
Molecular Mechanisms of Amylin Turnover, Misfolding and Toxicity in the Pancreas

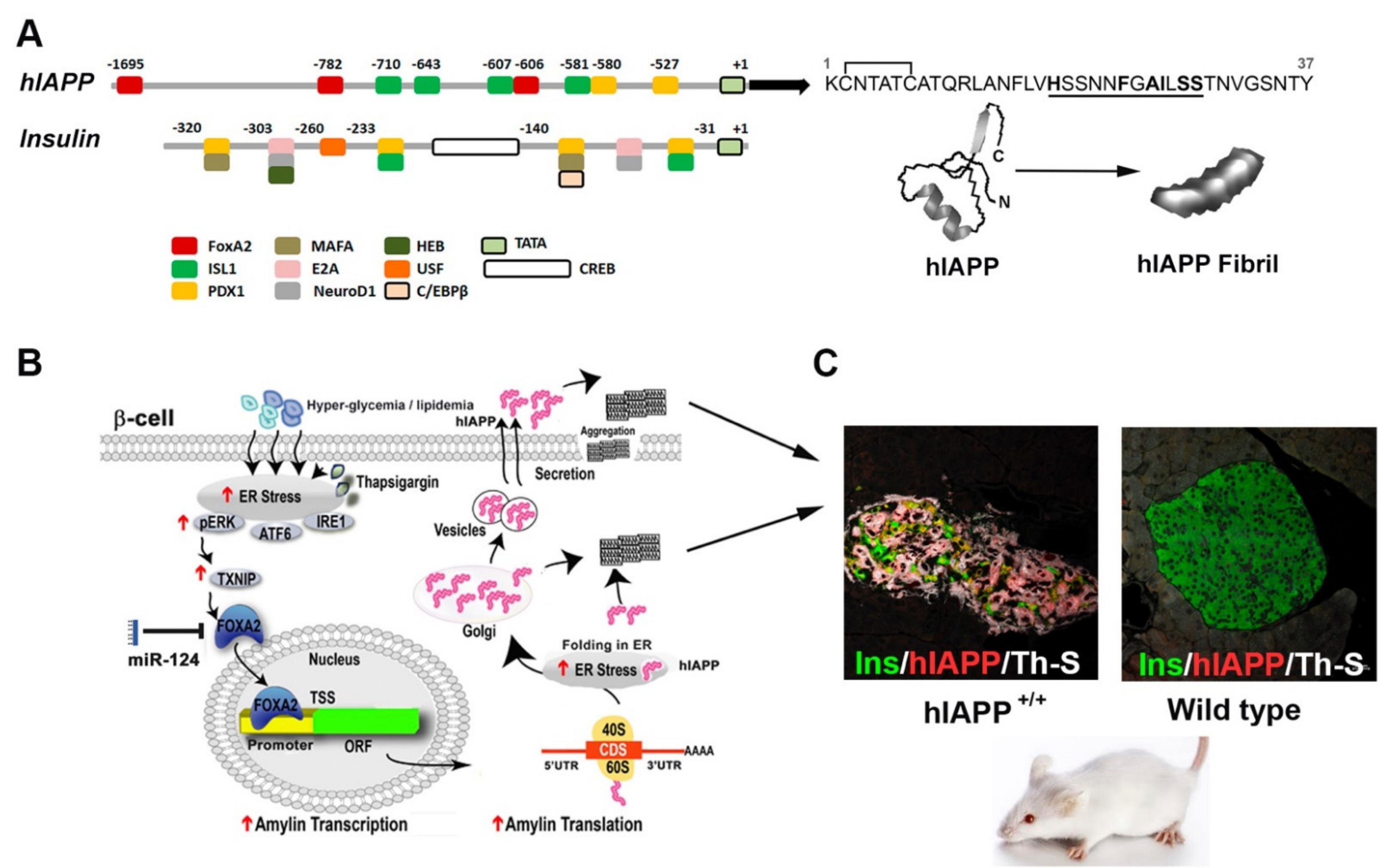{kind=link}
{kind=link}
{kind=link}
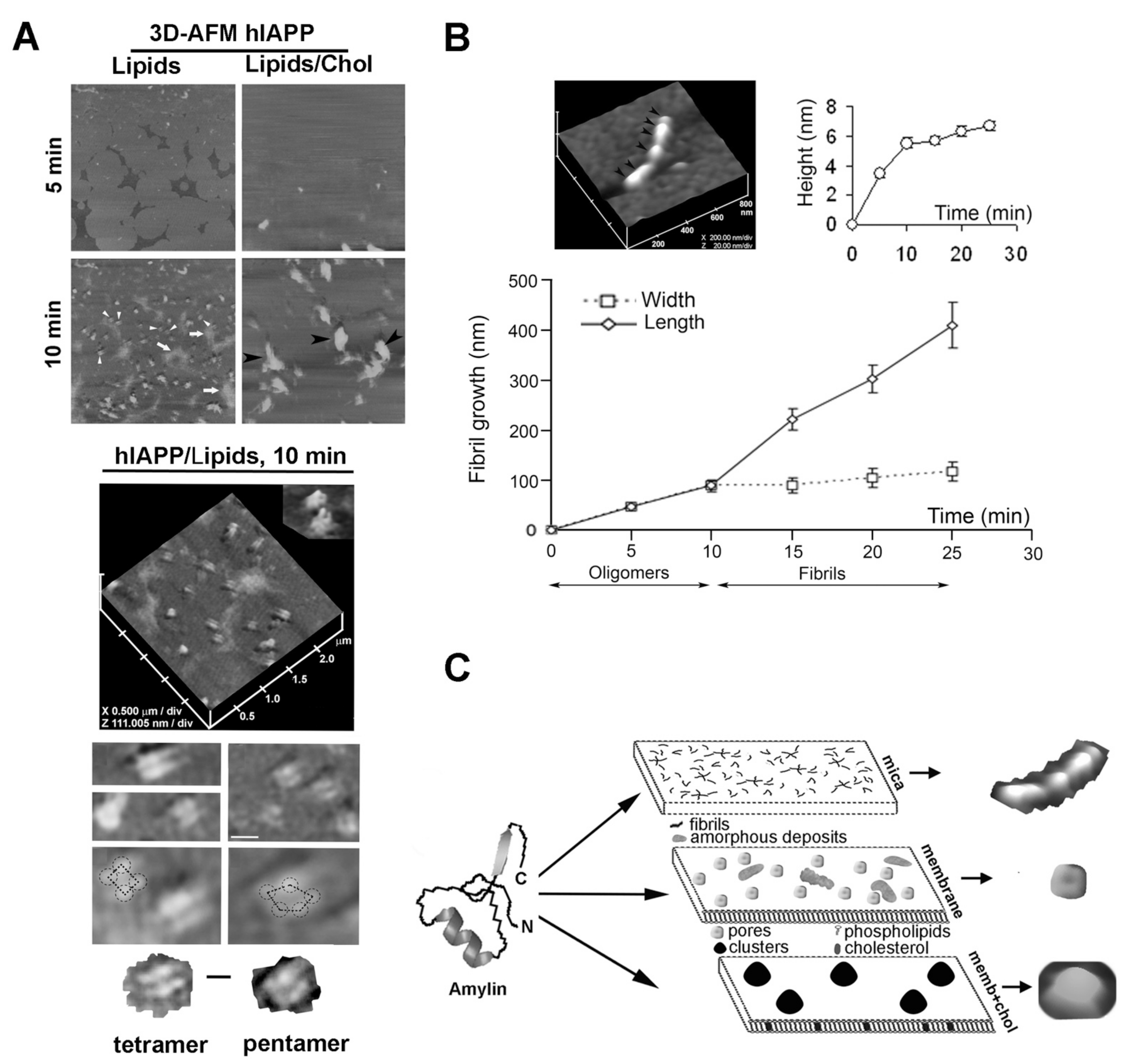{kind=link}
{kind=link}
Abstract
1. Introduction
2. Transcriptional Mechanisms Implicated in IAPP Synthesis in Pancreatic Cells
3. Proteolytic Pathways Regulate IAPP (Mis)Folding, Synthesis and Degradation
4. Lipids and Cholesterol as Modulators of Amylin Folding and Aggregation
5. Lipids and Cholesterol as Molecular Regulators of hIAPP Internalization, Aggregation and Toxicity
6. Small-Molecule Inhibitors of hIAPP Oligomerization and Fibrilization
7. hIAPP in Neurodegenerative and Metabolic Diseases: A Possible Synergy
8. Conclusions
9. Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef]
- Sipe, J.D.; Benson, M.D.; Buxbaum, J.N.; Ikeda, S.; Merlini, G.; Saraiva, M.J.; Westermark, P. Amyloid fibril protein nomenclature: 2010 recommendations from the nomenclature committee of the International Society of Amyloidosis. Amyloid Int. J. Exp. Clin. Investig. Off. J. Int. Soc. Amyloidosis 2010, 17, 101–104. [Google Scholar] [CrossRef]
- Granzotto, A.; Suwalsky, M.; Zatta, P. Physiological cholesterol concentration is a neuroprotective factor against beta-amyloid and beta-amyloid-metal complexes toxicity. J. Inorg. Biochem. 2011, 105, 1066–1072. [Google Scholar] [CrossRef] [PubMed]
- Lau, T.L.; Gehman, J.D.; Wade, J.D.; Perez, K.; Masters, C.L.; Barnham, K.J.; Separovic, F. Membrane interactions and the effect of metal ions of the amyloidogenic fragment Abeta(25-35) in comparison to Abeta(1-42). Biochim. Biophys. Acta 2007, 1768, 2400–2408. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rymer, D.L.; Good, T.A. The role of prion peptide structure and aggregation in toxicity and membrane binding. J. Neurochem. 2000, 75, 2536–2545. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Andersson, A.; Westermark, G.T. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol. Rev. 2011, 91, 795–826. [Google Scholar] [CrossRef] [PubMed]
- Milardi, D.; Gazit, E.; Radford, S.E.; Xu, Y.; Gallardo, R.U.; Caflisch, A.; Westermark, G.T.; Westermark, P.; Rosa, C.; Ramamoorthy, A. Proteostasis of islet amyloid polypeptide: A molecular perspective of risk factors and protective strategies for type II diabetes. Chem. Rev. 2021, 121, 1845–1893. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I.; et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Primers 2015, 1, 15019. [Google Scholar] [CrossRef]
- Kahn, S.E. Clinical review 135: The importance of beta-cell failure in the development and progression of type 2 diabetes. J. Clin. Endocrinol. Metab. 2001, 86, 4047–4058. [Google Scholar] [PubMed]
- Clark, A.; Nilsson, M.R. Islet amyloid: A complication of islet dysfunction or an aetiological factor in Type 2 diabetes? Diabetologia 2004, 47, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Zraika, S.; Hull, R.L.; Udayasankar, J.; Clark, A.; Utzschneider, K.M.; Tong, J.; Gerchman, F.; Kahn, S.E. Identification of the amyloid-degrading enzyme neprilysin in mouse islets and potential role in islet amyloidogenesis. Diabetes 2007, 56, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, J.; Dragunow, M.; Cooper, G.J. Fibrillogenic amylin evokes islet beta-cell apoptosis through linked activation of a caspase cascade and JNK1. J. Biol. Chem. 2003, 278, 52810–52819. [Google Scholar] [CrossRef] [PubMed]
- Ritzel, R.A.; Meier, J.J.; Lin, C.Y.; Veldhuis, J.D.; Butler, P.C. Human islet amyloid polypeptide oligomers disrupt cell coupling, induce apoptosis, and impair insulin secretion in isolated human islets. Diabetes 2007, 56, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Potter, K.J.; Scrocchi, L.A.; Warnock, G.L.; Ao, Z.; Younker, M.A.; Rosenberg, L.; Lipsett, M.; Verchere, C.B.; Fraser, P.E. Amyloid inhibitors enhance survival of cultured human islets. Biochim. Biophys. Acta 2009, 1790, 566–574. [Google Scholar] [CrossRef]
- Janson, J.; Soeller, W.C.; Roche, P.C.; Nelson, R.T.; Torchia, A.J.; Kreutter, D.K.; Butler, P.C. Spontaneous diabetes mellitus in transgenic mice expressing human islet amyloid polypeptide. Proc. Natl. Acad. Sci. USA 1996, 93, 7283–7288. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, J.; MacGibbon, G.; Dragunow, M.; Cooper, G.J. Increased expression and activation of c-Jun contributes to human amylin-induced apoptosis in pancreatic islet beta-cells. J. Mol. Biol. 2002, 324, 271–285. [Google Scholar] [CrossRef]
- Huang, C.J.; Lin, C.Y.; Haataja, L.; Gurlo, T.; Butler, A.E.; Rizza, R.A.; Butler, P.C. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated beta-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes 2007, 56, 2016–2027. [Google Scholar] [CrossRef]
- Casas, S.; Novials, A.; Reimann, F.; Gomis, R.; Gribble, F.M. Calcium elevation in mouse pancreatic beta cells evoked by extracellular human islet amyloid polypeptide involves activation of the mechanosensitive ion channel TRPV4. Diabetologia 2008, 51, 2252–2262. [Google Scholar] [CrossRef]
- Brender, J.R.; Hartman, K.; Reid, K.R.; Kennedy, R.T.; Ramamoorthy, A. A single mutation in the nonamyloidogenic region of islet amyloid polypeptide greatly reduces toxicity. Biochemistry 2008, 47, 12680–12688. [Google Scholar] [CrossRef]
- Nakamura, S.; Okabayashi, S.; Ageyama, N.; Koie, H.; Sankai, T.; Ono, F.; Fujimoto, K.; Terao, K. Transthyretin amyloidosis and two other aging-related amyloidoses in an aged vervet monkey. Vet. Pathol. 2008, 45, 67–72. [Google Scholar] [CrossRef]
- Guardado-Mendoza, R.; Davalli, A.M.; Chavez, A.O.; Hubbard, G.B.; Dick, E.J.; Majluf-Cruz, A.; Tene-Perez, C.E.; Goldschmidt, L.; Hart, J.; Perego, C.; et al. Pancreatic islet amyloidosis, beta-cell apoptosis, and alpha-cell proliferation are determinants of islet remodeling in type-2 diabetic baboons. Proc. Natl. Acad. Sci. USA 2009, 106, 13992–13997. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.; Wells, C.A.; Buley, I.D.; Cruickshank, J.K.; Vanhegan, R.I.; Matthews, D.R.; Cooper, G.J.; Holman, R.R.; Turner, R.C. Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: Quantitative changes in the pancreas in type 2 diabetes. Diabetes Res. 1988, 9, 151–159. [Google Scholar] [PubMed]
- Rocken, C.; Linke, R.P.; Saeger, W. Immunohistology of islet amyloid polypeptide in diabetes mellitus: Semi-quantitative studies in a post-mortem series. Virchows Arch. A Pathol. Anat. Histopathol. 1992, 421, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Morales-Scheihing, D.; Butler, P.C.; Soto, C. Type 2 diabetes as a protein misfolding disease. Trends Mol. Med. 2015, 21, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Wimalawansa, S.J. Amylin, calcitonin gene-related peptide, calcitonin, and adrenomedullin: A peptide superfamily. Crit. Rev. Neurobiol. 1997, 11, 167–239. [Google Scholar] [CrossRef]
- Christmanson, L.; Rorsman, F.; Stenman, G.; Westermark, P.; Betsholtz, C. The human islet amyloid polypeptide (IAPP) gene. Organization, chromosomal localization and functional identification of a promoter region. FEBS Lett. 1990, 267, 160–166. [Google Scholar] [CrossRef]
- Mosselman, S.; Hoppener, J.W.; Lips, C.J.; Jansz, H.S. The complete islet amyloid polypeptide precursor is encoded by two exons. FEBS Lett. 1989, 247, 154–158. [Google Scholar] [CrossRef]
- Owerbach, D.; Bell, G.I.; Rutter, W.J.; Shows, T.B. The insulin gene is located on chromosome 11 in humans. Nature 1980, 286, 82–84. [Google Scholar] [CrossRef]
- Sun, J.; Cui, J.; He, Q.; Chen, Z.; Arvan, P.; Liu, M. Proinsulin misfolding and endoplasmic reticulum stress during the development and progression of diabetes. Mol. Asp. Med. 2015, 42, 105–118. [Google Scholar] [CrossRef]
- Weiss, M.; Steiner, D.F.; Philipson, L.H. Insulin biosynthesis, secretion, structure, and structure-activity relationships. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., Dungan, K., Grossman, A., Hershman, J.M., Kaltsas, G., Koch, C., Kopp, P., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Chatterjee Bhowmick, D.; Burnett, L.; Kudaibergenova, Z.; Jeremic, A.M. FoxA2 and RNA Pol II mediate human islet amyloid polypeptide turnover in ER-stressed pancreatic beta-cells. Biochem. J. 2021, 478, 1261–1282. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Bhowmick, D.C.; Pany, S.; Joe, M.; Zaghlula, N.; Jeremic, A.M. Apoptosis signal regulating kinase-1 and NADPH oxidase mediate human amylin evoked redox stress and apoptosis in pancreatic beta-cells. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1721–1733. [Google Scholar] [CrossRef]
- Mulder, H.; Ahren, B.; Sundler, F. Islet amyloid polypeptide and insulin gene expression are regulated in parallel by glucose in vivo in rats. Am. J. Physiol. 1996, 271, E1008–E1014. [Google Scholar] [CrossRef] [PubMed]
- Shalev, A.; Pise-Masison, C.A.; Radonovich, M.; Hoffmann, S.C.; Hirshberg, B.; Brady, J.N.; Harlan, D.M. Oligonucleotide microarray analysis of intact human pancreatic islets: Identification of glucose-responsive genes and a highly regulated TGFbeta signaling pathway. Endocrinology 2002, 143, 3695–3698. [Google Scholar] [CrossRef] [PubMed]
- Gasa, R.; Gomis, R.; Casamitjana, R.; Novials, A. Signals related to glucose metabolism regulate islet amyloid polypeptide (IAPP) gene expression in human pancreatic islets. Regul. Pept. 1997, 68, 99–104. [Google Scholar] [CrossRef]
- Jing, G.; Westwell-Roper, C.; Chen, J.; Xu, G.; Verchere, C.B.; Shalev, A. Thioredoxin-interacting protein promotes islet amyloid polypeptide expression through miR-124a and FoxA2. J. Biol. Chem. 2014, 289, 11807–11815. [Google Scholar] [CrossRef]
- Shepherd, L.M.; Campbell, S.C.; Macfarlane, W.M. Transcriptional regulation of the IAPP gene in pancreatic beta-cells. Biochim. Biophys. Acta 2004, 1681, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, W.M.; Campbell, S.C.; Elrick, L.J.; Oates, V.; Bermano, G.; Lindley, K.J.; Aynsley-Green, A.; Dunne, M.J.; James, R.F.; Docherty, K. Glucose regulates islet amyloid polypeptide gene transcription in a PDX1- and calcium-dependent manner. J. Biol. Chem. 2000, 275, 15330–15335. [Google Scholar] [CrossRef] [PubMed]
- Watada, H.; Kajimoto, Y.; Kaneto, H.; Matsuoka, T.; Fujitani, Y.; Miyazaki, J.; Yamasaki, Y. Involvement of the homeodomain-containing transcription factor PDX-1 in islet amyloid polypeptide gene transcription. Biochem. Biophys. Res. Commun. 1996, 229, 746–751. [Google Scholar] [CrossRef] [PubMed]
- Hay, C.W.; Docherty, K. Comparative analysis of insulin gene promoters: Implications for diabetes research. Diabetes 2006, 55, 3201–3213. [Google Scholar] [CrossRef]
- Le Lay, J.; Matsuoka, T.A.; Henderson, E.; Stein, R. Identification of a novel PDX-1 binding site in the human insulin gene enhancer. J. Biol. Chem. 2004, 279, 22228–22235. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Drucker, D.J. Activation of amylin gene transcription by LIM domain homeobox gene isl-1. Mol. Endocrinol. 1996, 10, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Kaestner, K.H. The Foxa family of transcription factors in development and metabolism. Cell. Mol. Life Sci. 2006, 63, 2317–2328. [Google Scholar] [CrossRef] [PubMed]
- Lutz, T.A.; Meyer, U. Amylin at the interface between metabolic and neurodegenerative disorders. Front. Neurosci. 2015, 9, 216. [Google Scholar] [CrossRef]
- Cho, W.J.; Jena, B.P.; Jeremic, A.M. Nano-scale imaging and dynamics of amylin-membrane interactions and its implication in type II diabetes mellitus. Methods Cell Biol. 2008, 90, 267–286. [Google Scholar]
- Xu, G.; Chen, J.; Jing, G.; Shalev, A. Thioredoxin-interacting protein regulates insulin transcription through microRNA-204. Nat. Med. 2013, 19, 1141–1146. [Google Scholar] [CrossRef]
- Lee, S.; Min Kim, S.; Dotimas, J.; Li, L.; Feener, E.P.; Baldus, S.; Myers, R.B.; Chutkow, W.A.; Patwari, P.; Yoshioka, J.; et al. Thioredoxin-interacting protein regulates protein disulfide isomerases and endoplasmic reticulum stress. EMBO Mol. Med. 2014, 6, 732–743. [Google Scholar] [CrossRef]
- Oslowski, C.M.; Hara, T.; O’Sullivan-Murphy, B.; Kanekura, K.; Lu, S.; Hara, M.; Ishigaki, S.; Zhu, L.J.; Hayashi, E.; Hui, S.T.; et al. Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell Metab. 2012, 16, 265–273. [Google Scholar] [CrossRef]
- Chatterjee Bhowmick, D.; Jeremic, A. Functional proteasome complex is required for turnover of islet amyloid polypeptide in pancreatic beta-cells. J. Biol. Chem. 2018, 293, 14210–14223. [Google Scholar] [CrossRef]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef]
- Sun, Y.; Koo, S.; White, N.; Peralta, E.; Esau, C.; Dean, N.M.; Perera, R.J. Development of a micro-array to detect human and mouse microRNAs and characterization of expression in human organs. Nucleic Acids Res. 2004, 32, e188. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Kameswaran, V.; Bramswig, N.C.; McKenna, L.B.; Penn, M.; Schug, J.; Hand, N.J.; Chen, Y.; Choi, I.; Vourekas, A.; Won, K.J.; et al. Epigenetic regulation of the DLK1-MEG3 microRNA cluster in human type 2 diabetic islets. Cell Metab. 2014, 19, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Wernstedt, C.; Wilander, E.; Hayden, D.W.; O’Brien, T.D.; Johnson, K.H. Amyloid fibrils in human insulinoma and islets of Langerhans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc. Natl. Acad. Sci. USA 1987, 84, 3881–3885. [Google Scholar] [CrossRef] [PubMed]
- Jaisson, S.; Gillery, P. Impaired proteostasis: Role in the pathogenesis of diabetes mellitus. Diabetologia 2014, 57, 1517–1527. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.C.; Singh, S.; de Pablo, J.J. Effect of proline mutations on the monomer conformations of amylin. Biophys. J. 2013, 105, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, D.F.; Raleigh, D.P. Effects of sequential proline substitutions on amyloid formation by human amylin20-29. Biochemistry 1999, 38, 1811–1818. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Engstrom, U.; Johnson, K.H.; Westermark, G.T.; Betsholtz, C. Islet amyloid polypeptide: Pinpointing amino acid residues linked to amyloid fibril formation. Proc. Natl. Acad. Sci. USA 1990, 87, 5036–5040. [Google Scholar] [CrossRef]
- Abedini, A.; Raleigh, D.P. The role of His-18 in amyloid formation by human islet amyloid polypeptide. Biochemistry 2005, 44, 16284–16291. [Google Scholar] [CrossRef]
- Tu, L.H.; Raleigh, D.P. Role of aromatic interactions in amyloid formation by islet amyloid polypeptide. Biochemistry 2013, 52, 333–342. [Google Scholar] [CrossRef]
- Cao, P.; Tu, L.H.; Abedini, A.; Levsh, O.; Akter, R.; Patsalo, V.; Schmidt, A.M.; Raleigh, D.P. Sensitivity of amyloid formation by human islet amyloid polypeptide to mutations at residue 20. J. Mol. Biol. 2012, 421, 282–295. [Google Scholar] [CrossRef]
- Cao, P.; Abedini, A.; Wang, H.; Tu, L.H.; Zhang, X.; Schmidt, A.M.; Raleigh, D.P. Islet amyloid polypeptide toxicity and membrane interactions. Proc. Natl. Acad. Sci. USA 2013, 110, 19279–19284. [Google Scholar] [CrossRef] [PubMed]
- Janson, J.; Ashley, R.H.; Harrison, D.; McIntyre, S.; Butler, P.C. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes 1999, 48, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Konarkowska, B.; Aitken, J.F.; Kistler, J.; Zhang, S.; Cooper, G.J. The aggregation potential of human amylin determines its cytotoxicity towards islet beta-cells. FEBS J. 2006, 273, 3614–3624. [Google Scholar] [CrossRef]
- Trikha, S.; Jeremic, A.M. Clustering and internalization of toxic amylin oligomers in pancreatic cells require plasma membrane cholesterol. J. Biol. Chem. 2011, 286, 36086–36097. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, H.; Chuang, C.L.; Li, X.; Au, M.; Zhang, L.; Phillips, A.R.; Scott, D.W.; Cooper, G.J. The pathogenic mechanism of diabetes varies with the degree of overexpression and oligomerization of human amylin in the pancreatic islet beta cells. FASEB J. 2014, 28, 5083–5096. [Google Scholar] [CrossRef] [PubMed]
- Costes, S.; Huang, C.J.; Gurlo, T.; Daval, M.; Matveyenko, A.V.; Rizza, R.A.; Butler, A.E.; Butler, P.C. beta-cell dysfunctional ERAD/ubiquitin/proteasome system in type 2 diabetes mediated by islet amyloid polypeptide-induced UCH-L1 deficiency. Diabetes 2011, 60, 227–238. [Google Scholar] [CrossRef]
- Bhaumik, S.R.; Malik, S. Diverse regulatory mechanisms of eukaryotic transcriptional activation by the proteasome complex. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 419–433. [Google Scholar] [CrossRef]
- Kwak, J.; Workman, J.L.; Lee, D. The proteasome and its regulatory roles in gene expression. Biochim. Biophys. Acta 2011, 1809, 88–96. [Google Scholar] [CrossRef]
- Singh, S.; Trikha, S.; Sarkar, A.; Jeremic, A.M. Proteasome regulates turnover of toxic human amylin in pancreatic cells. Biochem. J. 2016, 473, 2655–2670. [Google Scholar] [CrossRef]
- Casas, S.; Gomis, R.; Gribble, F.M.; Altirriba, J.; Knuutila, S.; Novials, A. Impairment of the ubiquitin-proteasome pathway is a downstream endoplasmic reticulum stress response induced by extracellular human islet amyloid polypeptide and contributes to pancreatic beta-cell apoptosis. Diabetes 2007, 56, 2284–2294. [Google Scholar] [CrossRef] [PubMed]
- Costes, S.; Gurlo, T.; Rivera, J.F.; Butler, P.C. UCHL1 deficiency exacerbates human islet amyloid polypeptide toxicity in beta-cells: Evidence of interplay between the ubiquitin/proteasome system and autophagy. Autophagy 2014, 10, 1004–1014. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Cheon, H.; Jeong, Y.T.; Quan, W.; Kim, K.H.; Cho, J.M.; Lim, Y.M.; Oh, S.H.; Jin, S.M.; Kim, J.H.; et al. Amyloidogenic peptide oligomer accumulation in autophagy-deficient beta cells induces diabetes. J. Clin. Invest. 2014, 124, 3311–3324. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.F.; Costes, S.; Gurlo, T.; Glabe, C.G.; Butler, P.C. Autophagy defends pancreatic beta cells from human islet amyloid polypeptide-induced toxicity. J. Clin. Invest. 2014, 124, 3489–3500. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.F.; Gurlo, T.; Daval, M.; Huang, C.J.; Matveyenko, A.V.; Butler, P.C.; Costes, S. Human-IAPP disrupts the autophagy/lysosomal pathway in pancreatic beta-cells: Protective role of p62-positive cytoplasmic inclusions. Cell Death Differ. 2011, 18, 415–426. [Google Scholar] [CrossRef]
- Press, M.; Jung, T.; Konig, J.; Grune, T.; Hohn, A. Protein aggregates and proteostasis in aging: Amylin and beta-cell function. Mech. Ageing Dev. 2019, 177, 46–54. [Google Scholar] [CrossRef]
- Bennett, R.G.; Duckworth, W.C.; Hamel, F.G. Degradation of amylin by insulin-degrading enzyme. J. Biol. Chem. 2000, 275, 36621–36625. [Google Scholar] [CrossRef]
- Bennett, R.G.; Hamel, F.G.; Duckworth, W.C. An insulin-degrading enzyme inhibitor decreases amylin degradation, increases amylin-induced cytotoxicity, and increases amyloid formation in insulinoma cell cultures. Diabetes 2003, 52, 2315–2320. [Google Scholar] [CrossRef]
- Duckworth, W.C.; Bennett, R.G.; Hamel, F.G. Insulin degradation: Progress and potential. Endocr. Rev. 1998, 19, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Hogan, M.F.; Meier, D.T.; Zraika, S.; Templin, A.T.; Mellati, M.; Hull, R.L.; Leissring, M.A.; Kahn, S.E. Inhibition of insulin-degrading enzyme does not increase islet amyloid deposition in vitro. Endocrinology 2016, 157, 3462–3468. [Google Scholar] [CrossRef]
- Aston-Mourney, K.; Zraika, S.; Udayasankar, J.; Subramanian, S.L.; Green, P.S.; Kahn, S.E.; Hull, R.L. Matrix metalloproteinase-9 reduces islet amyloid formation by degrading islet amyloid polypeptide. J. Biol. Chem. 2013, 288, 3553–3559. [Google Scholar] [CrossRef] [PubMed]
- Zraika, S.; Aston-Mourney, K.; Marek, P.; Hull, R.L.; Green, P.S.; Udayasankar, J.; Subramanian, S.L.; Raleigh, D.P.; Kahn, S.E. Neprilysin impedes islet amyloid formation by inhibition of fibril formation rather than peptide degradation. J. Biol. Chem. 2010, 285, 18177–18183. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.J.; Trikha, S.; Jeremic, A.M. Cholesterol regulates assembly of human islet amyloid polypeptide on model membranes. J. Mol. Biol. 2009, 393, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.D.; Miranker, A.D. Phospholipid catalysis of diabetic amyloid assembly. J. Mol. Biol. 2004, 341, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, D.L.; de Koning, E.J.; Verbeek, J.S.; Morris, J.F.; Clark, A. Amyloid fibril formation is progressive and correlates with beta-cell secretion in transgenic mouse isolated islets. Diabetologia 1999, 42, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Brender, J.R.; Lee, E.L.; Cavitt, M.A.; Gafni, A.; Steel, D.G.; Ramamoorthy, A. Amyloid fiber formation and membrane disruption are separate processes localized in two distinct regions of IAPP, the type-2-diabetes-related peptide. J. Am. Chem. Soc. 2008, 130, 6424–6429. [Google Scholar] [CrossRef] [PubMed]
- Khemtemourian, L.; Killian, J.A.; Hoppener, J.W.; Engel, M.F. Recent insights in islet amyloid polypeptide-induced membrane disruption and its role in beta-cell death in type 2 diabetes mellitus. Exp. Diabetes Res. 2008, 2008, 421287. [Google Scholar] [CrossRef]
- Jayasinghe, S.A.; Langen, R. Lipid membranes modulate the structure of islet amyloid polypeptide. Biochemistry 2005, 44, 12113–12119. [Google Scholar] [CrossRef]
- Jayasinghe, S.A.; Langen, R. Membrane interaction of islet amyloid polypeptide. Biochim. Biophys. Acta 2007, 1768, 2002–2009. [Google Scholar] [CrossRef]
- Knight, J.D.; Hebda, J.A.; Miranker, A.D. Conserved and cooperative assembly of membrane-bound alpha-helical states of islet amyloid polypeptide. Biochemistry 2006, 45, 9496–9508. [Google Scholar] [CrossRef]
- Padrick, S.B.; Miranker, A.D. Islet amyloid: Phase partitioning and secondary nucleation are central to the mechanism of fibrillogenesis. Biochemistry 2002, 41, 4694–4703. [Google Scholar] [CrossRef] [PubMed]
- Almeida, Z.L.; Brito, R.M.M. Structure and aggregation mechanisms in amyloids. Molecules 2020, 25, 1195. [Google Scholar] [CrossRef]
- Quist, A.; Doudevski, I.; Lin, H.; Azimova, R.; Ng, D.; Frangione, B.; Kagan, B.; Ghiso, J.; Lal, R. Amyloid ion channels: A common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA 2005, 102, 10427–10432. [Google Scholar] [CrossRef] [PubMed]
- Haataja, L.; Gurlo, T.; Huang, C.J.; Butler, P.C. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr. Rev. 2008, 29, 303–316. [Google Scholar] [CrossRef]
- Mirzabekov, T.A.; Lin, M.C.; Kagan, B.L. Pore formation by the cytotoxic islet amyloid peptide amylin. J. Biol. Chem. 1996, 271, 1988–1992. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Hu, R.; Sciacca, M.F.; Brender, J.R.; Chen, H.; Ramamoorthy, A.; Zheng, J. Non-selective ion channel activity of polymorphic human islet amyloid polypeptide (amylin) double channels. Phys. Chem. Chem. Phys. 2014, 16, 2368–2377. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sciacca, M.F.; Lolicato, F.; Di Mauro, G.; Milardi, D.; D’Urso, L.; Satriano, C.; Ramamoorthy, A.; La Rosa, C. The role of cholesterol in driving IAPP-membrane interactions. Biophys. J. 2016, 111, 140–151. [Google Scholar] [CrossRef]
- Jaikaran, E.T.; Clark, A. Islet amyloid and type 2 diabetes: From molecular misfolding to islet pathophysiology. Biochim. Biophys. Acta 2001, 1537, 179–203. [Google Scholar] [CrossRef]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef]
- Chen, Z.; Rand, R.P. The influence of cholesterol on phospholipid membrane curvature and bending elasticity. Biophys. J. 1997, 73, 267–276. [Google Scholar] [CrossRef]
- Smith, P.E.; Brender, J.R.; Ramamoorthy, A. Induction of negative curvature as a mechanism of cell toxicity by amyloidogenic peptides: The case of islet amyloid polypeptide. J. Am. Chem. Soc. 2009, 131, 4470–4478. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.; Berglund, N.A.; Schiott, B. The effect of cholesterol on membrane-bound islet amyloid polypeptide. Front. Mol. Biosci. 2021, 8, 657946. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; St. Clair, J.R.; London, E.; Raleigh, D.P. Islet amyloid polypeptide membrane interactions: Effects of membrane composition. Biochemistry 2017, 56, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Engel, M.F.; Yigittop, H.; Elgersma, R.C.; Rijkers, D.T.; Liskamp, R.M.; de Kruijff, B.; Hoppener, J.W.; Antoinette Killian, J. Islet amyloid polypeptide inserts into phospholipid monolayers as monomer. J. Mol. Biol. 2006, 356, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guan, L.; Lu, T.; Li, H.; Li, Z.; Li, F. Interactions of the N-terminal domain of human islet amyloid polypeptide with lipid membranes: The effect of cholesterol. RSC Adv. 2016, 6, 96837–96846. [Google Scholar] [CrossRef]
- Zhang, X.; London, E.; Raleigh, D.P. Sterol structure strongly modulates membrane-islet amyloid polypeptide interactions. Biochemistry 2018, 57, 1868–1879. [Google Scholar] [CrossRef] [PubMed]
- Trikha, S.; Jeremic, A.M. Distinct internalization pathways of human amylin monomers and its cytotoxic oligomers in pancreatic cells. PLoS ONE 2013, 8, e73080. [Google Scholar] [CrossRef]
- Wijesekara, N.; Kaur, A.; Westwell-Roper, C.; Nackiewicz, D.; Soukhatcheva, G.; Hayden, M.R.; Verchere, C.B. ABCA1 deficiency and cellular cholesterol accumulation increases islet amyloidogenesis in mice. Diabetologia 2016, 59, 1242–1246. [Google Scholar] [CrossRef] [PubMed]
- Sciacca, M.F.; Lolicato, F.; Tempra, C.; Scollo, F.; Sahoo, B.R.; Watson, M.D.; Garcia-Vinuales, S.; Milardi, D.; Raudino, A.; Lee, J.C.; et al. Lipid-chaperone hypothesis: A common molecular mechanism of membrane disruption by intrinsically disordered proteins. ACS Chem. Neurosci. 2020, 11, 4336–4350. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, C.; Scalisi, S.; Lolicato, F.; Pannuzzo, M.; Raudino, A. Lipid-assisted protein transport: A diffusion-reaction model supported by kinetic experiments and molecular dynamics simulations. J. Chem. Phys. 2016, 144, 184901. [Google Scholar] [CrossRef]
- Scollo, F.; Tempra, C.; Lolicato, F.; Sciacca, M.F.M.; Raudino, A.; Milardi, D.; La Rosa, C. Phospholipids critical micellar concentrations trigger different mechanisms of intrinsically disordered proteins interaction with model membranes. J. Phys. Chem. Lett. 2018, 9, 5125–5129. [Google Scholar] [CrossRef] [PubMed]
- Poreba, M.; Rostoff, P.; Siniarski, A.; Mostowik, M.; Golebiowska-Wiatrak, R.; Nessler, J.; Undas, A.; Gajos, G. Relationship between polyunsaturated fatty acid composition in serum phospholipids, systemic low-grade inflammation, and glycemic control in patients with type 2 diabetes and atherosclerotic cardiovascular disease. Cardiovasc. Diabetol. 2018, 17, 29. [Google Scholar] [CrossRef] [PubMed]
- Larson, J.L.; Miranker, A.D. The mechanism of insulin action on islet amyloid polypeptide fiber formation. J. Mol. Biol. 2004, 335, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Brender, J.R.; Hartman, K.; Nanga, R.P.; Popovych, N.; de la Salud Bea, R.; Vivekanandan, S.; Marsh, E.N.; Ramamoorthy, A. Role of zinc in human islet amyloid polypeptide aggregation. J. Am. Chem. Soc. 2010, 132, 8973–8983. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Kakinen, A.; Gurzov, E.N.; Yang, W.; Pang, L.; Pilkington, E.H.; Govindan-Nedumpully, P.; Chen, P.; Separovic, F.; Davis, T.P.; et al. Zinc-coordination and C-peptide complexation: A potential mechanism for the endogenous inhibition of IAPP aggregation. Chem. Commun. 2017, 53, 9394–9397. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Lin, Y.; Cox, S.J.; Kinoshita, M.; Sahoo, B.R.; Ivanova, M.; Ramamoorthy, A. Zinc boosts EGCG’s hIAPP amyloid inhibition both in solution and membrane. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 529–536. [Google Scholar] [CrossRef]
- Nedumpully-Govindan, P.; Ding, F. Inhibition of IAPP aggregation by insulin depends on the insulin oligomeric state regulated by zinc ion concentration. Sci. Rep. 2015, 5, 8240. [Google Scholar] [CrossRef]
- Khemtemourian, L.; Antoniciello, F.; Sahoo, B.R.; Decossas, M.; Lecomte, S.; Ramamoorthy, A. Investigation of the effects of two major secretory granules components, insulin and zinc, on human-IAPP amyloid aggregation and membrane damage. Chem. Phys. Lipids 2021, 237, 105083. [Google Scholar] [CrossRef]
- Lee, E.C.; Ha, E.; Singh, S.; Legesse, L.; Ahmad, S.; Karnaukhova, E.; Donaldson, R.P.; Jeremic, A.M. Copper(II)-human amylin complex protects pancreatic cells from amylin toxicity. Phys. Chem. Chem. Phys. 2013, 15, 12558–12571. [Google Scholar] [CrossRef]
- Li, H.; Ha, E.; Donaldson, R.P.; Jeremic, A.M.; Vertes, A. Rapid assessment of human amylin aggregation and its inhibition by copper(II) ions by laser ablation electrospray ionization mass spectrometry with ion mobility separation. Anal. Chem. 2015, 87, 9829–9837. [Google Scholar] [CrossRef]
- He, L.; Wang, X.; Zhao, C.; Zhu, D.; Du, W. Inhibition of human amylin fibril formation by insulin-mimetic vanadium complexes. Metallomics 2014, 6, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- Alghrably, M.; Czaban, I.; Jaremko, L.; Jaremko, M. Interaction of amylin species with transition metals and membranes. J. Inorg. Biochem. 2019, 191, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Pithadia, A.; Brender, J.R.; Fierke, C.A.; Ramamoorthy, A. Inhibition of IAPP aggregation and toxicity by natural products and derivatives. J. Diabetes Res. 2016, 2016, 2046327. [Google Scholar] [CrossRef] [PubMed]
- Franko, A.; Rodriguez Camargo, D.C.; Boddrich, A.; Garg, D.; Rodriguez Camargo, A.; Rathkolb, B.; Janik, D.; Aichler, M.; Feuchtinger, A.; Neff, F.; et al. Epigallocatechin gallate (EGCG) reduces the intensity of pancreatic amyloid fibrils in human islet amyloid polypeptide (hIAPP) transgenic mice. Sci. Rep. 2018, 8, 1116. [Google Scholar] [CrossRef]
- Mo, Y.; Lei, J.; Sun, Y.; Zhang, Q.; Wei, G. Conformational ensemble of hIAPP dimer: Insight into the molecular mechanism by which a green tea extract inhibits hIAPP aggregation. Sci. Rep. 2016, 6, 33076. [Google Scholar] [CrossRef]
- Xu, Z.X.; Ma, G.L.; Zhang, Q.; Chen, C.H.; He, Y.M.; Xu, L.H.; Zhou, G.R.; Li, Z.H.; Yang, H.J.; Zhou, P. Inhibitory mechanism of epigallocatechin gallate on fibrillation and aggregation of amidated human islet amyloid polypeptide. ChemPhysChem 2017, 18, 1611–1619. [Google Scholar] [CrossRef]
- Lolicato, F.; Raudino, A.; Milardi, D.; La Rosa, C. Resveratrol interferes with the aggregation of membrane-bound human-IAPP: A molecular dynamics study. Eur. J. Med. Chem. 2015, 92, 876–881. [Google Scholar] [CrossRef]
- Sciacca, M.F.M.; Chillemi, R.; Sciuto, S.; Greco, V.; Messineo, C.; Kotler, S.A.; Lee, D.K.; Brender, J.R.; Ramamoorthy, A.; La Rosa, C.; et al. A blend of two resveratrol derivatives abolishes hIAPP amyloid growth and membrane damage. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1793–1802. [Google Scholar] [CrossRef]
- Yu, X.L.; Li, Y.N.; Zhang, H.; Su, Y.J.; Zhou, W.W.; Zhang, Z.P.; Wang, S.W.; Xu, P.X.; Wang, Y.J.; Liu, R.T. Rutin inhibits amylin-induced neurocytotoxicity and oxidative stress. Food Funct. 2015, 6, 3296–3306. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. Neuroanatomy and pathology of sporadic Alzheimer’s disease. Adv. Anat. Embryol. Cell Biol. 2015, 215, 1–162. [Google Scholar]
- Dickson, D.W.; Braak, H.; Duda, J.E.; Duyckaerts, C.; Gasser, T.; Halliday, G.M.; Hardy, J.; Leverenz, J.B.; Del Tredici, K.; Wszolek, Z.K.; et al. Neuropathological assessment of Parkinson’s disease: Refining the diagnostic criteria. Lancet Neurol. 2009, 8, 1150–1157. [Google Scholar] [CrossRef]
- Placido, A.I.; Pereira, C.M.; Duarte, A.I.; Candeias, E.; Correia, S.C.; Santos, R.X.; Carvalho, C.; Cardoso, S.; Oliveira, C.R.; Moreira, P.I. The role of endoplasmic reticulum in amyloid precursor protein processing and trafficking: Implications for Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1444–1453. [Google Scholar] [CrossRef] [PubMed]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [CrossRef] [PubMed]
- Eliezer, D.; Kutluay, E.; Bussell, R., Jr.; Browne, G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J. Mol. Biol. 2001, 307, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Iwai, A.; Masliah, E.; Yoshimoto, M.; Ge, N.; Flanagan, L.; de Silva, H.A.; Kittel, A.; Saitoh, T. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995, 14, 467–475. [Google Scholar] [CrossRef]
- Wang, Y.; Westermark, G.T. The amyloid forming peptides islet amyloid polypeptide and amyloid beta interact at the molecular level. Int. J. Mol. Sci. 2021, 22, 11153. [Google Scholar] [CrossRef]
- Jhamandas, J.H.; Mactavish, D. beta-Amyloid protein (Abeta) and human amylin regulation of apoptotic genes occurs through the amylin receptor. Apoptosis 2012, 17, 37–47. [Google Scholar] [CrossRef]
- Srodulski, S.; Sharma, S.; Bachstetter, A.B.; Brelsfoard, J.M.; Pascual, C.; Xie, X.S.; Saatman, K.E.; Van Eldik, L.J.; Despa, F. Neuroinflammation and neurologic deficits in diabetes linked to brain accumulation of amylin. Mol. Neurodegener. 2014, 9, 30. [Google Scholar] [CrossRef]
- Rosas, P.C.; Nagaraja, G.M.; Kaur, P.; Panossian, A.; Wickman, G.; Garcia, L.R.; Al-Khamis, F.A.; Asea, A.A. Hsp72 (HSPA1A) prevents human islet amyloid polypeptide aggregation and toxicity: A new approach for type 2 diabetes treatment. PLoS ONE 2016, 11, e0149409. [Google Scholar] [CrossRef]
- Schultz, S.W.; Nilsson, K.P.; Westermark, G.T. Drosophila melanogaster as a model system for studies of islet amyloid polypeptide aggregation. PLoS ONE 2011, 6, e20221. [Google Scholar] [CrossRef]
- Aldras, Y.; Singh, S.; Bode, K.; Bhowmick, D.C.; Jeremic, A.; O’Halloran, D.M. An inducible model of human amylin overexpression reveals diverse transcriptional changes. Neurosci. Lett. 2019, 704, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.; Barisone, G.A.; Diaz, E.; Jin, L.W.; DeCarli, C.; Despa, F. Amylin deposition in the brain: A second amyloid in Alzheimer disease? Ann. Neurol. 2013, 74, 517–526. [Google Scholar] [CrossRef]
- Lu, L.; Fu, D.L.; Li, H.Q.; Liu, A.J.; Li, J.H.; Zheng, G.Q. Diabetes and risk of Parkinson’s disease: An updated meta-analysis of case-control studies. PLoS ONE 2014, 9, e85781. [Google Scholar] [CrossRef]
- Martinez-Valbuena, I.; Valenti-Azcarate, R.; Amat-Villegas, I.; Marcilla, I.; Marti-Andres, G.; Caballero, M.C.; Riverol, M.; Tunon, M.T.; Fraser, P.E.; Luquin, M.R. Mixed pathologies in pancreatic beta cells from subjects with neurodegenerative diseases and their interaction with prion protein. Acta Neuropathol. Commun. 2021, 9, 64. [Google Scholar] [CrossRef] [PubMed]
- Peila, R.; Rodriguez, B.L.; Launer, L.J.; Honolulu-Asia Aging, S. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes 2002, 51, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Gomez, A.; Diaz, Y.; Duarte-Salles, T.; Compta, Y.; Marti, M.J. Prediabetes, type 2 diabetes mellitus and risk of Parkinson’s disease: A population-based cohort study. Parkinsonism Relat. Disord. 2021, 89, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Gotz, J.; Lim, Y.A.; Eckert, A. Lessons from two prevalent amyloidosis—What amylin and Abeta have in common. Front. Aging Neurosci. 2013, 5, 38. [Google Scholar] [CrossRef]
- Horvath, I.; Wittung-Stafshede, P. Cross-talk between amyloidogenic proteins in type-2 diabetes and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2016, 113, 12473–12477. [Google Scholar] [CrossRef]
- Mucibabic, M.; Steneberg, P.; Lidh, E.; Straseviciene, J.; Ziolkowska, A.; Dahl, U.; Lindahl, E.; Edlund, H. alpha-Synuclein promotes IAPP fibril formation in vitro and beta-cell amyloid formation in vivo in mice. Sci. Rep. 2020, 10, 20438. [Google Scholar] [CrossRef]
- Peng, C.; Gathagan, R.J.; Covell, D.J.; Medellin, C.; Stieber, A.; Robinson, J.L.; Zhang, B.; Pitkin, R.M.; Olufemi, M.F.; Luk, K.C.; et al. Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature 2018, 557, 558–563. [Google Scholar] [CrossRef]
- Ferreira, N.; Gram, H.; Sorrentino, Z.A.; Gregersen, E.; Schmidt, S.I.; Reimer, L.; Betzer, C.; Perez-Gozalbo, C.; Beltoja, M.; Nagaraj, M.; et al. Multiple system atrophy-associated oligodendroglial protein p25alpha stimulates formation of novel alpha-synuclein strain with enhanced neurodegenerative potential. Acta Neuropathol. 2021, 142, 87–115. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Rub, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Van Den Berge, N.; Ferreira, N.; Mikkelsen, T.W.; Alstrup, A.K.O.; Tamguney, G.; Karlsson, P.; Terkelsen, A.J.; Nyengaard, J.R.; Jensen, P.H.; Borghammer, P. Ageing promotes pathological alpha-synuclein propagation and autonomic dysfunction in wild-type rats. Brain 2021, 144, 1853–1868. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, N.; Goncalves, N.P.; Jan, A.; Jensen, N.M.; van der Laan, A.; Mohseni, S.; Vaegter, C.B.; Jensen, P.H. Trans-synaptic spreading of alpha-synuclein pathology through sensory afferents leads to sensory nerve degeneration and neuropathic pain. Acta Neuropathol. Commun. 2021, 9, 31. [Google Scholar] [CrossRef]
- Mukherjee, A.; Morales-Scheihing, D.; Salvadores, N.; Moreno-Gonzalez, I.; Gonzalez, C.; Taylor-Presse, K.; Mendez, N.; Shahnawaz, M.; Gaber, A.O.; Sabek, O.M.; et al. Induction of IAPP amyloid deposition and associated diabetic abnormalities by a prion-like mechanism. J. Exp. Med. 2017, 214, 2591–2610. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhowmick, D.C.; Kudaibergenova, Z.; Burnett, L.; Jeremic, A.M. Molecular Mechanisms of Amylin Turnover, Misfolding and Toxicity in the Pancreas. Molecules 2022, 27, 1021. https://doi.org/10.3390/molecules27031021
Bhowmick DC, Kudaibergenova Z, Burnett L, Jeremic AM. Molecular Mechanisms of Amylin Turnover, Misfolding and Toxicity in the Pancreas. Molecules. 2022; 27(3):1021. https://doi.org/10.3390/molecules27031021
Chicago/Turabian StyleBhowmick, Diti Chatterjee, Zhanar Kudaibergenova, Lydia Burnett, and Aleksandar M. Jeremic. 2022. "Molecular Mechanisms of Amylin Turnover, Misfolding and Toxicity in the Pancreas" Molecules 27, no. 3: 1021. https://doi.org/10.3390/molecules27031021
APA StyleBhowmick, D. C., Kudaibergenova, Z., Burnett, L., & Jeremic, A. M. (2022). Molecular Mechanisms of Amylin Turnover, Misfolding and Toxicity in the Pancreas. Molecules, 27(3), 1021. https://doi.org/10.3390/molecules27031021