
The AT1/AT2 Receptor Equilibrium Is a Cornerstone of the Regulation of the Renin Angiotensin System beyond the Cardiovascular System



Abstract
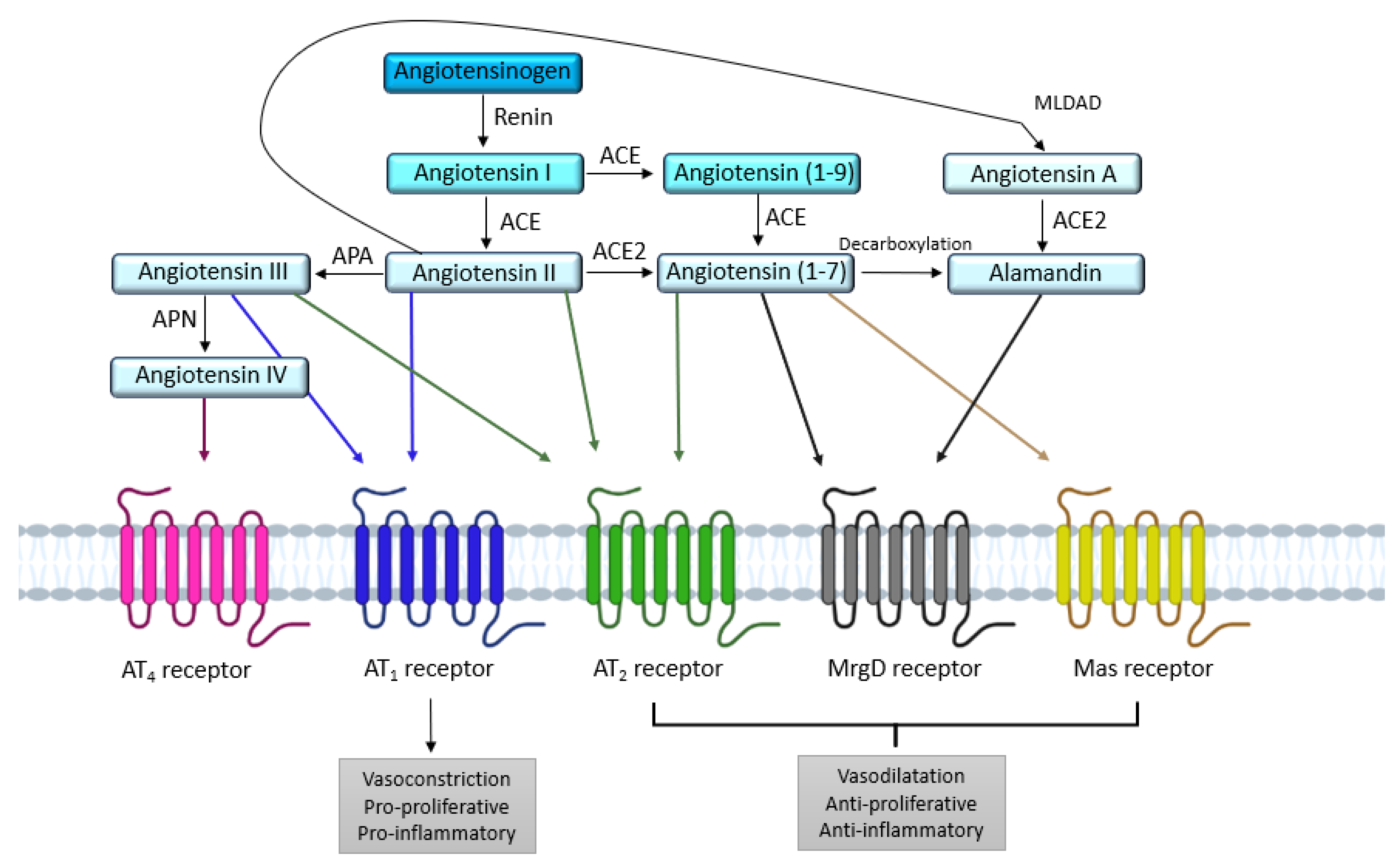
1. Introduction
2. AT1 and AT2 Angiotensin II Receptors
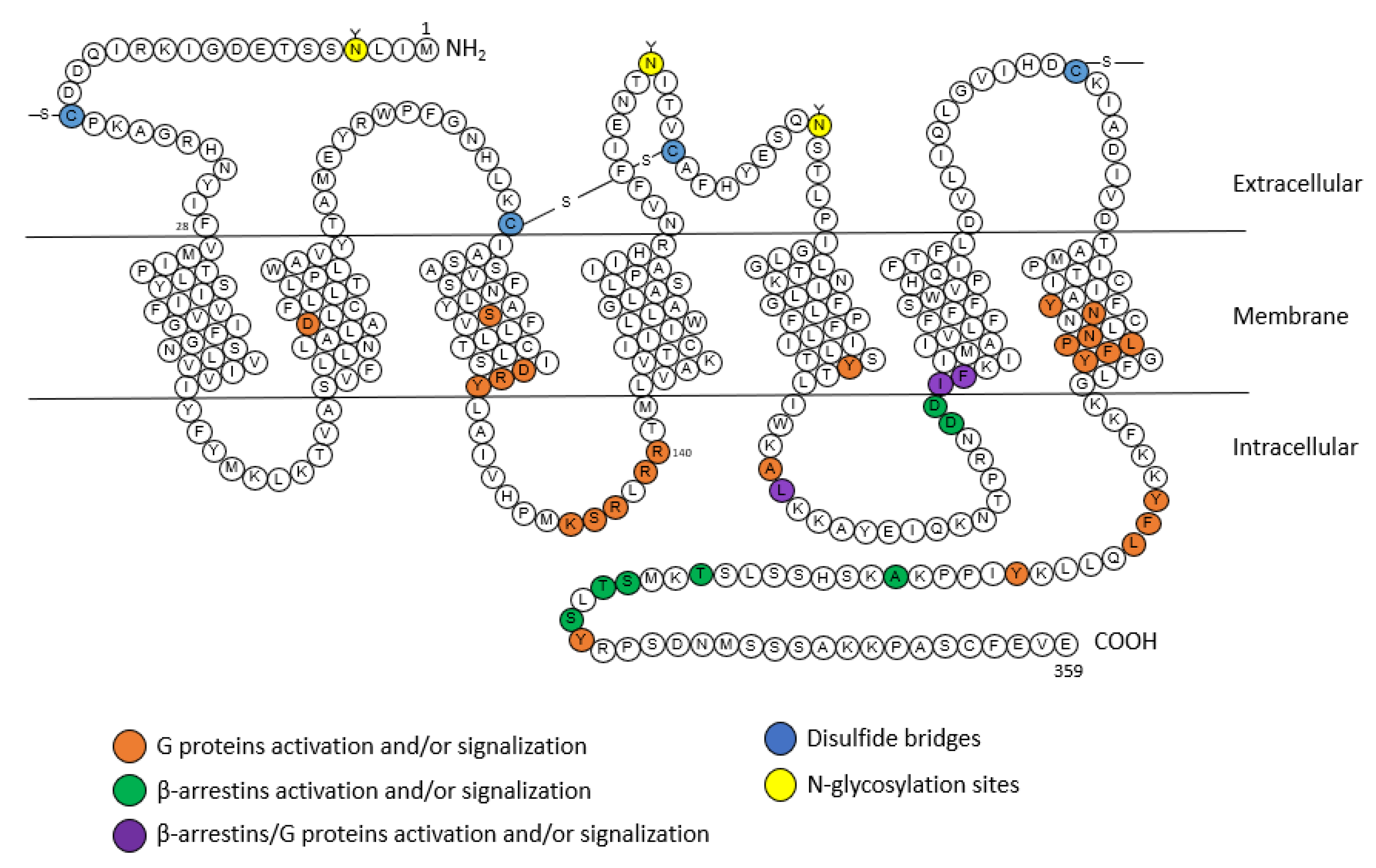
2.1. AT1 Receptor
2.1.1. Structure

2.1.2. Signaling
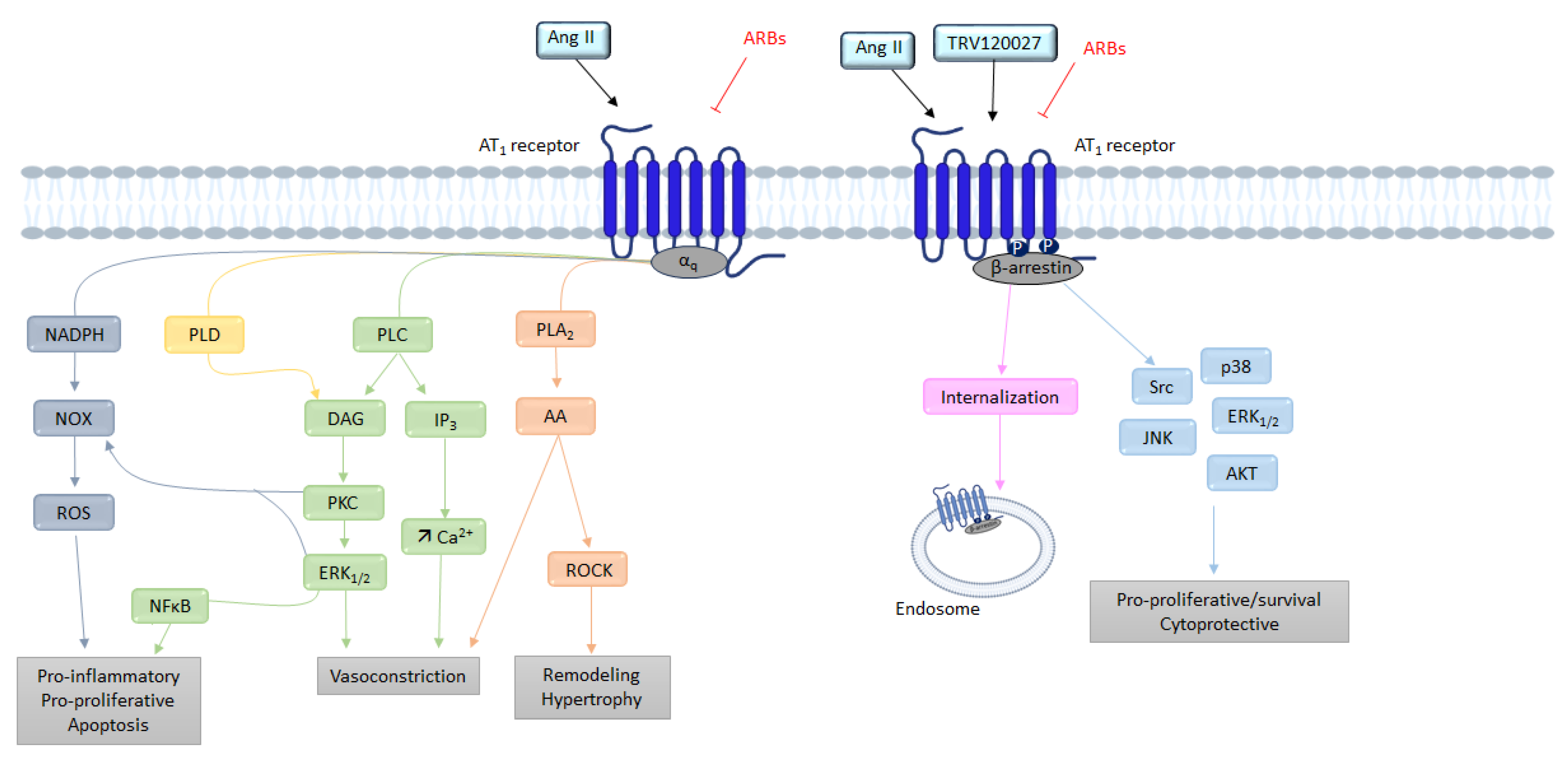
G Protein Pathway
β-Arrestins
NADPH

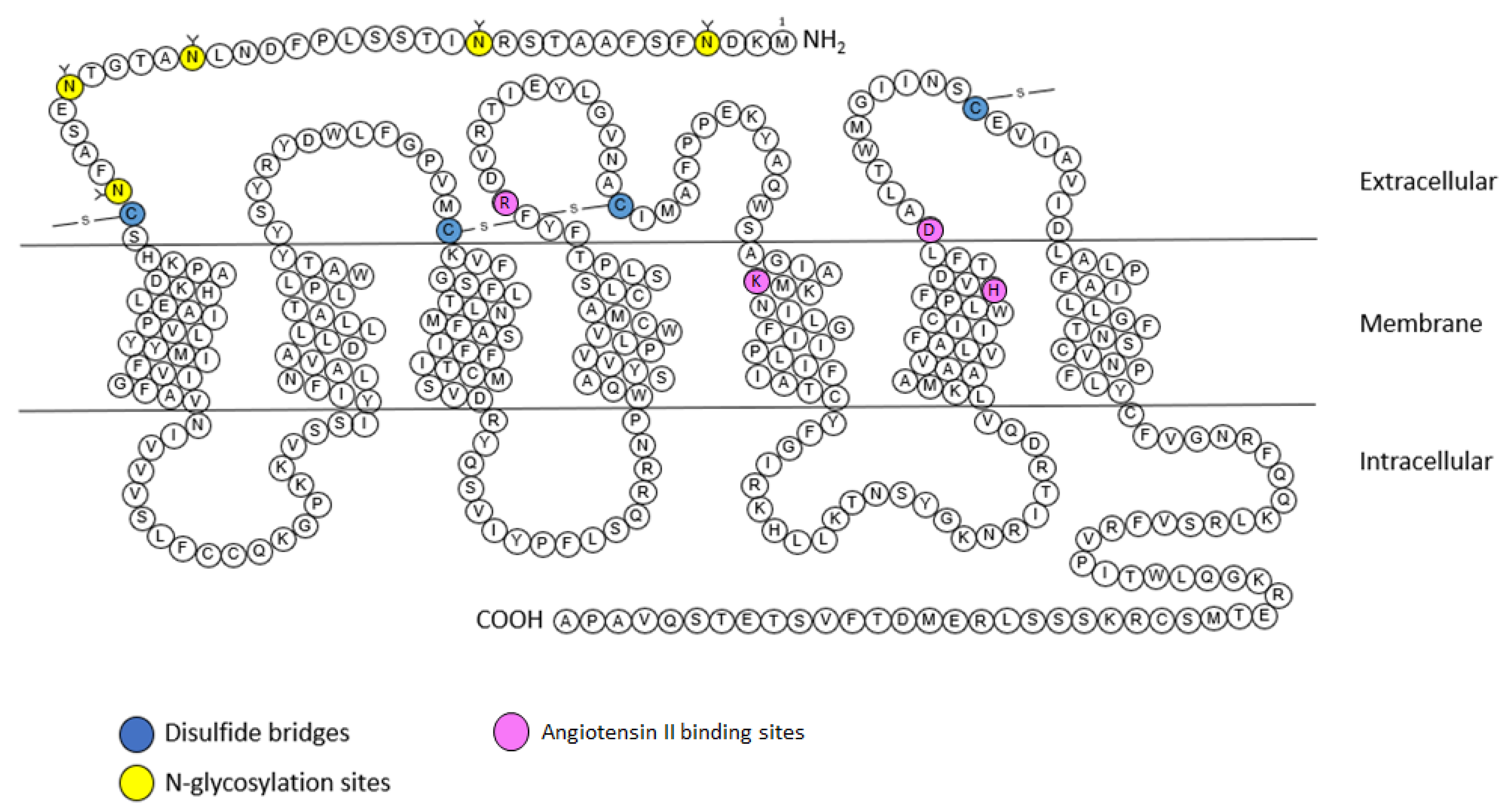
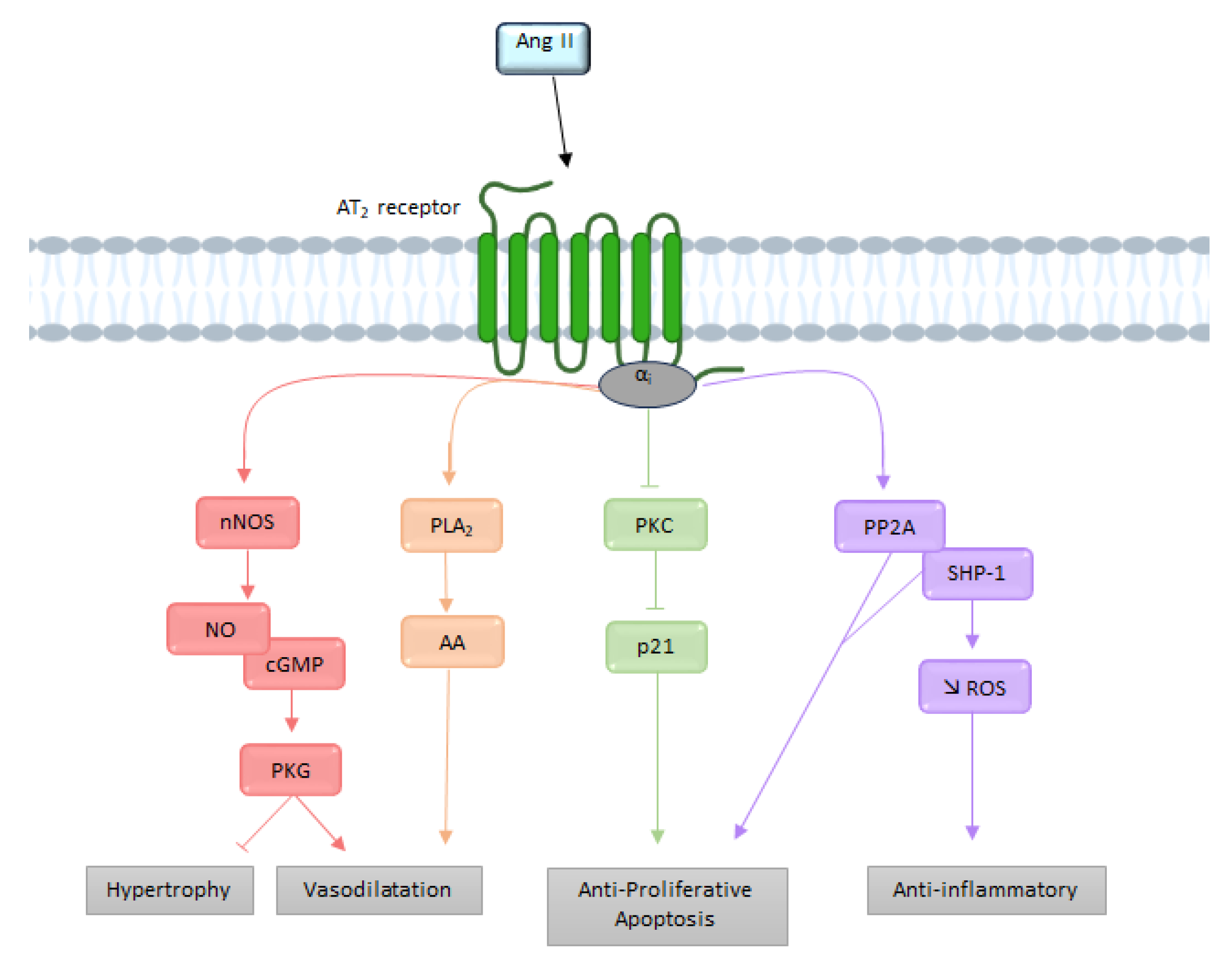
2.2. AT2 Receptor
2.2.1. Structure

2.2.2. Signaling
G Protein Pathway
Bradykinin

3. The Functional AT1/AT2 Receptors Balance
3.1. Systemic Cardiovascular Impact
3.2. AT1/AT2 Balance in the Brain and Cerebral Circulation
3.2.1. Cerebral Circulation
3.2.2. Cardiovascular Regulation
3.2.3. Neuroinflammation
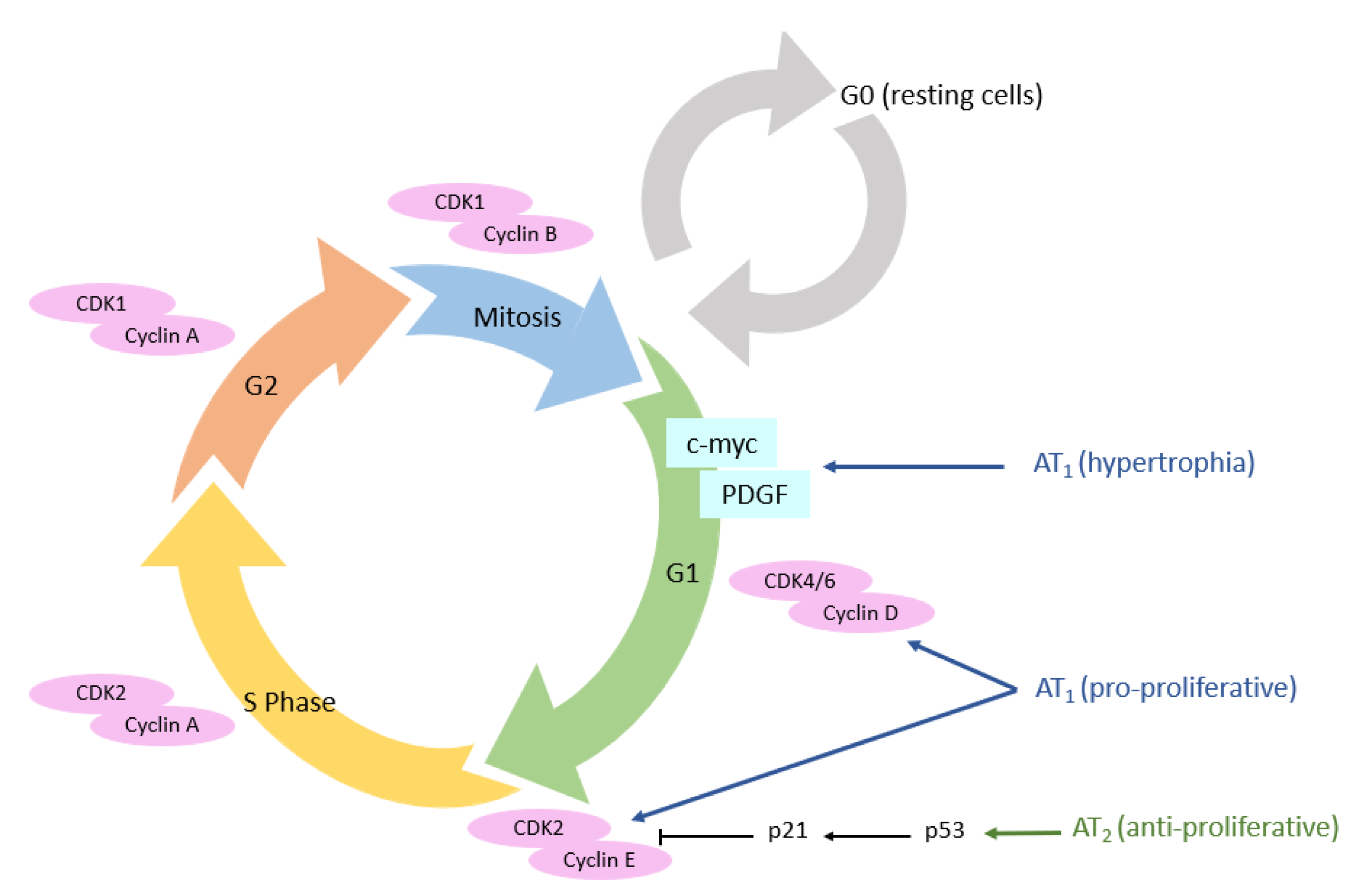
3.3. Cellular Cycle
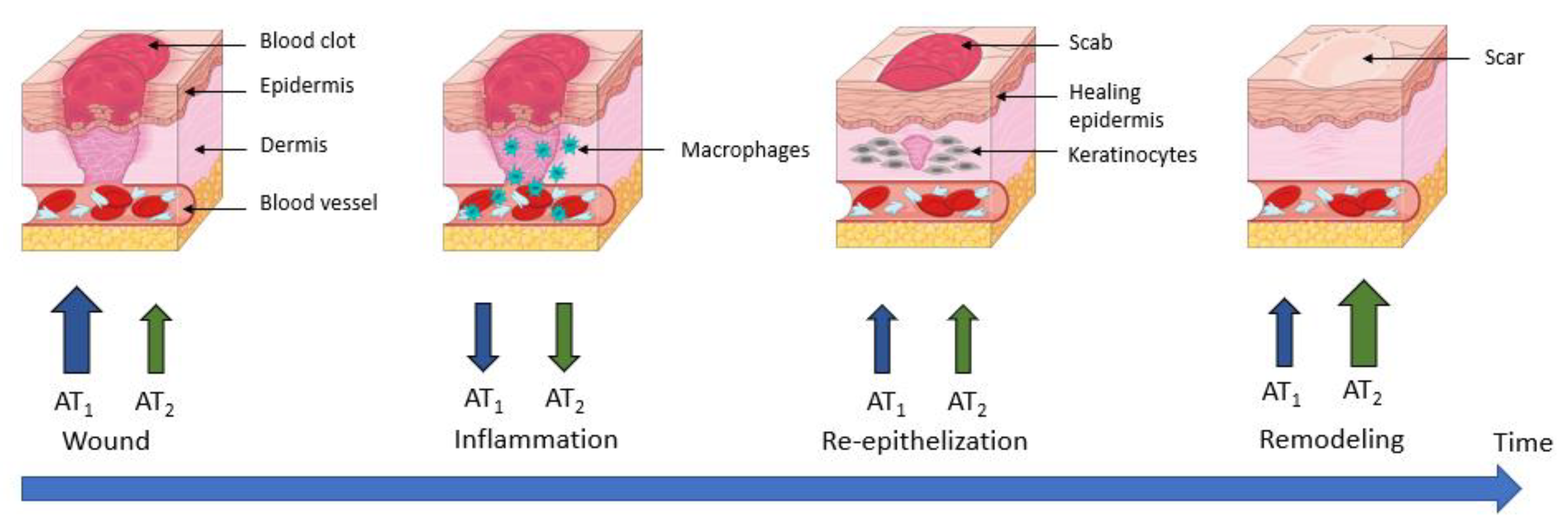
3.4. Wound Healing
4. Mechanisms Regulating the AT1/AT2 Functional Balance
4.1. Functional Opposition vs. Expression Level
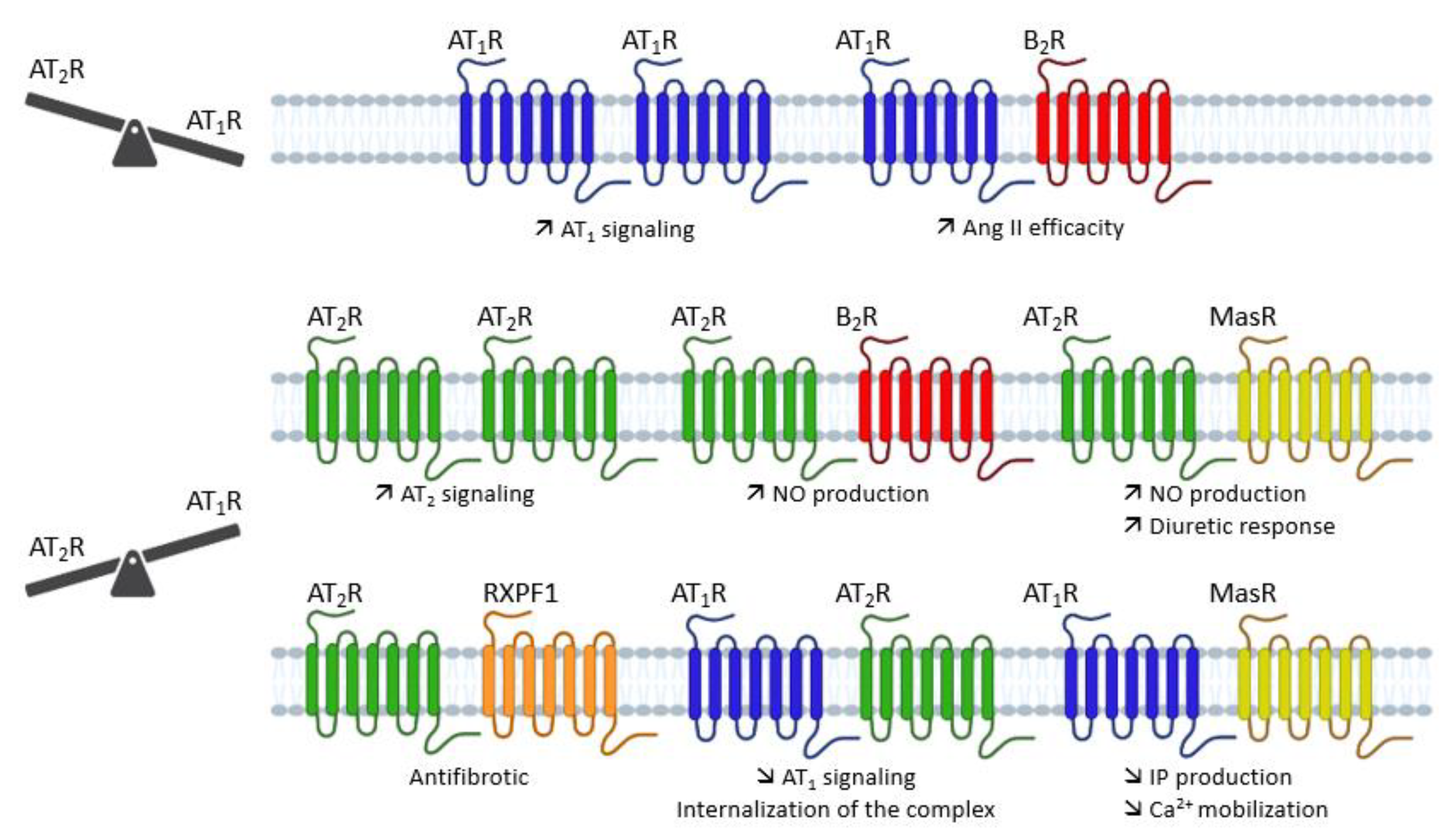
4.2. Direct AT1/AT2 Receptors Interactions
4.2.1. AT1 Receptor Dimerization
4.2.2. AT2 Receptor Dimerization

4.3. Post-Translational Modifications
4.3.1. N-Glycosylation
4.3.2. Phosphorylation
4.3.3. S-Nitrosation
5. Possible Ways to Tune the AT1/AT2 Functional Balance
5.1. Pushing the Balance Using Agonist/Antagonist Ligands

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agonists | |||
| Compounds | AT1 affinity | AT2 affinity | References |
| Ang II | pIC50 = 8.1 | pIC50 = 9.2 | [194] |
| Ang III | pIC50 = 7.6 | pIC50 = 9.2 | [194] |
| Ang IV | N.A. | pIC50 = 7.3 | [194] |
| Ang-(1-7) | pKi = 6.66 | pIC50 = 6.6 | [194,195] |
| SII | pKd = 6.5 | N.A. | [196] |
| TRV120023 | pEC50 = 7.4 | N.A. | [197] |
| TRV120026 | pEC50 = 7.6 | N.A. | [197] |
| TRV120027 | pEC50 = 7.7 | Ki = 7 nM | [197] |
| CGP42112A | N.A. | pIC50 = 9.6 | [194] |
| C21 | N.A. | pIC50 = 8.6 | [194] |
| Antagonists | |||
| Compounds | AT1 affinity | AT2 affinity | References |
| Losartan | pIC50 = 7.4–8.7 | N.A. | [198] |
| Candesartan | pIC50 = 9.5–9.7 | N.A. | [199] |
| Valsartan | pIC50 = 8.6 | N.A. | [200] |
| Telmisartan | pIC50 = 8.4 | N.A. | [201] |
| PD123177 | N.A. | pIC50 = 8.5–9.5 | [202] |
| PD123319 | N.A. | pIC50 = 8.25 | [194] |
5.2. Selective Activation of the β-Arrestin Pathway
5.3. Post-Translational Regulation
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACE | Angiotensin-converting enzyme |
| Ang II | angiotensin II |
| ARB | AT1 receptor blocker |
| AT1 | angiotensin II type 1 receptor |
| AT2 | angiotensin II type 2 receptor |
| B2 | bradykinin receptor |
| BK | bradykinin |
| C21 | compound 21 |
| GPCR | G protein-coupled receptor |
| GRKs | G protein-coupled receptor kinases |
| KO | knock-out |
| MAPK | mitogen-activated protein kinases |
| MasR | Mas receptor |
| NF-kβ | nuclear factor kβ |
| PKC | protein kinase C |
| PLA2 | phospholipase A2 |
| PLC | phospholipase C |
| RAS | renin-angiotensin system |
| ROS | reactive oxygen species |
| VSMC | vascular smooth muscle cells |
References
- Henrion, D.; Chillon, J.-M.; Capdeville-Atkinson, C.; Vinceneux-Feugier, M.; Atkinson, J. Chronic treatment with the angiotensin I converting enzyme inhibitor, perindopril, protects in vitro carbachol-induced vasorelaxation in a rat model of vascular calcium overload. Br. J. Pharmacol. 1991, 104, 966–972. [Google Scholar] [CrossRef][Green Version]
- Lartaud, I.; Bray-des-Boscs, L.; Chillon, J.M.; Atkinson, J.; Capdeville-Atkinson, C. In vivo cerebrovascular reactivity in Wistar and Fischer 344 rat strains during aging. Am. J. Physiol. Heart Circ. Physiol. 1993, 264, H851–H858. [Google Scholar] [CrossRef]
- Régrigny, O.; Atkinson, J.; Capdeville-Atkinson, C.; Limiñana, P.; Chillon, J.-M. Effect of Lovastatin on Cerebral Circulation in Spontaneously Hypertensive Rats. Hypertension 2000, 35, 1105–1110. [Google Scholar] [CrossRef]
- Atkinson, J. Stroke, high blood pressure and the renin–angiotensin–aldosterone system—New developments. Front. Pharm. 2011, 2, 22. [Google Scholar] [CrossRef]
- Tigerstedt, R.; Bergman, P.Q. Niere und Kreislauf 1. Skand. Arch. Für Physiol. 1898, 8, 223–271. [Google Scholar] [CrossRef]
- Guyton, A.C. Blood Pressure Control—Special Role of the Kidneys and Body Fluids. Science 1991, 252, 1813–1816. [Google Scholar] [CrossRef] [PubMed]
- Skeggs, L.T.; Lentz, K.E.; Gould, A.B.; Hochstrasser, H.; Kahn, J.R. Biochemistry and kinetics of the renin-angiotensin system. Fed. Proc. 1967, 26, 42–47. [Google Scholar]
- Murphy, T.J.; Alexander, R.W.; Griendling, K.K.; Runge, M.S.; Bernstein, K.E. Isolation of a cDNA encoding the vascular type-1 angiotensin II receptor. Nature 1991, 351, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Mukoyama, M.; Nakajima, M.; Horiuchi, M.; Sasamura, H.; Pratt, R.E.; Dzau, V.J. Expression cloning of type 2 angiotensin II receptor reveals a unique class of seven-transmembrane receptors. J. Biol. Chem. 1993, 268, 24539–24542. [Google Scholar] [CrossRef]
- Sevá Pessôa, B.; van der Lubbe, N.; Verdonk, K.; Roks, A.J.M.; Hoorn, E.J.; Danser, A.H.J. Key developments in renin–angiotensin–aldosterone system inhibition. Nat. Rev. Nephrol. 2013, 9, 26–36. [Google Scholar] [CrossRef]
- Simões e Silva, A.; Silveira, K.; Ferreira, A.; Teixeira, M. ACE2, angiotensin-(1-7) and Mas receptor axis in inflammation and fibrosis: Angiotensin-(1-7) in inflammation and fibrosis. Br. J. Pharmacol. 2013, 169, 477–492. [Google Scholar] [CrossRef] [PubMed]
- Povlsen, A.; Grimm, D.; Wehland, M.; Infanger, M.; Krüger, M. The Vasoactive Mas Receptor in Essential Hypertension. JCM 2020, 9, 267. [Google Scholar] [CrossRef]
- Schleifenbaum, J. Alamandine and Its Receptor MrgD Pair Up to Join the Protective Arm of the Renin-Angiotensin System. Front. Med. 2019, 6, 107. [Google Scholar] [CrossRef] [PubMed]
- Chai, S.Y.; Fernando, R.; Peck, G.; Ye, S.-Y.; Mendelsohn, F.A.O.; Jenkins, T.A.; Albiston, A.L. What’s new in the renin-angiotensin system?: The angiotensin IV/AT4 receptor. CMLS Cell. Mol. Life Sci. 2004, 61, 2728–2737. [Google Scholar] [CrossRef]
- Kramár, E.A.; Krishnan, R.; Harding, J.W.; Wright, J.W. Role of nitric oxide in angiotensin IV-induced increases in cerebral blood flow. Regul. Pept. 1998, 74, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, T.A.; Handa, R.K.; Harding, J.W.; Wright, J.W. A role for the angiotensin IV/AT4 system in mediating natriuresis in the rat. Peptides 2001, 22, 935–944. [Google Scholar] [CrossRef]
- Wilson, W.L.; Munn, C.; Ross, R.C.; Harding, J.W.; Wright, J.W. The role of the AT4 and cholinergic systems in the Nucleus Basalis Magnocellularis (NBM): Effects on spatial memory. Brain Res. 2009, 1272, 25–31. [Google Scholar] [CrossRef]
- Royea, J.; Hamel, E. Brain angiotensin II and angiotensin IV receptors as potential Alzheimer’s disease therapeutic targets. GeroScience 2020, 42, 1237–1256. [Google Scholar] [CrossRef]
- Horiuchi, M.; Akishita, M.; Dzau, V.J. Recent Progress in Angiotensin II Type 2 Receptor Research in the Cardiovascular System. Hypertension 1999, 33, 613–621. [Google Scholar] [CrossRef]
- Gasparo, M.D.; Catt, K.J.; Inagami, T.; Wright, J.W.; Unger, T. International Union of Pharmacology. XXIII. The Angiotensin II Receptors. Pharmacol. Rev. 2000, 52, 415–472. [Google Scholar]
- Campbell, D.J.; Habener, J.F. Angiotensinogen gene is expressed and differentially regulated in multiple tissues of the rat. J. Clin. Investig. 1986, 78, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Delaitre, C.; Boisbrun, M.; Lecat, S.; Dupuis, F. Targeting the Angiotensin II Type 1 Receptor in Cerebrovascular Diseases: Biased Signaling Raises New Hopes. Int. J. Mol. Sci. 2021, 22, 6738. [Google Scholar] [CrossRef] [PubMed]
- Steckelings, U.M.; Widdop, R.E.; Sturrock, E.D.; Lubbe, L.; Hussain, T.; Kaschina, E.; Unger, T.; Hallberg, A.; Carey, R.M.; Sumners, C. The Angiotensin AT2 Receptor: From a Binding Site to a Novel Therapeutic Target. Pharmacol. Rev. 2022, 74, 1051–1135. [Google Scholar] [CrossRef] [PubMed]
- Hunyady, L.; Catt, K.J. Pleiotropic AT1 Receptor Signaling Pathways Mediating Physiological and Pathogenic Actions of Angiotensin II. Mol. Endocrinol. 2006, 20, 953–970. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef]
- Pueyo, M.E.; N’Diaye, N.; Michel, J.-B. Angiotensin Il-elicited signal transduction via AT1 receptors in endothelial cells. Br. J. Pharmacol. 1996, 118, 79–84. [Google Scholar] [CrossRef]
- Busche, S.; Gallinat, S.; Bohle, R.-M.; Reinecke, A.; Seebeck, J.; Franke, F.; Fink, L.; Zhu, M.; Sumners, C.; Unger, T. Expression of Angiotensin AT1 and AT2 Receptors in Adult Rat Cardiomyocytes after Myocardial Infarction. Am. J. Pathol. 2000, 157, 605–611. [Google Scholar] [CrossRef]
- Siragy, H.M. AT1 and AT2 receptor in the kidney: Role in health and disease. Semin. Nephrol. 2004, 24, 93–100. [Google Scholar] [CrossRef]
- Lenkei, Z.; Palkovits, M.; Corvol, P.; Llorens-Cortès, C. Expression of Angiotensin Type-1 (AT1) and Type-2 (AT2) Receptor mRNAs in the Adult Rat Brain: A Functional Neuroanatomical Review. Front. Neuroendocrinol. 1997, 18, 383–439. [Google Scholar] [CrossRef]
- Johren, O.; Saavedra, J.M. Expression of AT1A and AT1B angiotensin II receptor messenger RNA in forebrain of 2-wk-old rats. Am. J. Physiol. Endocrinol. Metab. 1996, 271, E104–E112. [Google Scholar] [CrossRef]
- Zhang, H.; Unal, H.; Gati, C.; Han, G.W.; Liu, W.; Zatsepin, N.A.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; et al. Structure of the Angiotensin Receptor Revealed by Serial Femtosecond Crystallography. Cell 2015, 161, 833–844. [Google Scholar] [CrossRef]
- Ohyama, K.; Yamano, Y.; Sano, T.; Nakagomi, Y.; Hamakubo, T.; Morishima, I.; Inagami, T. Disulfide bridges in extracellular domains of angiotensin II receptor type IA. Regul. Pept. 1995, 57, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Wingler, L.M.; Skiba, M.A.; McMahon, C.; Staus, D.P.; Kleinhenz, A.L.W.; Suomivuori, C.-M.; Latorraca, N.R.; Dror, R.O.; Lefkowitz, R.J.; Kruse, A.C. Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR. Science 2020, 367, 888–892. [Google Scholar] [CrossRef] [PubMed]
- Suomivuori, C.-M.; Latorraca, N.R.; Wingler, L.M.; Eismann, S.; King, M.C.; Kleinhenz, A.L.W.; Skiba, M.A.; Staus, D.P.; Kruse, A.C.; Lefkowitz, R.J.; et al. Molecular mechanism of biased signaling in a prototypical G protein–coupled receptor. Science. 2020, 367, 881–887. [Google Scholar] [CrossRef]
- Aplin, M.; Bonde, M.M.; Hansen, J.L. Molecular determinants of angiotensin II type 1 receptor functional selectivity. J. Mol. Cell. Cardiol. 2009, 46, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Balakumar, P.; Jagadeesh, G. Structural determinants for binding, activation, and functional selectivity of the angiotensin AT1 receptor. J. Mol. Endocrinol. 2014, 53, R71–R92. [Google Scholar] [CrossRef]
- Kaschina, E.; Unger, T. Angiotensin AT1/AT2 Receptors: Regulation, Signalling and Function. Blood Press. 2003, 12, 70–88. [Google Scholar] [CrossRef]
- Freeman, E.J.; Chisolm, G.M.; Tallant, E.A. Role of calcium and protein kinase C in the activation of phospholipase D by angiotensin II in vascular smooth muscle cells. Arch. Biochem. Biophys. 1995, 319, 84–92. [Google Scholar] [CrossRef]
- Lassegue, B.; Alexander, R.W.; Clark, M.; Akers, M.; Griendling, K.K. Phosphatidylcholine is a major source of phosphatidic acid and diacylglycerol in angiotensin Il-stimulated vascular smooth-muscle cells. Biochem. J. 1993, 292, 509–517. [Google Scholar] [CrossRef]
- Vinturache, A.E.; Smith, F.G. Angiotensin type 1 and type 2 receptors during ontogeny: Cardiovascular and renal effects. Vasc. Pharmacol. 2014, 63, 145–154. [Google Scholar] [CrossRef]
- Rao, G.N.; Lassegue, B.; Alexander, R.W.; Griendling, K.K. Angiotensin 11 stimulates phosphorylation of high-molecular-mass cytosolic phospholipase A2 in vascular smooth-muscle cells. Biochem. J. 1994, 299, 197–201. [Google Scholar] [CrossRef]
- Touyz, R.M.; Schiffrin, E.L. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol. Rev. 2000, 52, 639–672. [Google Scholar] [PubMed]
- Ohtsu, H.; Suzuki, H.; Nakashima, H.; Dhobale, S.; Frank, G.D.; Motley, E.D.; Eguchi, S. Angiotensin II Signal Transduction Through Small GTP-Binding Proteins: Mechanism and Significance in Vascular Smooth Muscle Cells. Hypertension 2006, 48, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Forrester, S.J.; O’Brien, S.; Baggett, A.; Rizzo, V.; Eguchi, S. AT1 receptor signaling pathways in the cardiovascular system. Pharmacol. Res. 2017, 125, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Violin, J.D.; Lefkowitz, R.J. β-Arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol. Sci. 2007, 28, 416–422. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Lefkowitz, R.J. The role of β-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 2002, 115, 455–465. [Google Scholar] [CrossRef]
- Tohgo, A.; Pierce, K.L.; Choy, E.W.; Lefkowitz, R.J.; Luttrell, L.M. β-Arrestin Scaffolding of the ERK Cascade Enhances Cytosolic ERK Activity but Inhibits ERK-mediated Transcription following Angiotensin AT1a Receptor Stimulation. J. Biol. Chem. 2002, 277, 9429–9436. [Google Scholar] [CrossRef]
- Peterson, Y.K.; Luttrell, L.M. The Diverse Roles of Arrestin Scaffolds in G Protein–Coupled Receptor Signaling. Pharmacol. Rev. 2017, 69, 256–297. [Google Scholar] [CrossRef]
- Griendling, K.K.; Minieri, C.A.; Ollerenshaw, J.D.; Alexander, R.W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994, 74, 1141–1148. [Google Scholar] [CrossRef]
- Touyz, R.M.; Briones, A.M. Reactive oxygen species and vascular biology: Implications in human hypertension. Hypertens. Res. 2011, 34, 5–14. [Google Scholar] [CrossRef]
- Tsutsumi, K.; Saavedra, J.M. Characterization and development of angiotensin II receptor subtypes (AT1 and AT2) in rat brain. Am. J. Physiol. 1991, 261, R209–R216. [Google Scholar] [CrossRef] [PubMed]
- Ichiki, T.; Herold, C.L.; Kambayashi, Y.; Bardhan, S.; Inagami, T. Cloning of the cDNA and the genomic DNA of the mouse angiotensin II type 2 receptor. Biochim. Biophys. Acta (BBA) Biomembr. 1994, 1189, 247–250. [Google Scholar] [CrossRef]
- Rompe, F.; Artuc, M.; Hallberg, A.; Alterman, M.; Ströder, K.; Thöne-Reineke, C.; Reichenbach, A.; Schacherl, J.; Dahlöf, B.; Bader, M.; et al. Direct Angiotensin II Type 2 Receptor Stimulation Acts Anti-Inflammatory Through Epoxyeicosatrienoic Acid and Inhibition of Nuclear Factor κB. Hypertension 2010, 55, 924–931. [Google Scholar] [CrossRef]
- Zhang, H.; Han, G.W.; Batyuk, A.; Ishchenko, A.; White, K.L.; Patel, N.; Sadybekov, A.; Zamlynny, B.; Rudd, M.T.; Hollenstein, K.; et al. Structural basis for selectivity and diversity in angiotensin II receptors. Nature 2017, 544, 327–332. [Google Scholar] [CrossRef]
- Griendling, K.K.; Lassègue, B.; Alexander, R.W. Angiotensin receptors and their therapeutic implications. Annu. Rev. Pharmacol. Toxicol. 1996, 36, 281–306. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Pratt, R.E. The AT2 Receptor Selectively Associates with Giα2 and Giα3 in the Rat Fetus. J. Biol. Chem. 1996, 271, 15026–15033. [Google Scholar] [CrossRef]
- Feng, Y.-H.; Saad, Y.; Karnik, S.S. Reversible inactivation of AT2 angiotensin II receptor from cysteine-disulfide bond exchange. FEBS Lett. 2000, 484, 133–138. [Google Scholar] [CrossRef]
- Heerding, J.N.; Yee, D.K.; Jacobs, S.L.; Fluharty, S.J. Mutational analysis of the angiotensin II type 2 receptor: Contribution of conserved extracellular amino acids. Regul. Pept. 1997, 72, 97–103. [Google Scholar] [CrossRef]
- Yee, D.K.; Kisley, L.R.; Heerding, J.N.; Fluharty, S.J. Mutation of a conserved fifth transmembrane domain lysine residue (Lys215) attenuates ligand binding in the angiotensin II type 2 receptor. Brain Res. Mol. Brain Res. 1997, 51, 238–241. [Google Scholar] [CrossRef]
- Turner, C.A.; Cooper, S.; Pulakat, L. Role of the His273 located in the sixth transmembrane domain of the Angiotensin II receptor subtype AT2 in ligand–receptor interaction. Biochem. Biophys. Res. Commun. 1999, 257, 704–707. [Google Scholar] [CrossRef]
- Steckelings, U.M.; Kaschina, E.; Unger, T. The AT2 receptor—A matter of love and hate. Peptides 2005, 26, 1401–1409. [Google Scholar] [CrossRef]
- Turu, G.; Szidonya, L.; Gáborik, Z.; Buday, L.; Spät, A.; Clark, A.J.L.; Hunyady, L. Differential beta-arrestin binding of AT1 and AT2 angiotensin receptors. FEBS Lett. 2006, 580, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Padia, S.H.; Carey, R.M. AT2 receptors: Beneficial counter-regulatory role in cardiovascular and renal function. Pflug. Arch. Eur. J. Physiol. 2013, 465, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Bottari, S.P.; Taylor, V.; King, I.N.; Bogdal, Y.; Whitebread, S.; de Gasparo, M. Angiotensin II AT2 receptors do not interact with guanine nucleotide binding proteins. Eur. J. Pharmacol. Mol. Pharmacol. 1991, 207, 157–163. [Google Scholar] [CrossRef]
- Stennett, A.K.; Qiao, X.; Falone, A.E.; Koledova, V.V.; Khalil, R.A. Increased vascular angiotensin type 2 receptor expression and NOS-mediated mechanisms of vascular relaxation in pregnant rats. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H745–H755. [Google Scholar] [CrossRef] [PubMed]
- Alexander, L.D.; Ding, Y.; Alagarsamy, S.; Cui, X. Angiotensin II stimulates fibronectin protein synthesis via a Gβγ/arachidonic acid-dependent pathway. Am. J. Physiol. Ren. Physiol. 2014, 307, F287–F302. [Google Scholar] [CrossRef]
- Savoia, C.; Tabet, F.; Yao, G.; Schiffrin, E.L.; Touyz, R.M. Negative regulation of RhoA/Rho kinase by angiotensin II type 2 receptor in vascular smooth muscle cells: Role in angiotensin II-induced vasodilation in stroke-prone spontaneously hypertensive rats. J. Hypertens. 2005, 23, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Savoia, C.; Ebrahimian, T.; He, Y.; Gratton, J.-P.; Schiffrin, E.L.; Touyz, R.M. Angiotensin II/AT2 receptor-induced vasodilation in stroke-prone spontaneously hypertensive rats involves nitric oxide and cGMP-dependent protein kinase. J. Hypertens. 2006, 24, 2417–2422. [Google Scholar] [CrossRef] [PubMed]
- Siragy, H.M.; Carey, R.M. The subtype 2 (AT2) angiotensin receptor mediates renal production of nitric oxide in conscious rats. J. Clin. Investig. 1997, 100, 264–269. [Google Scholar] [CrossRef]
- Tsutsumi, Y.; Matsubara, H.; Masaki, H.; Kurihara, H.; Murasawa, S.; Takai, S.; Miyazaki, M.; Nozawa, Y.; Ozono, R.; Nakagawa, K.; et al. Angiotensin II type 2 receptor overexpression activates the vascular kinin system and causes vasodilation. J. Clin. Investig. 1999, 104, 925–935. [Google Scholar] [CrossRef]
- Yayama, K.; Hiyoshi, H.; Imazu, D.; Okamoto, H. Angiotensin II Stimulates Endothelial NO Synthase Phosphorylation in Thoracic Aorta of Mice With Abdominal Aortic Banding Via Type 2 Receptor. Hypertension 2006, 48, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Abadir, P.M.; Periasamy, A.; Carey, R.M.; Siragy, H.M. Angiotensin II Type 2 Receptor–Bradykinin B2 Receptor Functional Heterodimerization. Hypertension 2006, 48, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Dimitropoulou, C.; White, R.E.; Fuchs, L.; Zhang, H.; Catravas, J.D.; Carrier, G.O. Angiotensin II Relaxes Microvessels Via the AT2 Receptor and Ca2+-Activated K+ (BK Ca) Channels. Hypertension 2001, 37, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, J.M.; Armando, I. Angiotensin II AT2 Receptors Contribute to Regulate the Sympathoadrenal and Hormonal Reaction to Stress Stimuli. Cell. Mol. Neurobiol. 2018, 38, 85–108. [Google Scholar] [CrossRef] [PubMed]
- Foulquier, S.; Steckelings, U.M.; Unger, T. Perspective: A tale of two receptors. Nature 2013, 493, S9. [Google Scholar] [CrossRef]
- Anderson, W.P.; Selig, S.E.; Korner, P.I. Role of Angiotensin II in the Hypertension Induced by Renal Artery Stenosis. Clin. Exp. Hypertens. Part A Theory Pract. 1984, 6, 299–314. [Google Scholar] [CrossRef]
- Foulquier, S.; Steckelings, U.M.; Unger, T. Impact of the AT2 Receptor Agonist C21 on Blood Pressure and Beyond. Curr. Hypertens. Rep. 2012, 14, 403–409. [Google Scholar] [CrossRef]
- Fischer-Ferraro, C.; Nahmod, V.E.; Goldstein, D.J.; Finkielman, S. Angiotensin and renin in rat and dog brain. J. Exp. Med. 1971, 133, 353–361. [Google Scholar] [CrossRef]
- Barnes, J.M.; Steward, L.J.; Barber, P.C.; Barnes, N.M. Identification and characterisation of angiotensin II receptor subtypes in human brain. Eur. J. Pharmacol. 1993, 230, 251–258. [Google Scholar] [CrossRef]
- Vincent, J.-M.; Kwan, Y.W.; Lung Chan, S.; Perrin-Sarrado, C.; Atkinson, J.; Chillon, J.-M. Constrictor and Dilator Effects of Angiotensin II on Cerebral Arterioles. Stroke 2005, 36, 2691–2695. [Google Scholar] [CrossRef]
- Dupuis, F.; Atkinson, J.; Limiñana, P.; Chillon, J.-M. Comparative effects of the angiotensin II receptor blocker, telmisartan, and the angiotensin-converting enzyme inhibitor, ramipril, on cerebrovascular structure in spontaneously hypertensive rats. J. Hypertens. 2005, 23, 1061–1066. [Google Scholar] [CrossRef]
- Foulquier, S.; Dupuis, F.; Perrin-Sarrado, C.; Maguin Gatè, K.; Leroy, P.; Liminana, P.; Atkinson, J.; Capdeville-Atkinson, C.; Lartaud, I. Differential Effects of Short-Term Treatment with Two AT1 Receptor Blockers on Diameter of Pial Arterioles in SHR. PLoS ONE 2012, 7, e42469. [Google Scholar] [CrossRef]
- Stenman, E.; Edvinsson, L. Cerebral Ischemia Enhances Vascular Angiotensin AT1 Receptor–Mediated Contraction in Rats. Stroke 2004, 35, 970–974. [Google Scholar] [CrossRef]
- Vikman, P.; Edvinsson, L. Gene expression profiling in the human middle cerebral artery after cerebral ischemia. Eur. J. Neurol. 2006, 13, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Jancovski, N.; Bassi, J.K.; Nguyen-Huu, T.-P.; Choong, Y.-T.; Palma-Rigo, K.; Davern, P.J.; Gurley, S.B.; Thomas, W.G.; Head, G.A.; et al. Angiotensin Type 1A Receptors in C1 Neurons of the Rostral Ventrolateral Medulla Modulate the Pressor Response to Aversive Stress. J. Neurosci. 2012, 32, 2051–2061. [Google Scholar] [CrossRef] [PubMed]
- De Kloet, A.D.; Wang, L.; Ludin, J.A.; Smith, J.A.; Pioquinto, D.J.; Hiller, H.; Steckelings, U.M.; Scheuer, D.A.; Sumners, C.; Krause, E.G. Reporter mouse strain provides a novel look at angiotensin type-2 receptor distribution in the central nervous system. Brain Struct. Funct. 2016, 221, 891–912. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.D.; Wang, G.; Waters, E.M.; Gonzales, K.L.; Speth, R.C.; Van Kempen, T.A.; Marques-Lopes, J.; Young, C.N.; Butler, S.D.; Davisson, R.L.; et al. Distribution of angiotensin type 1a receptor-containing cells in the brains of bacterial artificial chromosome transgenic mice. Neuroscience 2012, 226, 489–509. [Google Scholar] [CrossRef] [PubMed]
- Elsaafien, K.; de Kloet, A.D.; Krause, E.G.; Sumners, C. Brain Angiotensin Type-1 and Type-2 Receptors in Physiological and Hypertensive Conditions: Focus on Neuroinflammation. Curr. Hypertens. Rep. 2020, 22, 48. [Google Scholar] [CrossRef] [PubMed]
- De Kloet, A.D.; Wang, L.; Pitra, S.; Hiller, H.; Smith, J.A.; Tan, Y.; Nguyen, D.; Cahill, K.M.; Sumners, C.; Stern, J.E.; et al. A Unique “Angiotensin-Sensitive” Neuronal Population Coordinates Neuroendocrine, Cardiovascular, and Behavioral Responses to Stress. J. Neurosci. 2017, 37, 3478–3490. [Google Scholar] [CrossRef]
- De Kloet, A.D.; Pitra, S.; Wang, L.; Hiller, H.; Pioquinto, D.J.; Smith, J.A.; Sumners, C.; Stern, J.E.; Krause, E.G. Angiotensin Type-2 Receptors Influence the Activity of Vasopressin Neurons in the Paraventricular Nucleus of the Hypothalamus in Male Mice. Endocrinology 2016, 157, 3167–3180. [Google Scholar] [CrossRef]
- de Oliveira-Sales, E.B.; Nishi, E.E.; Boim, M.A.; Dolnikoff, M.S.; Bergamaschi, C.T.; Campos, R.R. Upregulation of AT1R and iNOS in the Rostral Ventrolateral Medulla (RVLM) Is Essential for the Sympathetic Hyperactivity and Hypertension in the 2K-1C Wistar Rat Model. Am. J. Hypertens. 2010, 23, 708–715. [Google Scholar] [CrossRef]
- Li, Z.; Iwai, M.; Wu, L.; Shiuchi, T.; Jinno, T.; Cui, T.-X.; Horiuchi, M. Role of AT2 receptor in the brain in regulation of blood pressure and water intake. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H116–H121. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Wang, W.-Z.; Wang, W.; Zucker, I.H. Imbalance of Angiotensin Type 1 Receptor and Angiotensin II Type 2 Receptor in the Rostral Ventrolateral Medulla: Potential Mechanism for Sympathetic Overactivity in Heart Failure. Hypertension 2008, 52, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Valero-Esquitino, V.; Lucht, K.; Namsolleck, P.; Monnet-Tschudi, F.; Stubbe, T.; Lucht, F.; Liu, M.; Ebner, F.; Brandt, C.; Danyel, L.A.; et al. Direct angiotensin type 2 receptor (AT2R) stimulation attenuates T-cell and microglia activation and prevents demyelination in experimental autoimmune encephalomyelitis in mice. Clin. Sci. 2015, 128, 95–109. [Google Scholar] [CrossRef]
- Kim, J.-H.; Afridi, R.; Cho, E.; Yoon, J.H.; Lim, Y.-H.; Lee, H.-W.; Ryu, H.; Suk, K. Soluble ANPEP Released From Human Astrocytes as a Positive Regulator of Microglial Activation and Neuroinflammation: Brain Renin–Angiotensin System in Astrocyte–Microglia Crosstalk. Mol. Cell. Proteom. 2022, 21, 100424. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Perez, A.I.; Borrajo, A.; Rodriguez-Pallares, J.; Guerra, M.J.; Labandeira-Garcia, J.L. Interaction between NADPH-oxidase and Rho-kinase in angiotensin II-induced microglial activation: NADPH-Oxidase and Rho-Kinase Interaction. Glia 2015, 63, 466–482. [Google Scholar] [CrossRef]
- Villar-Cheda, B.; Valenzuela, R.; Rodriguez-Perez, A.I.; Guerra, M.J.; Labandeira-Garcia, J.L. Aging-related changes in the nigral angiotensin system enhances proinflammatory and pro-oxidative markers and 6-OHDA-induced dopaminergic degeneration. Neurobiol. Aging 2012, 33, 204.e1–204.e11. [Google Scholar] [CrossRef]
- Labandeira-Garcia, J.L.; Rodríguez-Perez, A.I.; Garrido-Gil, P.; Rodriguez-Pallares, J.; Lanciego, J.L.; Guerra, M.J. Brain Renin-Angiotensin System and Microglial Polarization: Implications for Aging and Neurodegeneration. Front. Aging Neurosci. 2017, 9, 129. [Google Scholar] [CrossRef]
- Campbell, G.J.; Hands, E.L.; Van de Pette, M. The Role of CDKs and CDKIs in Murine Development. Int. J. Mol. Sci. 2020, 21, 5343. [Google Scholar] [CrossRef]
- Han, H.J.; Han, J.Y.; Heo, J.S.; Lee, S.H.; Lee, M.Y.; Kim, Y.H. ANG II-stimulated DNA synthesis is mediated by ANG II receptor-dependent Ca2+/PKC as well as EGF receptor-dependent PI3K/Akt/mTOR/p70S6K1 signal pathways in mouse embryonic stem cells. J. Cell. Physiol. 2007, 211, 618–629. [Google Scholar] [CrossRef]
- Diep, Q.N.; El Mabrouk, M.; Touyz, R.M.; Schiffrin, E.L. Expression of Cell Cycle Proteins in Blood Vessels of Angiotensin II–Infused Rats: Role of AT1 Receptors. Hypertension 2001, 37, 604–608. [Google Scholar] [CrossRef]
- Gingras, B.; Rodier, G.; Giasson, E.; Coulombe, P.; Chassagne, C.; Meloche, S. Expression of angiotensin type II receptor downregulates Cdk4 synthesis and inhibits cell-cycle progression. Oncogene 2003, 22, 2633–2642. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Peluso, A.A.; Kempf, S.J.; Verano-Braga, T.; Rodrigues-Ribeiro, L.; Johansen, L.E.; Hansen, M.R.; Kitlen, G.; Haugaard, A.H.; Sumners, C.; Ditzel, H.J.; et al. Quantitative Phosphoproteomics of the Angiotensin AT2-Receptor Signaling Network Identifies HDAC1 (Histone-Deacetylase-1) and p53 as Mediators of Antiproliferation and Apoptosis. Hypertension 2022, 79, 2530–2541. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Rojas, M.; Schneider, T.; Bartrons, R.; Ventura, F.; Rosa, J.L. NEURL4 regulates the transcriptional activity of tumor suppressor protein p53 by modulating its oligomerization. Oncotarget 2017, 8, 61824–61836. [Google Scholar] [CrossRef]
- Yu, L.; Meng, W.; Ding, J.; Cheng, M. Klotho inhibits angiotensin II-induced cardiomyocyte hypertrophy through suppression of the AT1R/beta catenin pathway. Biochem. Biophys. Res. Commun. 2016, 473, 455–461. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, Y. Wnt/β-catenin signaling and renin–angiotensin system in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2016, 25, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Geisterfer, A.A.; Peach, M.J.; Owens, G.K. Angiotensin II induces hypertrophy, not hyperplasia, of cultured rat aortic smooth muscle cells. Circ. Res. 1988, 62, 749–756. [Google Scholar] [CrossRef]
- Naftilan, A.J.; Pratt, R.E.; Dzau, V.J. Induction of Platelet-derived Growth Factor A-chain and c-myc Gene Expressions by Angiotensin 11 in Cultured Rat Vascular Smooth Muscle Cells. J. Clin. Investig. 1989, 83, 1419–1424. [Google Scholar] [CrossRef]
- Wolf, G.; Wenzel, U.O. Angiotensin II and Cell Cycle Regulation. Hypertension 2004, 43, 693–698. [Google Scholar] [CrossRef]
- Fujihara, S.; Morishita, A.; Ogawa, K.; Tadokoro, T.; Chiyo, T.; Kato, K.; Kobara, H.; Mori, H.; Iwama, H.; Masaki, T. The angiotensin II type 1 receptor antagonist telmisartan inhibits cell proliferation and tumor growth of esophageal adenocarcinoma via the AMPKα/mTOR pathway in vitro and in vivo. Oncotarget 2017, 8, 8536–8549. [Google Scholar] [CrossRef]
- Samukawa, E.; Fujihara, S.; Oura, K.; Iwama, H.; Yamana, Y.; Tadokoro, T.; Chiyo, T.; Kobayashi, K.; Morishita, A.; Nakahara, M.; et al. Angiotensin receptor blocker telmisartan inhibits cell proliferation and tumor growth of cholangiocarcinoma through cell cycle arrest. Int. J. Oncol. 2017, 51, 1674–1684. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Liang, Z.; Zhang, Y.; Jie, F.; Li, J.; Fei, Y.; Huang, Z.; Pei, N.; Wang, S.; Li, A.; et al. Effects of Angiotensin II Type 2 Receptor Overexpression on the Growth of Hepatocellular Carcinoma Cells In Vitro and In Vivo. PLoS ONE 2013, 8, e83754. [Google Scholar] [CrossRef] [PubMed]
- Arrieta, O.; Pineda-Olvera, B.; Guevara-Salazar, P.; Hernández-Pedro, N.; Morales-Espinosa, D.; Cerón-Lizarraga, T.L.; González-De la Rosa, C.H.; Rembao, D.; Segura-Pacheco, B.; Sotelo, J. Expression of AT1 and AT2 angiotensin receptors in astrocytomas is associated with poor prognosis. Br. J. Cancer 2008, 99, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Dolley-Hitze, T.; Jouan, F.; Martin, B.; Mottier, S.; Edeline, J.; Moranne, O.; Le Pogamp, P.; Belaud-Rotureau, M.-A.; Patard, J.-J.; Rioux-Leclercq, N.; et al. Angiotensin-2 receptors (AT1-R and AT2-R), new prognostic factors for renal clear-cell carcinoma? Br. J. Cancer 2010, 103, 1698–1705. [Google Scholar] [CrossRef]
- Perini, M.V.; Dmello, R.S.; Nero, T.L.; Chand, A.L. Evaluating the benefits of renin-angiotensin system inhibitors as cancer treatments. Pharmacol. Ther. 2020, 211, 107527. [Google Scholar] [CrossRef]
- Suganuma, T.; Ino, K.; Shibata, K.; Kajiyama, H.; Nagasaka, T.; Mizutani, S.; Kikkawa, F. Functional Expression of the Angiotensin II Type1 Receptor in Human Ovarian Carcinoma Cells and Its Blockade Therapy Resulting in Suppression of Tumor Invasion, Angiogenesis, and Peritoneal Dissemination. Clin. Cancer Res. 2005, 11, 2686–2694. [Google Scholar] [CrossRef]
- Rhodes, D.R.; Ateeq, B.; Cao, Q.; Tomlins, S.A.; Mehra, R.; Laxman, B.; Kalyana-Sundaram, S.; Lonigro, R.J.; Helgeson, B.E.; Bhojani, M.S.; et al. AGTR1 overexpression defines a subset of breast cancer and confers sensitivity to losartan, an AGTR1 antagonist. Proc. Natl. Acad. Sci. USA 2009, 106, 10284–10289. [Google Scholar] [CrossRef]
- Tamura, M.; Yan, H.; Zegarra-Moro, O.; Edl, J.; Oursler, S.; Chard-Bergstrom, C.; Andrews, G.; Kanehira, T.; Takekoshi, S.; Mernaugh, R. Specific single chain variable fragment (ScFv) antibodies to angiotensin II AT2 receptor: Evaluation of the angiotensin II receptor expression in normal and tumor-bearing mouse lung. J. Mol. Hist. 2008, 39, 351–358. [Google Scholar] [CrossRef]
- Pickel, L.; Matsuzuka, T.; Doi, C.; Ayuzawa, R.; Maurya, D.K.; Xie, S.-X.; Berkland, C.; Tamura, M. Over-expression of angiotensin II type 2 receptor gene induces cell death in lung adenocarcinoma cells. Cancer Biol. Ther. 2010, 9, 277–285. [Google Scholar] [CrossRef][Green Version]
- Pei, N.; Mao, Y.; Wan, P.; Chen, X.; Li, A.; Chen, H.; Li, J.; Wan, R.; Zhang, Y.; Du, H.; et al. Angiotensin II type 2 receptor promotes apoptosis and inhibits angiogenesis in bladder cancer. J. Exp. Clin. Cancer Res. 2017, 36, 77. [Google Scholar] [CrossRef]
- Ito, Y.; Naiki-Ito, A.; Kato, H.; Suzuki, S.; Kuno, T.; Ishiguro, Y.; Takahashi, S.; Uemura, H. Chemopreventive effects of angiotensin II receptor type 2 agonist on prostate carcinogenesis by the down-regulation of the androgen receptor. Oncotarget 2018, 9, 13859–13869. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Lützen, U.; Fritsch, J.; Zuhayra, M.; Schütze, S.; Steckelings, U.M.; Recanti, C.; Namsoleck, P.; Unger, T.; Culman, J. Activation of intracellular angiotensin AT₂ receptors induces rapid cell death in human uterine leiomyosarcoma cells. Clin. Sci. 2015, 128, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Aleksiejczuk, M.; Gromotowicz-Poplawska, A.; Marcinczyk, N.; Przylipiak, A.; Chabielska, E. The expression of the renin-angiotensin-aldosterone system in the skin and its effects on skin physiology and pathophysiology. J. Physiol. Pharmacol. 2019, 70. [Google Scholar] [CrossRef]
- Silva, I.M.S.; Assersen, K.B.; Willadsen, N.N.; Jepsen, J.; Artuc, M.; Steckelings, U.M. The role of the renin-angiotensin system in skin physiology and pathophysiology. Exp. Dermatol. 2020, 29, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, M.; Saavedra, J.M. Expression of angiotensin II AT2 receptors in the rat skin during experimental wound healing. Peptides 1992, 13, 783–786. [Google Scholar] [CrossRef] [PubMed]
- Steckelings, U.M.; Henz, B.M.; Wiehstutz, S.; Unger, T.; Artuc, M. Differential expression of angiotensin receptors in human cutaneous wound healing. Br. J. Dermatol. 2005, 153, 887–893. [Google Scholar] [CrossRef]
- Takeda, H.; Katagata, Y.; Kondo, S. Immunohistochemical study of angiotensin receptors in human anagen hair follicles and basal cell carcinoma. Br. J. Dermatol. 2002, 147, 276–280. [Google Scholar] [CrossRef]
- Nehme, A.; Zouein, F.A.; Zayeri, Z.D.; Zibara, K. An Update on the Tissue Renin Angiotensin System and Its Role in Physiology and Pathology. J. Cardiovasc. Dev. Dis. 2019, 6, 14. [Google Scholar] [CrossRef]
- Jiang, X.; Wu, F.; Xu, Y.; Yan, J.-X.; Wu, Y.-D.; Li, S.-H.; Liao, X.; Liang, J.-X.; Li, Z.-H.; Liu, H.-W. A novel role of angiotensin II in epidermal cell lineage determination: Angiotensin II promotes the differentiation of mesenchymal stem cells into keratinocytes through the p38 MAPK, JNK and JAK2 signalling pathways. Exp. Dermatol. 2019, 28, 59–65. [Google Scholar] [CrossRef]
- Rha, E.Y.; Kim, J.W.; Kim, J.H.; Yoo, G. Angiotensin-Converting Enzyme Inhibitor, Captopril, Improves Scar Healing in Hypertensive Rats. Int. J. Med. Sci. 2021, 18, 975–983. [Google Scholar] [CrossRef]
- Jadhav, S.S.; Sharma, N.; Meeks, C.J.; Mordwinkin, N.M.; Espinoza, T.B.; Roda, N.R.; DiZerega, G.S.; Hill, C.K.; Louie, S.G.; Rodgers, K.E. Effects of combined radiation and burn injury on the renin-angiotensin system: CRBI and renin-angiotensin system. Wound Repair. Regen. 2013, 21, 131–140. [Google Scholar] [CrossRef]
- Kamiñska, M.; Mogielnicki, A.; Stankiewicz, A.; Kramkowski, K.; Domaniewski, T.; Buczko, W.; Chabielska, E. Angiotensin Ii Via At1 Receptor Accelerates Arterial Thrombosis In Renovascular Hypertensive Rats. J. Physiol. Pharmacol. 2005, 56, 571–585. [Google Scholar] [PubMed]
- Bernasconi, R.; Nyström, A. Balance and circumstance: The renin angiotensin system in wound healing and fibrosis. Cell. Signal. 2018, 51, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Yahata, Y.; Shirakata, Y.; Tokumaru, S.; Yang, L.; Dai, X.; Tohyama, M.; Tsuda, T.; Sayama, K.; Iwai, M.; Horiuchi, M.; et al. A Novel Function of Angiotensin II in Skin Wound Healing. J. Biol. Chem. 2006, 281, 13209–13216. [Google Scholar] [CrossRef] [PubMed]
- Faghih, M.; Hosseini, S.M.; Smith, B.; Ansari, A.M.; Lay, F.; Ahmed, A.K.; Inagami, T.; Marti, G.P.; Harmon, J.W.; Walston, J.D.; et al. Knockout of Angiotensin AT2 receptors accelerates healing but impairs quality. Aging 2015, 7, 1185–1197. [Google Scholar] [CrossRef]
- Hedayatyanfard, K.; Haddadi, N.; Ziai, S.A.; Karim, H.; Niazi, F.; Steckelings, U.M.; Habibi, B.; Modarressi, A.; Dehpour, A. The renin-angiotensin system in cutaneous hypertrophic scar and keloid formation. Exp. Dermatol. 2020, 29, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Karppinen, S.-M.; Heljasvaara, R.; Gullberg, D.; Tasanen, K.; Pihlajaniemi, T. Toward understanding scarless skin wound healing and pathological scarring. F1000Res 2019, 8, 787. [Google Scholar] [CrossRef]
- Murphy, A.M.; Wong, A.L.; Bezuhly, M. Modulation of angiotensin II signaling in the prevention of fibrosis. Fibrogenes. Tissue Repair. 2015, 8, 7. [Google Scholar] [CrossRef]
- Murphy, A.; LeVatte, T.; Boudreau, C.; Midgen, C.; Gratzer, P.; Marshall, J.; Bezuhly, M. Angiotensin II Type I Receptor Blockade Is Associated with Decreased Cutaneous Scar Formation in a Rat Model. Plast. Reconstr. Surg. 2019, 144, 803e–813e. [Google Scholar] [CrossRef]
- Huang, Y.; Li, J.; Wang, Y.; Chen, D.; Huang, J.; Dai, W.; Peng, P.; Guo, L.; Lei, Y. Intradermal delivery of an angiotensin II receptor blocker using a personalized microneedle patch for treatment of hypertrophic scars. Biomater. Sci. 2023, 11, 583–595. [Google Scholar] [CrossRef]
- Ito, M.; Oliverio, M.I.; Mannon, P.J.; Best, C.F.; Maeda, N.; Smithies, O.; Coffman, T.M. Regulation of blood pressure by the type 1A angiotensin II receptor gene. Proc. Natl. Acad. Sci. USA 1995, 92, 3521–3525. [Google Scholar] [CrossRef]
- Siragy, H.M.; Inagami, T.; Ichiki, T.; Carey, R.M. Sustained hypersensitivity to angiotensin II and its mechanism in mice lacking the subtype-2 (AT2) angiotensin receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 6506–6510. [Google Scholar] [CrossRef] [PubMed]
- Daviet, L.; Lehtonen, J.Y.; Tamura, K.; Griese, D.P.; Horiuchi, M.; Dzau, V.J. Cloning and characterization of ATRAP, a novel protein that interacts with the angiotensin II type 1 receptor. J. Biol. Chem. 1999, 274, 17058–17062. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Ilasaca, M.; Liu, X.; Tamura, K.; Dzau, V.J. The angiotensin II type I receptor-associated protein, ATRAP, is a transmembrane protein and a modulator of angiotensin II signaling. Mol. Biol. Cell. 2003, 14, 5038–5050. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Iwai, M.; Horiuchi, M. Emerging Concepts of Regulation of Angiotensin II Receptors: New Players and Targets for Traditional Receptors. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2532–2539. [Google Scholar] [CrossRef]
- Tanaka, Y.; Tamura, K.; Koide, Y.; Sakai, M.; Tsurumi, Y.; Noda, Y.; Umemura, M.; Ishigami, T.; Uchino, K.; Kimura, K.; et al. The novel angiotensin II type 1 receptor (AT1R)-associated protein ATRAP downregulates AT1R and ameliorates cardiomyocyte hypertrophy. FEBS Lett. 2005, 579, 1579–1586. [Google Scholar] [CrossRef]
- Guo, D.-F.; Chenier, I.; Tardif, V.; Orlov, S.N.; Inagami, T. Type 1 angiotensin II receptor-associated protein ARAP1 binds and recycles the receptor to the plasma membrane. Biochem. Biophys. Res. Commun. 2003, 310, 1254–1265. [Google Scholar] [CrossRef]
- Guo, D.-F.; Chenier, I.; Lavoie, J.L.; Chan, J.S.D.; Hamet, P.; Tremblay, J.; Chen, X.M.; Wang, D.H.; Inagami, T. Development of hypertension and kidney hypertrophy in transgenic mice overexpressing ARAP1 gene in the kidney. Hypertension 2006, 48, 453–459. [Google Scholar] [CrossRef][Green Version]
- Wruck, C.J.; Funke-Kaiser, H.; Pufe, T.; Kusserow, H.; Menk, M.; Schefe, J.H.; Kruse, M.L.; Stoll, M.; Unger, T. Regulation of transport of the angiotensin AT2 receptor by a novel membrane-associated Golgi protein. Arterioscl. Thromb. Vasc. Biol. 2005, 25, 57–64. [Google Scholar] [CrossRef]
- Reinemund, J.; Seidel, K.; Steckelings, U.M.; Zaade, D.; Klare, S.; Rompe, F.; Katerbaum, M.; Schacherl, J.; Li, Y.; Menk, M.; et al. Poly(ADP-ribose) polymerase-1 (PARP-1) transcriptionally regulates angiotensin AT2 receptor (AT2R) and AT2R binding protein (ATBP) genes. Biochem. Pharmacol. 2009, 77, 1795–1805. [Google Scholar] [CrossRef]
- Senbonmatsu, T.; Saito, T.; Landon, E.J.; Watanabe, O.; Price, E.J.; Roberts, R.L.; Imboden, H.; Fitzgerald, T.G.; Gaffney, F.A.; Inagami, T. A novel angiotensin II type 2 receptor signaling pathway: Possible role in cardiac hypertrophy. EMBO J. 2003, 22, 6471–6482. [Google Scholar] [CrossRef]
- Foulquier, S.; Dupuis, F.; Perrin-Sarrado, C.; Maguin Gaté, K.; Merhi-Soussi, F.; Liminana, P.; Kwan, Y.-W.; Capdeville-Atkinson, C.; Lartaud, I.; Atkinson, J. High salt intake abolishes AT2-mediated vasodilation of pial arterioles in rats. J. Hypertens. 2011, 29, 1392–1399. [Google Scholar] [CrossRef]
- AbdAlla, S.; Lother, H.; Langer, A.; el Faramawy, Y.; Quitterer, U. Factor XIIIA Transglutaminase Crosslinks AT1 Receptor Dimers of Monocytes at the Onset of Atherosclerosis. Cell 2004, 119, 343–354. [Google Scholar] [CrossRef]
- Oro, C.; Qian, H.; Thomas, W.G. Type 1 angiotensin receptor pharmacology: Signaling beyond G proteins. Pharmacol. Ther. 2007, 113, 210–226. [Google Scholar] [CrossRef]
- AbdAlla, S.; Lother, H.; Quitterer, U. AT1-receptor heterodimers show enhanced G-protein activation and altered receptor sequestration. Nature 2000, 407, 94–98. [Google Scholar] [CrossRef]
- Bokslag, A.; van Weissenbruch, M.; Mol, B.W.; de Groot, C.J.M. Preeclampsia; short and long-term consequences for mother and neonate. Early Hum. Dev. 2016, 102, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Quitterer, U.; AbdAlla, S. Pathological AT1R-B2R Protein Aggregation and Preeclampsia. Cells 2021, 10, 2609. [Google Scholar] [CrossRef] [PubMed]
- Kostenis, E.; Milligan, G.; Christopoulos, A.; Sanchez-Ferrer, C.F.; Heringer-Walther, S.; Sexton, P.M.; Gembardt, F.; Kellett, E.; Martini, L.; Vanderheyden, P.; et al. G-Protein–Coupled Receptor Mas Is a Physiological Antagonist of the Angiotensin II Type 1 Receptor. Circulation 2005, 111, 1806–1813. [Google Scholar] [CrossRef]
- Miura, S.; Karnik, S.S.; Saku, K. Constitutively Active Homo-oligomeric Angiotensin II Type 2 Receptor Induces Cell Signaling Independent of Receptor Conformation and Ligand Stimulation. J. Biol. Chem. 2005, 280, 18237–18244. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Pfleger, K.D.G.; Seeber, R.M.; Qian, H.; Oro, C.; Abogadie, F.; Delbridge, L.M.D.; Thomas, W.G. Heteromerization of angiotensin receptors changes trafficking and arrestin recruitment profiles. Cell. Signal. 2011, 23, 1767–1776. [Google Scholar] [CrossRef]
- Zha, D.; Cheng, H.; Li, W.; Wu, Y.; Li, X.; Zhang, L.; Feng, Y.-H.; Wu, X. High glucose instigates tubulointerstitial injury by stimulating hetero-dimerization of adiponectin and angiotensin II receptors. Biochem. Biophys. Res. Commun. 2017, 493, 840–846. [Google Scholar] [CrossRef]
- Abadir, P.M.; Carey, R.M.; Siragy, H.M. Angiotensin AT2 Receptors Directly Stimulate Renal Nitric Oxide in Bradykinin B2-Receptor–Null Mice. Hypertension 2003, 42, 600–604. [Google Scholar] [CrossRef]
- Zhao, Y.; Biermann, T.; Luther, C.; Unger, T.; Culman, J.; Gohlke, P. Contribution of bradykinin and nitric oxide to AT2 receptor-mediated differentiation in PC12 W cells: Angiotensin II and PC12 W cell differentiation. J. Neurochem. 2003, 85, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.N.; Ali, Q.; Samuel, P.; Steckelings, U.M.; Hussain, T. Angiotensin II Type 2 Receptor and Receptor Mas Are Colocalized and Functionally Interdependent in Obese Zucker Rat Kidney. Hypertension 2017, 70, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Walters, P.E.; Gaspari, T.A.; Widdop, R.E. Angiotensin-(1–7) Acts as a Vasodepressor Agent Via Angiotensin II Type 2 Receptors in Conscious Rats. Hypertension 2005, 45, 960–966. [Google Scholar] [CrossRef]
- Durand, M.J.; Raffai, G.; Weinberg, B.D.; Lombard, J.H. Angiotensin-(1-7) and low-dose angiotensin II infusion reverse salt-induced endothelial dysfunction via different mechanisms in rat middle cerebral arteries. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1024–H1033. [Google Scholar] [CrossRef]
- Roks, A.J.; Nijholt, J.; van Buiten, A.; van Gilst, W.H.; de Zeeuw, D.; Henning, R.H. Low sodium diet inhibits the local counter-regulator effect of angiotensin-(1-7) on angiotensin II. J. Hypertens. 2004, 22, 2355–2361. [Google Scholar] [CrossRef][Green Version]
- Leonhardt, J.; Villela, D.C.; Teichmann, A.; Münter, L.-M.; Mayer, M.C.; Mardahl, M.; Kirsch, S.; Namsolleck, P.; Lucht, K.; Benz, V.; et al. Evidence for Heterodimerization and Functional Interaction of the Angiotensin Type 2 Receptor and the Receptor MAS. Hypertension 2017, 69, 1128–1135. [Google Scholar] [CrossRef]
- Lemos, V.S.; Silva, D.M.R.; Walther, T.; Alenina, N.; Bader, M.; Santos, R.A.S. The Endothelium-Dependent Vasodilator Effect of the Nonpeptide Ang(1-7) Mimic AVE 0991 Is Abolished in the Aorta of Mas-Knockout Mice. J. Cardiovasc. Pharmacol. 2005, 46, 274–279. [Google Scholar] [CrossRef]
- AbdAlla, S.; Lother, H.; Abdel-tawab, A.M.; Quitterer, U. The Angiotensin II AT2 Receptor Is an AT1Receptor Antagonist. J. Biol. Chem. 2001, 276, 39721–39726. [Google Scholar] [CrossRef] [PubMed]
- Inuzuka, T.; Fujioka, Y.; Tsuda, M.; Fujioka, M.; Satoh, A.O.; Horiuchi, K.; Nishide, S.; Nanbo, A.; Tanaka, S.; Ohba, Y. Attenuation of ligand-induced activation of angiotensin II type 1 receptor signaling by the type 2 receptor via protein kinase C. Sci Rep 2016, 6, 21613. [Google Scholar] [CrossRef] [PubMed]
- Rivas-Santisteban, R.; Rodriguez-Perez, A.I.; Muñoz, A.; Reyes-Resina, I.; Labandeira-García, J.L.; Navarro, G.; Franco, R. Angiotensin AT1 and AT2 receptor heteromer expression in the hemilesioned rat model of Parkinson’s disease that increases with levodopa-induced dyskinesia. J. Neuroinflamm. 2020, 17, 243. [Google Scholar] [CrossRef] [PubMed]
- Lanctôt, P.M.; Leclerc, P.C.; Escher, E.; Leduc, R.; Guillemette, G. Role of N-glycosylation in the expression and functional properties of human AT1 receptor. Biochemistry 1999, 38, 8621–8627. [Google Scholar] [CrossRef]
- Lanctot, P.M.; Leclerc, P.C.; Clément, M.; Auger-Messier, M.; Escher, E.; Leduc, R.; Guillemette, G. Importance of N-glycosylation positioning for cell-surface expression, targeting, affinity and quality control of the human AT1 receptor. Biochem. J. 2005, 390, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Servant, G.; Dudley, D.T.; Escher, E.; Guillemette, G. The marked disparity between the sizes of angiotensin type 2 receptors from different tissues is related to different degrees of N-glycosylation. Mol. Pharmacol. 1994, 45, 1112–1118. [Google Scholar]
- Servant, G.; Dudley, D.T.; Escher, E.; Guillemette, G. Analysis of the role of N-glycosylation in cell-surface expression and binding properties of angiotensin II type-2 receptor of rat pheochromocytoma cells. Biochem. J. 1996, 313, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Qian, H. Association of -Arrestin 1 with the Type 1A Angiotensin II Receptor Involves Phosphorylation of the Receptor Carboxyl Terminus and Correlates with Receptor Internalization. Mol. Endocrinol. 2001, 15, 1706–1719. [Google Scholar] [CrossRef]
- Kule, C.E.; Karoor, V.; Day, J.N.E.; Thomas, W.G.; Baker, K.M.; Dinh, D.; Acker, K.A.; Booz, G.W. Agonist-dependent internalization of the angiotensin II type one receptor (AT1): Role of C-terminus phosphorylation in recruitment of β-arrestins. Regul. Pept. 2004, 120, 141–148. [Google Scholar] [CrossRef]
- Chen, K.; Fu, C.; Chen, C.; Jose, P.A.; Zeng, C. Role of GRK4 in the Regulation of Arterial AT1 Receptor in Hypertension. J. Am. Soc. Hypertens. 2016, 10, e3. [Google Scholar] [CrossRef][Green Version]
- Olivares-Reyes, J.A.; Smith, R.D.; Hunyady, L.; Shah, B.H.; Catt, K.J. Agonist-induced Signaling, Desensitization, and Internalization of a Phosphorylation-deficient AT1A Angiotensin Receptor. J. Biol. Chem. 2001, 276, 37761–37768. [Google Scholar] [CrossRef]
- Zhang, F.; Lei, L.; Huang, J.; Wang, W.; Su, Q.; Yan, H.; Chen, C.; Zheng, S.; Ren, H.; Li, Z.; et al. G-protein-coupled receptor kinase 4 causes renal angiotensin II type 2 receptor dysfunction by increasing its phosphorylation. Clin. Sci. 2022, 136, 989–1003. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, P.C.; Lanctot, P.M.; Auger-Messier, M.; Escher, E.; Leduc, R.; Guillemette, G. S-nitrosylation of cysteine 289 of the AT1 receptor decreases its binding affinity for angiotensin II: S -nitrosylation of the AT1 receptor. Br. J. Pharmacol. 2006, 148, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.H.; Chun, J.N.; Godo, S.; Wu, G.; Shimokawa, H.; Jin, C.Z.; Jeon, J.H.; Kim, S.J.; Jin, Z.H.; Zhang, Y.H. ROS and endothelial nitric oxide synthase (eNOS)-dependent trafficking of angiotensin II type 2 receptor begets neuronal NOS in cardiac myocytes. Basic Res. Cardiol. 2015, 110, 21. [Google Scholar] [CrossRef]
- Sica, D.A.; Gehr, T.W.B.; Ghosh, S. Clinical Pharmacokinetics of Losartan. Clin. Pharmacokinet. 2005, 44, 797–814. [Google Scholar] [CrossRef]
- Barber, M.N.; Sampey, D.B.; Widdop, R.E. AT2 Receptor Stimulation Enhances Antihypertensive Effect of AT1 Receptor Antagonist in Hypertensive Rats. Hypertension 1999, 34, 1112–1116. [Google Scholar] [CrossRef]
- Ewert, S.; Laesser, M.; Johansson, B.; Holm, M.; Aneman, A.; Fandriks, L. The angiotensin II receptor type 2 agonist CGP 42112A stimulates NO production in the porcine jejunal mucosa. BMC Pharmacol. 2003, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Wallinder, C.; Plouffe, B.; Beaudry, H.; Mahalingam, A.K.; Wu, X.; Johansson, B.; Holm, M.; Botoros, M.; Karlén, A.; et al. Design, Synthesis, and Biological Evaluation of the First Selective Nonpeptide AT2 Receptor Agonist. J. Med. Chem. 2004, 47, 5995–6008. [Google Scholar] [CrossRef]
- Carey, R. Role of the angiotensin AT2 receptor in blood pressure regulation and therapeutic implications. Am. J. Hypertens. 2001, 14, S98–S102. [Google Scholar] [CrossRef]
- Sumners, C.; de Kloet, A.D.; Krause, E.G.; Unger, T.; Steckelings, U.M. Angiotensin type 2 receptors: Blood pressure regulation and end organ damage. Curr. Opin. Pharmacol. 2015, 21, 115–121. [Google Scholar] [CrossRef]
- Zizzo, M.G.; Caldara, G.; Bellanca, A.; Nuzzo, D.; Di Carlo, M.; Serio, R. PD123319, angiotensin II type II receptor antagonist, inhibits oxidative stress and inflammation in 2, 4-dinitrobenzene sulfonic acid-induced colitis in rat and ameliorates colonic contractility. Inflammopharmacology 2020, 28, 187–199. [Google Scholar] [CrossRef]
- Wagenaar, G.T.M.; Sengers, R.M.A.; Laghmani, E.H.; Chen, X.; Lindeboom, M.P.H.A.; Roks, A.J.M.; Folkerts, G.; Walther, F.J. Angiotensin II type 2 receptor ligand PD123319 attenuates hyperoxia-induced lung and heart injury at a low dose in newborn rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L261–L272. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, F.; Vincent, J.-M.; Limiñana, P.; Chillon, J.-M.; Capdeville-Atkinson, C.; Atkinson, J. Effects of suboptimal doses of the AT1 receptor blocker, telmisartan, with the angiotensin-converting enzyme inhibitor, ramipril, on cerebral arterioles in spontaneously hypertensive rat. J. Hypertens. 2010, 28, 1566–1573. [Google Scholar] [CrossRef] [PubMed]
- Budzyn, K.; Sobey, C.G.; Drummond, G.R. Reduced cerebrovascular remodeling and functional impairment in spontaneously hypertensive rats following combined treatment with suboptimal doses of telmisartan and ramipril: Is less really more? J. Hypertens. 2010, 28, 1384–1389. [Google Scholar] [CrossRef] [PubMed]
- Bosnyak, S.; Jones, E.S.; Christopoulos, A.; Aguilar, M.-I.; Thomas, W.G.; Widdop, R.E. Relative affinity of angiotensin peptides and novel ligands at AT1 and AT2 receptors. Clin. Sci. 2011, 121, 297–303. [Google Scholar] [CrossRef]
- Teixeira, L.B.; Parreiras-e-Silva, L.T.; Bruder-Nascimento, T.; Duarte, D.A.; Simões, S.C.; Costa, R.M.; Rodríguez, D.Y.; Ferreira, P.A.B.; Silva, C.A.A.; Abrao, E.P.; et al. Ang-(1-7) is an endogenous β-arrestin-biased agonist of the AT1 receptor with protective action in cardiac hypertrophy. Sci. Rep. 2017, 7, 11903. [Google Scholar] [CrossRef]
- Wei, H.; Ahn, S.; Shenoy, S.K.; Karnik, S.S.; Hunyady, L.; Luttrell, L.M.; Lefkowitz, R.J. Independent β-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc. Natl. Acad. Sci. USA 2003, 100, 10782–10787. [Google Scholar] [CrossRef]
- Violin, J.D.; DeWire, S.M.; Yamashita, D.; Rominger, D.H.; Nguyen, L.; Schiller, K.; Whalen, E.J.; Gowen, M.; Lark, M.W. Selectively engaging β-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J. Pharmacol. Exp. Ther. 2010, 335, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Timmermans, P.B.; Carini, D.J.; Chiu, A.T.; Duncia, J.V.; Price, W.A.J.; Wells, G.J.; Wong, P.C.; Johnson, A.L.; Wexler, R.R. The discovery of a new class of highly specific nonpeptide angiotensin II receptor antagonists. Am. J. Hypertens. 1991, 4, 275S–281S. [Google Scholar] [CrossRef]
- Vanderheyden, P.M.L.; Fierens, F.L.P.; De Backer, J.P.; Fraeyman, N.; Vauquelin, G. Distinction between surmountable and insurmountable selective AT1 receptor antagonists by use of CHO-K1 cells expressing human angiotensin II AT1 receptors: Insurmountable antagonism of candesartan. Br. J. Pharmacol. 1999, 126, 1057–1065. [Google Scholar] [CrossRef][Green Version]
- De Gasparo, M.; Whitebread, S. Binding of valsartan to mammalian angiotensin AT1 receptors. Regul. Pept. 1995, 59, 303–311. [Google Scholar] [CrossRef]
- McClellan, K.J.; Markham, A. Telmisartan. Drugs 1998, 56, 1039–1044; discussion 1045–1046. [Google Scholar] [CrossRef]
- Dudley, D.T.; Summerfelt, R.M. Regulated expression of angiotensin II (AT2) binding sites in R3T3 cells. Regul. Pept. 1993, 44, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Kassmann, M.; Nickel, S.; Zhang, C.; Alenina, N.; Anistan, Y.M.; Schleifenbaum, J.; Bader, M.; Welsh, D.G.; Huang, Y.; et al. Myogenic Vasoconstriction Requires Canonical Gq/11 Signaling of the Angiotensin II Type 1 Receptor. J. Am. Heart Assoc. 2022, 11, e022070. [Google Scholar] [CrossRef]
- Jean-Charles, P.-Y.; Kaur, S.; Shenoy, S.K. G Protein–Coupled Receptor Signaling Through β-Arrestin–Dependent Mechanisms. J. Cardiovasc. Pharmacol. 2017, 70, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Viswanathan, G.; Sellig, M.; Jassal, C.; Choi, I.; Garikipati, A.; Xiong, X.; Nazo, N.; Rajagopal, S. β-Arrestin–Mediated Angiotensin II Type 1 Receptor Activation Promotes Pulmonary Vascular Remodeling in Pulmonary Hypertension. JACC Basic Transl. Sci. 2021, 6, 854–869. [Google Scholar] [CrossRef] [PubMed]
- Boerrigter, G.; Soergel, D.G.; Lark, M.W.; Burnett, J.C. TRV120027, a Novel Beta-Arrestin Biased Ligand at the Angiotensin II Type I Receptor, Unloads the Heart and Maintains Renal Function When Added to Furosemide in Experimental Heart Failure. J. Card. Fail. 2011, 17, S63–S64. [Google Scholar] [CrossRef]
- Kanki, H.; Sasaki, T.; Matsumura, S.; Yokawa, S.; Yukami, T.; Shimamura, M.; Sakaguchi, M.; Furuno, T.; Suzuki, T.; Mochizuki, H. β-arrestin-2 in PAR-1-biased signaling has a crucial role in endothelial function via PDGF-β in stroke. Cell Death Dis. 2019, 10, 100. [Google Scholar] [CrossRef]
- Bouressam, M.; Lecat, S.; Raoul, A.; Gaucher, C.; Perrin-Sarrado, C.; Lartaud, I.; Dupuis, F. S-nitrosoglutathione inhibits cerebrovascular angiotensin II-dependent and -independent AT1 receptor responses: A possible role of S-nitrosation. Br. J. Pharmacol. 2019, 176, 2049–2062. [Google Scholar] [CrossRef]
- Pinheiro, L.C.; Oliveira-Paula, G.H.; Ferreira, G.C.; Dal-Cin de Paula, T.; Duarte, D.A.; Costa-Neto, C.M.; Tanus-Santos, J.E. Oral nitrite treatment increases S-nitrosylation of vascular protein kinase C and attenuates the responses to angiotensin II. Redox Biol. 2021, 38, 101769. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colin, M.; Delaitre, C.; Foulquier, S.; Dupuis, F. The AT1/AT2 Receptor Equilibrium Is a Cornerstone of the Regulation of the Renin Angiotensin System beyond the Cardiovascular System. Molecules 2023, 28, 5481. https://doi.org/10.3390/molecules28145481
Colin M, Delaitre C, Foulquier S, Dupuis F. The AT1/AT2 Receptor Equilibrium Is a Cornerstone of the Regulation of the Renin Angiotensin System beyond the Cardiovascular System. Molecules. 2023; 28(14):5481. https://doi.org/10.3390/molecules28145481
Chicago/Turabian StyleColin, Mélissa, Céline Delaitre, Sébastien Foulquier, and François Dupuis. 2023. "The AT1/AT2 Receptor Equilibrium Is a Cornerstone of the Regulation of the Renin Angiotensin System beyond the Cardiovascular System" Molecules 28, no. 14: 5481. https://doi.org/10.3390/molecules28145481
APA StyleColin, M., Delaitre, C., Foulquier, S., & Dupuis, F. (2023). The AT1/AT2 Receptor Equilibrium Is a Cornerstone of the Regulation of the Renin Angiotensin System beyond the Cardiovascular System. Molecules, 28(14), 5481. https://doi.org/10.3390/molecules28145481