resistance in Acute Lymphoblastic Leukemia: Repurposed Drugs Can Improve the Protocol. Front. Oncol. 2021, 11, 617937. [Google Scholar] [CrossRef]
- Rice, J.B.; White, A.G.; Scarpati, L.M.; Wan, G.; Nelson, W.W. Long-Term Systemic Corticosteroid Exposure: A Systematic Literature Review. Clin. Ther. 2017, 39, 2216–2229. [Google Scholar] [CrossRef]
- Timmermans, S.; Souffriau, J.; Libert, C. A General Introduction to Glucocorticoid Biology. Front. Immunol. 2019, 10, 1545. [Google Scholar] [CrossRef]
- Ramos-Ramírez, P.; Tliba, O. Glucocorticoid Receptor β (GRβ): Beyond Its Dominant-Negative Function. Int. J. Mol. Sci. 2021, 22, 3649. [Google Scholar] [CrossRef]
- Tai, F.W.D.; McAlindon, M.E. Non-Steroidal Anti-Inflammatory Drugs and the Gastrointestinal Tract. Clin. Med. 2021, 21, 131–134. [Google Scholar] [CrossRef]
- Schjerning, A.-M.; McGettigan, P.; Gislason, G. Cardiovascular Effects and Safety of (Non-Aspirin) NSAIDs. Nat. Rev. Cardiol. 2020, 17, 574–584. [Google Scholar] [CrossRef]
- Gouda, S.; Das, G.; Sen, S.K.; Shin, H.-S.; Patra, J.K. Endophytes: A Treasure House of Bioactive Compounds of Medicinal Importance. Front. Microbiol. 2016, 7, 1538. [Google Scholar] [CrossRef] [PubMed]
- Tadych, M.; White, J. Endophytic Microbes. In Reference Module in Life Sciences; Elsevier: Amsterdam, The Netherlands, 2019; pp. 123–136. ISBN 978-0-12-811737-8. [Google Scholar]
- Rajamanikyam, M.; Vadlapudi, V.; Amanchy, R.; Upadhyayula, S.M. Endophytic Fungi as Novel Resources of Natural Therapeutics. Braz. Arch. Biol. Technol. 2017, 60, e17160542. [Google Scholar] [CrossRef]
- Liu, J.; Liu, G. Analysis of Secondary Metabolites from Plant Endophytic Fungi. In Plant Pathogenic Fungi and Oomycetes: Methods and Protocols; Ma, W., Wolpert, T., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2018; pp. 25–38. ISBN 978-1-4939-8724-5. [Google Scholar]
- Joseph, B.; Priya, R.M. Bioactive Compounds from Endophytes and Their Potential in Pharmaceutical Effect: A Review. Am. J. Biochem. Mol. Biol. 2011, 1, 291–309. [Google Scholar] [CrossRef]
- Conrado, R.; Gomes, T.C.; Roque, G.S.C.; De Souza, A.O. Overview of Bioactive Fungal Secondary Metabolites: Cytotoxic and Antimicrobial Compounds. Antibiotics 2022, 11, 1604. [Google Scholar] [CrossRef]
- Deshmukh, S.K.; Dufossé, L.; Chhipa, H.; Saxena, S.; Mahajan, G.B.; Gupta, M.K. Fungal Endophytes: A Potential Source of Antibacterial Compounds. J. Fungi 2022, 8, 164. [Google Scholar] [CrossRef]
- Hridoy, M.; Gorapi, M.Z.H.; Noor, S.; Chowdhury, N.S.; Rahman, M.M.; Muscari, I.; Masia, F.; Adorisio, S.; Delfino, D.V.; Mazid, M.A. Putative Anticancer Compounds from Plant-Derived Endophytic Fungi: A Review. Molecules 2022, 27, 296. [Google Scholar] [CrossRef] [PubMed]
- Bogas, A.C.; Cruz, F.P.N.; Lacava, P.T.; Sousa, C.P. Endophytic Fungi: An Overview on Biotechnological and Agronomic Potential. Braz. J. Biol. 2022, 84, e258557. [Google Scholar] [CrossRef] [PubMed]
- Khiralla, A.; Mohamed, I.; Thomas, J.; Mignard, B.; Spina, R.; Yagi, S.; Laurain-Mattar, D. A Pilot Study of Antioxidant Potential of Endophytic Fungi from Some Sudanese Medicinal Plants. Asian Pac. J. Trop. Med. 2015, 8, 701–704. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Stack, M.E.; Mazzola, E.P.; Page, S.W.; Pohland, A.E.; Highet, R.J.; Tempesta, M.S.; Corley, D.G. Mutagenic Perylenequinone Metabolites of Alternaria Alternata: Altertoxins I, II, and III. J. Nat. Prod. 1986, 49, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Liebermann, B.; Ellinger, R.; Günther, W.; Ihn, W.; Gallander, H. Tricycloalternarenes Produced by Alternaria Alternata Related to ACTG-Toxins. Phytochemistry 1997, 46, 297–303. [Google Scholar] [CrossRef]
- Kono, Y.; Gardner, J.M.; Takeuchi, S. Studies on Host-Selective Toxins Produced by a Pathotype of Alternaria Citri Causing Brown Spot Disease of Mandarins. Agric. Biol. Chem. 1986, 50, 1597–1606. [Google Scholar] [CrossRef]
- Hussain, T.; Tan, B.; Yin, Y.; Blachier, F.; Tossou, M.C.B.; Rahu, N. Oxidative Stress and Inflammation: What Polyphenols Can Do for Us? Oxid. Med. Cell Longev. 2016, 2016, 7432797. [Google Scholar] [CrossRef]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Origins and Hallmarks of Macrophages: Development, Homeostasis, and Disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.-A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage Plasticity, Polarization, and Function in Health and Disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- de Souza, J.J.; Vieira, I.J.C.; Rodrigues-Filho, E.; Braz-Filho, R. Terpenoids from Endophytic Fungi. Molecules 2011, 16, 10604–10618. [Google Scholar] [CrossRef]
- Gershenzon, J.; Dudareva, N. The Function of Terpene Natural Products in the Natural World. Nat. Chem. Biol. 2007, 3, 408–414. [Google Scholar] [CrossRef]
- Pasanen, A.L.; Yli-Pietila, K.; Pasanen, P.; Kalliokoski, P.; Tarhanen, J. Ergosterol Content in Various Fungal Species and Biocontaminated Building Materials. Appl. Environ. Microbiol. 1999, 65, 138–142. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and Community Curation of Mass Spectrometry Data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.D.; Wu, C.-H.; Moree, W.J.; Lamsa, A.; Medema, M.H.; Zhao, X.; Gavilan, R.G.; Aparicio, M.; Atencio, L.; Jackson, C.; et al. MS/MS Networking Guided Analysis of Molecule and Gene Cluster Families. Proc. Natl. Acad. Sci. USA 2013, 110, E2611–E2620. [Google Scholar] [CrossRef] [PubMed]
- Subhan, M.; Faryal, R.; Macreadie, I. Exploitation of Aspergillus Terreus for the Production of Natural Statins. J. Fungi 2016, 2, 13. [Google Scholar] [CrossRef] [PubMed]
- Tawfike, A.F.; Tate, R.; Abbott, G.; Young, L.; Viegelmann, C.; Schumacher, M.; Diederich, M.; Edrada-Ebel, R. Metabolomic Tools to Assess the Chemistry and Bioactivity of Endophytic Aspergillus Strain. Chem. Biodivers. 2017, 14, e1700040. [Google Scholar] [CrossRef]
- Mamgain, A.; Roychowdhury, R.; Tah, J. Alternaria Pathogenicity and Its Strategic Controls. Res. Rev. J. Biol. 2013, 1, 1–9. [Google Scholar]
- Thomma, B.P.H.J. Alternaria Spp.: From General Saprophyte to Specific Parasite. Mol. Plant Pathol. 2003, 4, 225–236. [Google Scholar] [CrossRef]
- Escrivá, L.; Oueslati, S.; Font, G.; Manyes, L. Alternaria Mycotoxins in Food and Feed: An Overview. J. Food Qual. 2017, 2017, e1569748. [Google Scholar] [CrossRef]
- Kustrzeba-Wójcicka, I.; Siwak, E.; Terlecki, G.; Wolańczyk-Mędrala, A.; Mędrala, W. Alternaria Alternata and Its Allergens: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2014, 47, 354–365. [Google Scholar] [CrossRef]
- Aichinger, G.; Krüger, F.; Puntscher, H.; Preindl, K.; Warth, B.; Marko, D. Naturally Occurring Mixtures of Alternaria Toxins: Anti-Estrogenic and Genotoxic Effects in Vitro. Arch. Toxicol. 2019, 93, 3021–3031. [Google Scholar] [CrossRef]
- Lou, J.; Fu, L.; Peng, Y.; Zhou, L. Metabolites from Alternaria Fungi and Their Bioactivities. Molecules 2013, 18, 5891–5935. [Google Scholar] [CrossRef]
- Dalinova, A.A.; Salimova, D.R.; Berestetskiy, A.O. Fungi of the Genera Alternaria as Producers of Biological Active Compounds and Mycoherbicides. Appl. Biochem. Microbiol. 2020, 56, 256–272. [Google Scholar] [CrossRef]
- Zwickel, T.; Kahl, S.M.; Rychlik, M.; Müller, M.E.H. Chemotaxonomy of Mycotoxigenic Small-Spored Alternaria Fungi—Do Multitoxin Mixtures Act as an Indicator for Species Differentiation? Front. Microbiol. 2018, 9, 1368. [Google Scholar] [CrossRef] [PubMed]
- Escrivá, L.; Font, G.; Manyes, L.; Berrada, H. Studies on the Presence of Mycotoxins in Biological Samples: An Overview. Toxins 2017, 9, 251. [Google Scholar] [CrossRef] [PubMed]
- Perdoncini, M.R.F.G.; Sereia, M.J.; Scopel, F.H.P.; Formigoni, M.; Rigobello, E.S.; Beneti, S.C.; Cardoso, F.A.R.; Marchi, L.B.; Junior, C.G.d.S.; Fernandes, P.G.M.; et al. Growth of Fungal Cells and the Production of Mycotoxins. In Cell Growth; IntechOpen: London, UK, 2019; ISBN 978-1-78984-557-0. [Google Scholar]
- Cantani, A.; Ciaschi, V. Epidemiology of Alternaria Alternata Allergy: A Prospective Study in 6840 Italian Asthmatic Children. Eur. Rev. Med. Pharmacol. Sci. 2005, 8, 289–294. [Google Scholar]
- Randriamanantany, Z.A.; Annesi-Maesano, I.; Moreau, D.; Raherison, C.; Charpin, D.; Kopferschmitt, C.; Lavaud, F.; Taytard, A.; De Blay, F.; Caillaud, D. Alternaria Sensitization and Allergic Rhinitis with or without Asthma in the French Six Cities Study. Allergy 2010, 65, 368–375. [Google Scholar] [CrossRef]
- Meena, M.; Swapnil, P.; Upadhyay, R.S. Isolation, Characterization and Toxicological Potential of Alternaria-Mycotoxins (TeA, AOH and AME) in Different Alternaria Species from Various Regions of India. Sci. Rep. 2017, 7, 8777. [Google Scholar] [CrossRef]
- Qin, J.-C.; Zhang, Y.-M.; Hu, L.; Ma, Y.-T.; Gao, J.-M. Cytotoxic Metabolites Produced by Alternaria No.28, an Endophytic Fungus Isolated from Ginkgo Biloba. Nat. Prod. Commun. 2009, 4, 1473–1476. [Google Scholar] [CrossRef]
- Bashyal, B.P.; Wellensiek, B.P.; Ramakrishnan, R.; Faeth, S.H.; Ahmad, N.; Gunatilaka, A.A.L. Altertoxins with Potent Anti-HIV Activity from Alternaria Tenuissima QUE1Se, a Fungal Endophyte of Quercus Emoryi. Bioorg. Med. Chem. 2014, 22, 6112–6116. [Google Scholar] [CrossRef]
- Khiralla, A.; Mohammed, A.O.; Yagi, S. Fungal Perylenequinones. Mycol. Progress 2022, 21, 38. [Google Scholar] [CrossRef]
- Lee, C.; Li, W.; Bang, S.; Lee, S.J.; Kang, N.; Kim, S.; Kim, T.I.; Go, Y.; Shim, S.H. Secondary Metabolites of The Endophytic Fungus Alternaria Alternata JS0515 Isolated from Vitex Rotundifolia and Their Effects on Pyruvate Dehydrogenase Activity. Molecules 2019, 24, 4450. [Google Scholar] [CrossRef]
- Del Favero, G.; Zaharescu, R.; Marko, D. Functional Impairment Triggered by Altertoxin II (ATXII) in Intestinal Cells in Vitro: Cross-Talk between Cytotoxicity and Mechanotransduction. Arch. Toxicol. 2018, 92, 3535–3547. [Google Scholar] [CrossRef] [PubMed]
- Del Favero, G.; Hohenbichler, J.; Mayer, R.M.; Rychlik, M.; Marko, D. Mycotoxin Altertoxin II Induces Lipid Peroxidation Connecting Mitochondrial Stress Response to NF-ΚB Inhibition in THP-1 Macrophages. Chem. Res. Toxicol. 2020, 33, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Li, J.; Liu, J.; Xiao, S.; Yang, S.; Mei, J.; Ren, M.; Wu, S.; Zhang, H.; Yang, X. Secondary Metabolites of Alternaria: A Comprehensive Review of Chemical Diversity and Pharmacological Properties. Front. Microbiol. 2023, 13, 1085666. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Huang, Z.-H.; Liang, X.; Ma, X.; Qi, S.-H. Study of Tricycloalternarenes from the Deep-Sea Derived Fungus Alternaria Tenuissma DFFSCS013 and Their Antimicrobial Activity. Nat. Prod. Res. Dev. 2018, 30, 1166–1169+1201. [Google Scholar]
- Zhang, H.; Zhao, Z.; Chen, J.; Bai, X.; Wang, H. Tricycloalternarene Analogs from a Symbiotic Fungus Aspergillus Sp. D and Their Antimicrobial and Cytotoxic Effects. Molecules 2018, 23, 855. [Google Scholar] [CrossRef] [PubMed]
- Khiralla, A.; Mohamed, I.E.; Tzanova, T.; Schohn, H.; Slezack-Deschaumes, S.; Hehn, A.; André, P.; Carre, G.; Spina, R.; Lobstein, A.; et al. Endophytic Fungi Associated with Sudanese Medicinal Plants Show Cytotoxic and Antibiotic Potential. FEMS Microbiol. Lett. 2016, 363, fnw089. [Google Scholar] [CrossRef]
- Roumani, M.; Ropars, A.; Robin, C.; Duval, R.E.; Frippiat, J.-P.; Boisbrun, M.; Larbat, R. Characterization of Biological Properties of Individual Phenolamides and Phenolamide-Enriched Leaf Tomato Extracts. Molecules 2023, 28, 1552. [Google Scholar] [CrossRef]
- Kiprop, A.K.; Pourtier, E.; Kimutai, S.K. GC-MS and ESI-MS Detection of Catechol. Int. J. Educ. Res. 2013, 1, 1–12. [Google Scholar]
- Ernst, M.; Kang, K.B.; Caraballo-Rodríguez, A.M.; Nothias, L.-F.; Wandy, J.; Chen, C.; Wang, M.; Rogers, S.; Medema, M.H.; Dorrestein, P.C.; et al. MolNetEnhancer: Enhanced Molecular Networks by Integrating Metabolome Mining and Annotation Tools. Metabolites 2019, 9, 144. [Google Scholar] [CrossRef]
- Djoumbou Feunang, Y.; Eisner, R.; Knox, C.; Chepelev, L.; Hastings, J.; Owen, G.; Fahy, E.; Steinbeck, C.; Subramanian, S.; Bolton, E.; et al. ClassyFire: Automated Chemical Classification with a Comprehensive, Computable Taxonomy. J. Cheminform. 2016, 8, 61. [Google Scholar] [CrossRef]
- Elmi, A.; Mohamed Abdoul-Latif, F.; Spina, R.; Dupire, F.; Philippot, S.; Marie-France, C.; Jacobs, H.; Laurain-Mattar, D. Aloe Djiboutiensis: Antioxidant Activity, Molecular Networking-Based Approach and In Vivo Toxicity of This Endemic Species in Djibouti. Molecules 2021, 26, 3046. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Concentration (µg/mL) | % of Viable Cell | S.D. | Sample Concentration (µg/mL) | % of Viable Cells | S.D. | Sample Concentration (µg/mL) | % of Viable Cells | S.D. |
|---|---|---|---|---|---|---|---|---|
| Control | 100.00 | 0 | LPS | 99.20 | 6.78 | |||
| Solv,5 | 104.76 | 9.40 | LPS+Solv,5 | 99.48 | 3.48 | Solv,5+LPS | 101.26 | 9.33 |
| Solv,10 | 105.56 | 8.05 | LPS+Solv,10 | 100.57 | 7.18 | Solv,10+LPS | 99.96 | 6.32 |
| Solv,20 | 106.86 | 7.32 | LPS+Solv,20 | 97.62 | 7.27 | Solv,20+LPS | 100.43 | 8.78 |
| Solv,40 | 104.39 | 8.52 | LPS+Solv,40 | 99.25 | 8.47 | Solv,40+LPS | 99.48 | 5.54 |
| Solv,60 | 99.18 | 4.53 | LPS+Solv,60 | 97.61 | 7.70 | Solv,60+LPS | 100.59 | 6.53 |
| A,5 | 104.42 | 8.84 | LPS+A,5 | 101.13 | 10.48 | A,5+LPS | 101.05 | 9.02 |
| A,10 | 99.41 | 6.26 | LPS+A,10 | 102.26 | 8.92 | A,10+LPS | 103.24 | 4.20 |
| A,20 | 101.37 | 4.76 | LPS+A,20 | 98.44 | 8.13 | A,20+LPS | 98.20 | 8.93 |
| A,40 *,$$$ | 79.71 | 6.76 | LPS+A,40 *,$$ | 78.96 | 9.44 | A,40+LPS *,$$ | 80.50 | 9.22 |
| B,5 | 104.98 | 5.76 | LPS+B,5 | 101.32 | 6.14 | B,5+LPS | 101.38 | 8.51 |
| B,10 | 99.19 | 8.17 | LPS+B,10 | 98.07 | 6.15 | B,10+LPS | 99.74 | 7.84 |
| B,20 | 95.06 | 3.41 | LPS+B,20 | 93.78 | 5.84 | B,20+LPS | 94.06 | 6.09 |
| B,40 | 92.44 | 3.23 | LPS+B,40 | 92.34 | 7.37 | B,40+LPS | 92.79 | 7.75 |
| B,60 *,$ | 78.97 | 2.47 | LPS+B,60 *,$ | 77.10 | 5.81 | B,60+LPS *,$ | 77.48 | 7.02 |
| C,5 | 103.90 | 9.54 | LPS+C,5 | 102.45 | 8.99 | C,5+LPS | 99.93 | 7.34 |
| C,10 | 95.76 | 7.68 | LPS+C,10 | 97.11 | 8.35 | C,10+LPS | 102.68 | 5.86 |
| C,20 | 103.64 | 9.39 | LPS+C,20 | 100.36 | 9.63 | C,20+LPS | 104.14 | 7.36 |
| C,40 | 101.28 | 5.03 | LPS+C,40 | 98.34 | 9.05 | C,40+LPS | 100.17 | 8.96 |
| C,60 | 99.74 | 6.00 | LPS+C,60 | 99.04 | 9.08 | C,60+LPS | 99.82 | 7.67 |
| D,5 | 100.76 | 9.07 | LPS+D,5 | 98.38 | 6.86 | D,5+LPS | 100.14 | 9.09 |
| D,10 | 104.59 | 5.59 | LPS+D,10 | 99.12 | 2.36 | D,10+LPS | 98.03 | 7.28 |
| D,20 | 92.76 | 4.75 | LPS+D,20 | 87.77 | 8.47 | D,20+LPS | 91.84 | 7.68 |
| D,40 *,$ | 85.07 | 8.77 | LPS+D,40 *,$ | 83.89 | 9.00 | D,40+LPS *,$ | 83.10 | 8.73 |
| E,5 | 100.08 | 9.15 | LPS+E,5 | 103.69 | 5.54 | E,5+LPS | 99.75 | 8.06 |
| E,10 | 101.09 | 9.36 | LPS+E,10 | 99.52 | 7.78 | E,10+LPS | 98.09 | 9.64 |
| E,20 | 98.61 | 5.33 | LPS+E,20 | 97.32 | 9.41 | E,20+LPS | 97.89 | 9.62 |
| E,40 | 93.59 | 9.34 | LPS+E,40 | 92.04 | 9.15 | E,40+LPS | 93.95 | 8.66 |
| E,60 *,$ | 87.29 | 6.53 | LPS+E,60 *,$ | 85.69 | 8.82 | E,60+LPS *,$$ | 85.30 | 4.90 |
| Compound Name | Chemical Formula | MW | rt (min) | RI | Similarity % | Extracts | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| A | B | C | D | E | ||||||
| 3-t-Pentylcyclopentanone | C10H18O | 154 | 11.31 | 1145 | 83 | - | - | + | + | - |
| Thujopsene | C15H24 | 204 | 14.15 | 1416 | 85 | + | - | - | - | - |
| Methyl 2-nonynoate | C10H16O2 | 168 | 14.56 | 1200 | 83 | - | - | + | - | - |
| Dodecanal | C12H24O | 184 | 17.5 | 1402 | 89 | - | - | - | - | + |
| 3-Isobutylhexahydropyrrolo[1,2-a]pyrazine-1,4-dione or cyclo (Pro-Leu) | C11H18N2O2 | 210 | 19.8 | 1699 | 92 | + | - | - | - | + |
| Palmitic acid | C16H32O2 | 256 | 20.04 | 1968 | 95 | + | + | - | + | + |
| 11,14-Eicosadienoic acid, methyl ester | C21H38O2 | 322 | 21.6 | 2292 | 88 | - | - | + | - | + |
| Linoleic acid | C18H32O2 | 280 | 21.7 | 2183 | 93 | + | + | + | + | + |
| Oleic Acid | C18H34O2 | 282 | 21.72 | 2175 | 87 | + | + | - | + | + |
| Stearic acid | C18H36O2 | 284 | 21.95 | 2167 | 88 | + | - | - | - | + |
| 3-Benzylhexahydropyrrolo[1,2-a]pyrazine-1,4-dione or cyclo (Phe-Pro) | C14H16N2O2 | 244 | 23.9 | 2138 | 80 | - | - | - | - | + |
| trans-Squalene | C30H50 | 410 | 27.08 | 2914 | 87 | + | - | - | - | + |
| Lovastatin | C24H36O5 | 404 | 28.02 | 3091 | 77 | - | + | + | + | - |
| Ergosterol | C28H44O | 396 | 29.74 | 2679 | 80 | - | - | - | - | + |
| Hippuric-benzaldehyde azalactone | C16H11NO2 | 249 | 32.94 | 2266 | 82 | - | + | + | + | - |
| Total | 7 | 6 | 6 | 7 | 9 | |||||
| Compound Name | Chemical Formula | MW | rt (min) | RI | Similarity % | Extracts | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| A | B | C | D | E | ||||||
| 2-Ketoisocaproic acid, trimethylsilyl ester | C9H18O3Si | 202 | 9.06 | 1065 | 86 | + | - | - | - | - |
| Benzeneacetic acid, trimethylsilyl ester | C11H16O2Si | 208 | 12.15 | 1269 | 90 | + | - | - | - | + |
| Succinic acid, di(trimethylsilyl) ester | C10H22O4Si2 | 262 | 12.4 | 1170 | 81 | + | - | - | - | + |
| Acide itaconique (tms) | C11H22O4Si2 | 274 | 12.80 | 1236 | 80 | - | - | + | - | - |
| Fumaric acid, bis(trimethylsilyl) ester | C10H20O4Si2 | 260 | 12.94 | 1178 | 81 | - | - | - | - | + |
| Malic acid, O-trimethylsilyl-, bis(trimethylsilyl) ester | C13H30O5Si3 | 350 | 14.74 | 1390 | 87 | + | - | - | - | + |
| Benzoic acid, 4-[(trimethylsilyl)oxy]-, trimethylsilyl ester | C13H22O3Si2 | 282 | 16.44 | 1467 | 85 | - | - | + | - | + |
| Benzeneacetic acid, 4-[(trimethylsilyl)oxy]-, trimethylsilyl ester | C14H24O3Si2 | 296 | 16.6 | 1566 | 82 | + | - | - | - | - |
| Phenylpyruvic acid, bis(trimethylsilyl) | C15H24O3Si2 | 308 | 17.33 | 1637 | 83 | + | - | - | - | - |
| 2-Propenoic acid, 2-[(trimethylsilyl)oxy] | C18H32O4Si3 | 396 | 20.8 | 1935 | 76 | - | + | - | - | - |
| Palmitic acid, trimethylsilyl ester | C16H32O2 | 256 | 20.9 | 1987 | 91 | + | + | - | + | + |
| Linoleic acid trimethylsilyl ester | C21H40O2Si | 352 | 22.34 | 2202 | 93 | + | + | + | + | + |
| Oleic acid, trimethylsilyl ester | C21H42O2Si | 354 | 22.40 | 2194 | 90 | + | + | - | + | + |
| Stearic acid, trimethylsilyl ester | C21H44O2Si | 356 | 22.63 | 2186 | 90 | + | - | - | - | + |
| 1-Monooleoylglycerol trimethylsilyl ether | C27H56O4Si2 | 500 | 26.65 | 2788 | 81 | - | - | - | - | + |
| Lovastatin | C24H36O5 | 404 | 29.01 | 3012 | 96 | - | + | + | + | - |
| Bis(trimethylsilyl) 3-methyl-3-trimethylsilyloxypentanedioate | C15H34O5Si3 | 378 | 29.68 | 1568 | 70 | - | - | + | + | - |
| (22E)-3-[(trimethylsilyl)oxy]ergosta-5,7,22-triene | C31H52OSi | 468 | 29.81 | 2708 | 70 | - | - | + | - | + |
| Total | 10 | 4 | 6 | 5 | 11 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spina, R.; Ropars, A.; Bouazzi, S.; Dadi, S.; Lemiere, P.; Dupire, F.; Khiralla, A.; Yagi, S.; Frippiat, J.-P.; Laurain-Mattar, D. Screening of Anti-Inflammatory Activity and Metabolomics Analysis of Endophytic Fungal Extracts; Identification and Characterization of Perylenequinones and Terpenoids from the Interesting Active Alternaria Endophyte. Molecules 2023, 28, 6531. https://doi.org/10.3390/molecules28186531
Spina R, Ropars A, Bouazzi S, Dadi S, Lemiere P, Dupire F, Khiralla A, Yagi S, Frippiat J-P, Laurain-Mattar D. Screening of Anti-Inflammatory Activity and Metabolomics Analysis of Endophytic Fungal Extracts; Identification and Characterization of Perylenequinones and Terpenoids from the Interesting Active Alternaria Endophyte. Molecules. 2023; 28(18):6531. https://doi.org/10.3390/molecules28186531
Chicago/Turabian StyleSpina, Rosella, Armelle Ropars, Sihem Bouazzi, Safa Dadi, Pascal Lemiere, François Dupire, Afra Khiralla, Sakina Yagi, Jean-Pol Frippiat, and Dominique Laurain-Mattar. 2023. "Screening of Anti-Inflammatory Activity and Metabolomics Analysis of Endophytic Fungal Extracts; Identification and Characterization of Perylenequinones and Terpenoids from the Interesting Active Alternaria Endophyte" Molecules 28, no. 18: 6531. https://doi.org/10.3390/molecules28186531
APA StyleSpina, R., Ropars, A., Bouazzi, S., Dadi, S., Lemiere, P., Dupire, F., Khiralla, A., Yagi, S., Frippiat, J.-P., & Laurain-Mattar, D. (2023). Screening of Anti-Inflammatory Activity and Metabolomics Analysis of Endophytic Fungal Extracts; Identification and Characterization of Perylenequinones and Terpenoids from the Interesting Active Alternaria Endophyte. Molecules, 28(18), 6531. https://doi.org/10.3390/molecules28186531