

Comparative Interaction Studies of Quercetin with 2-Hydroxyl-propyl-β-cyclodextrin and 2,6-Methylated-β-cyclodextrin

 , , , ,
, , , ,  ,
,  , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
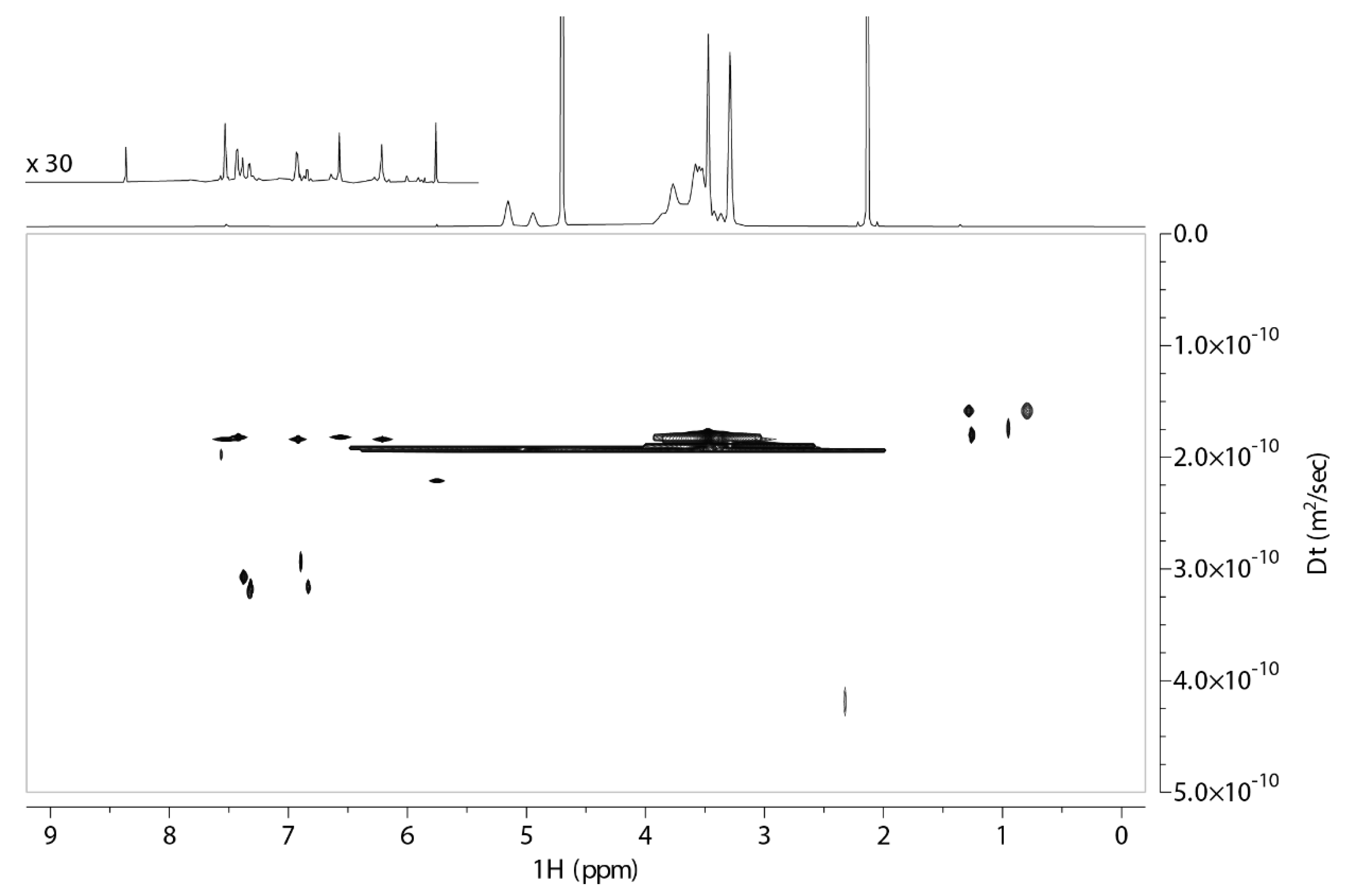
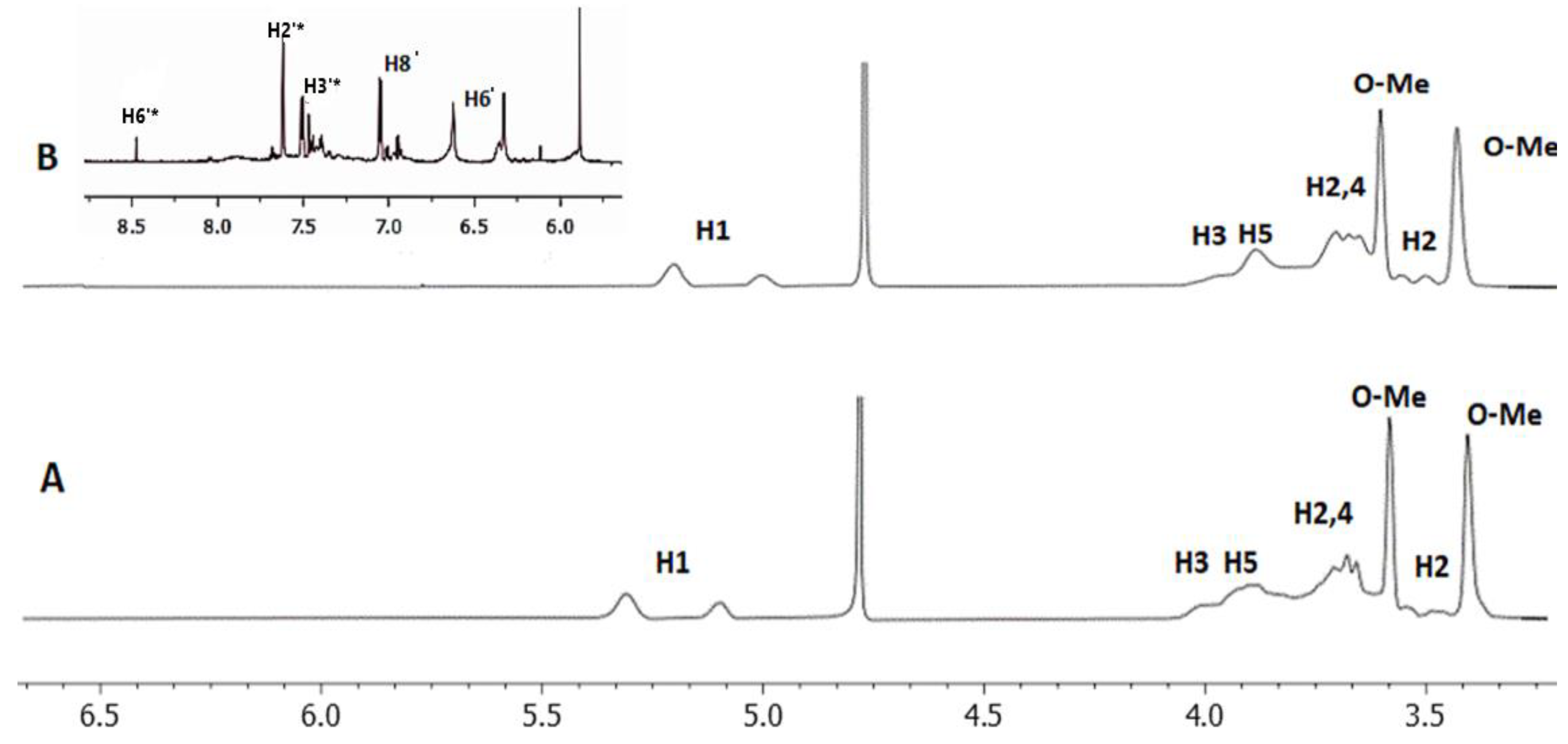
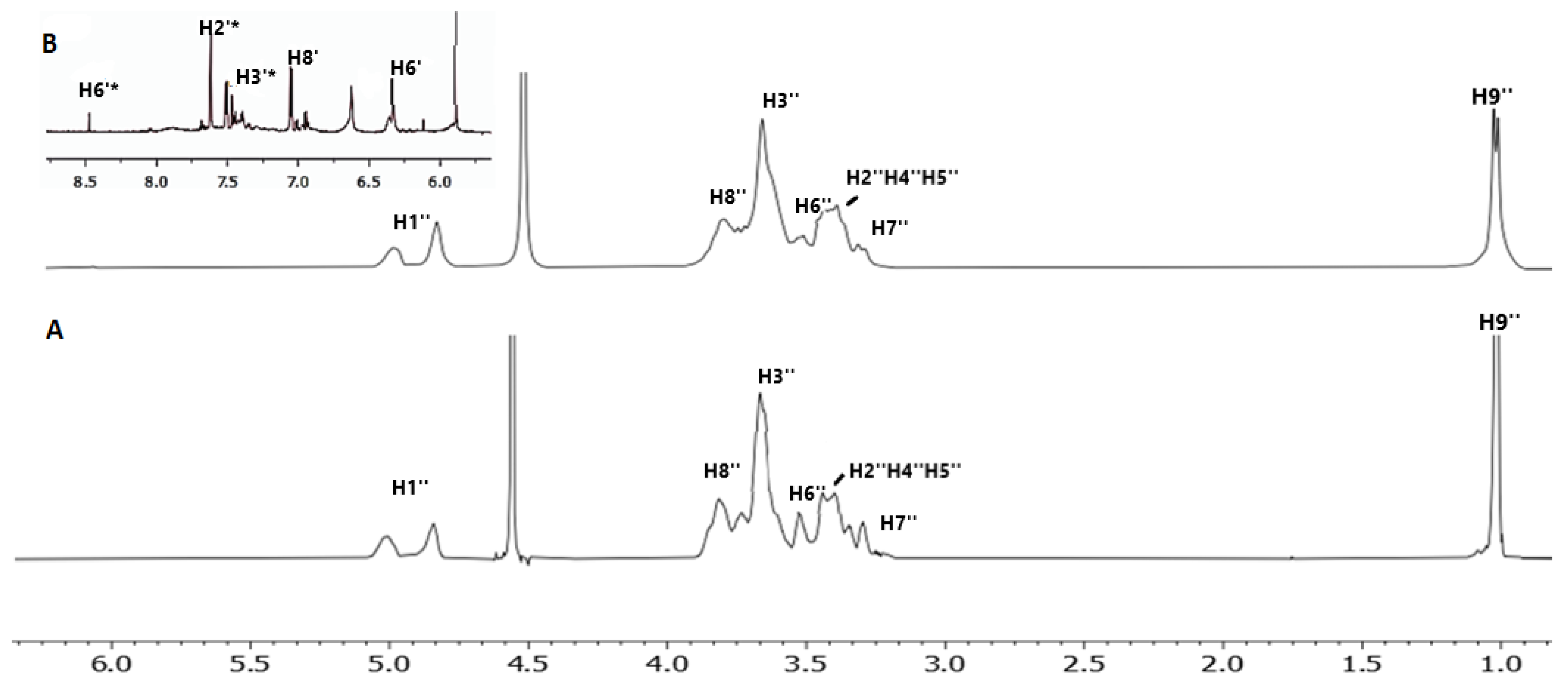
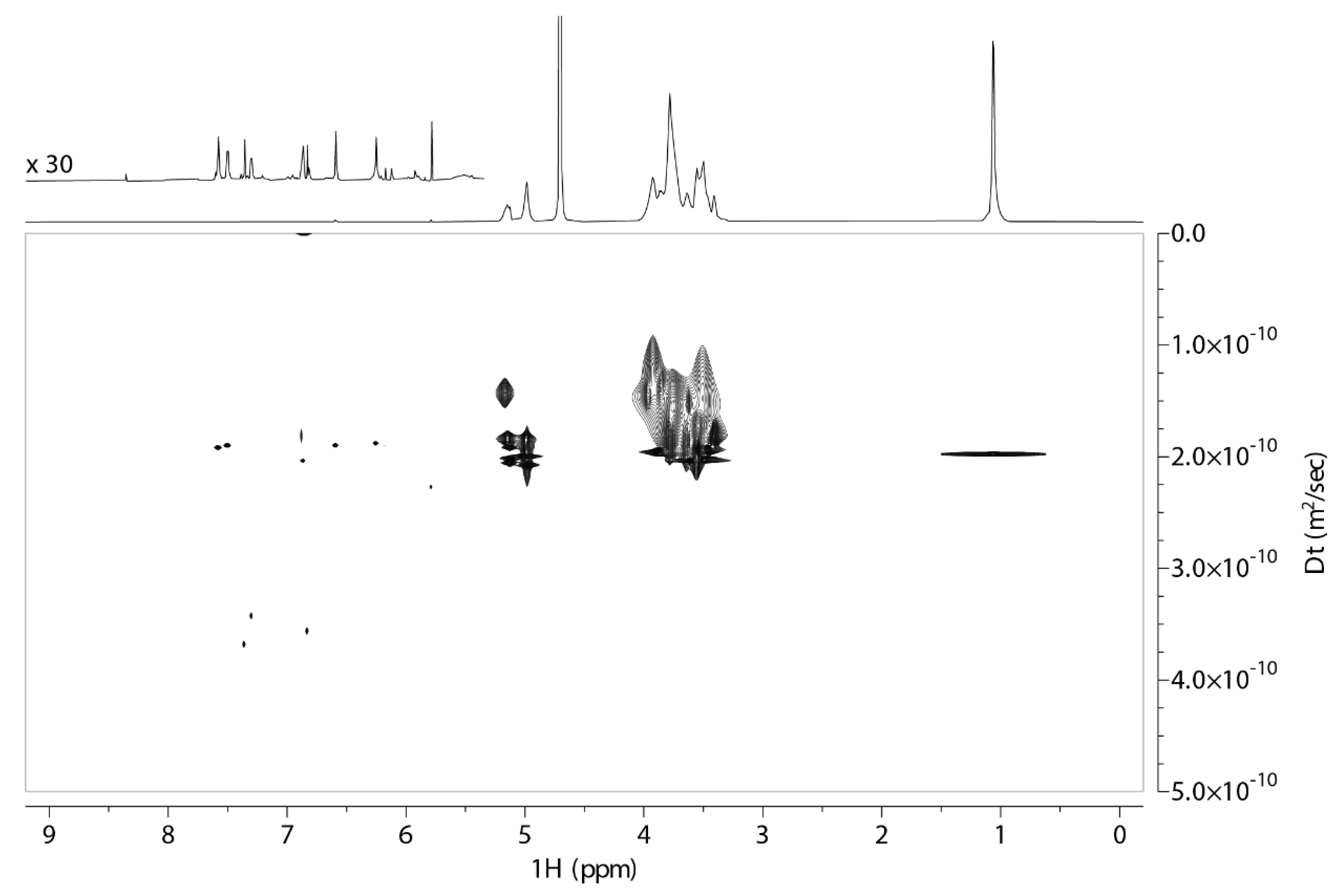
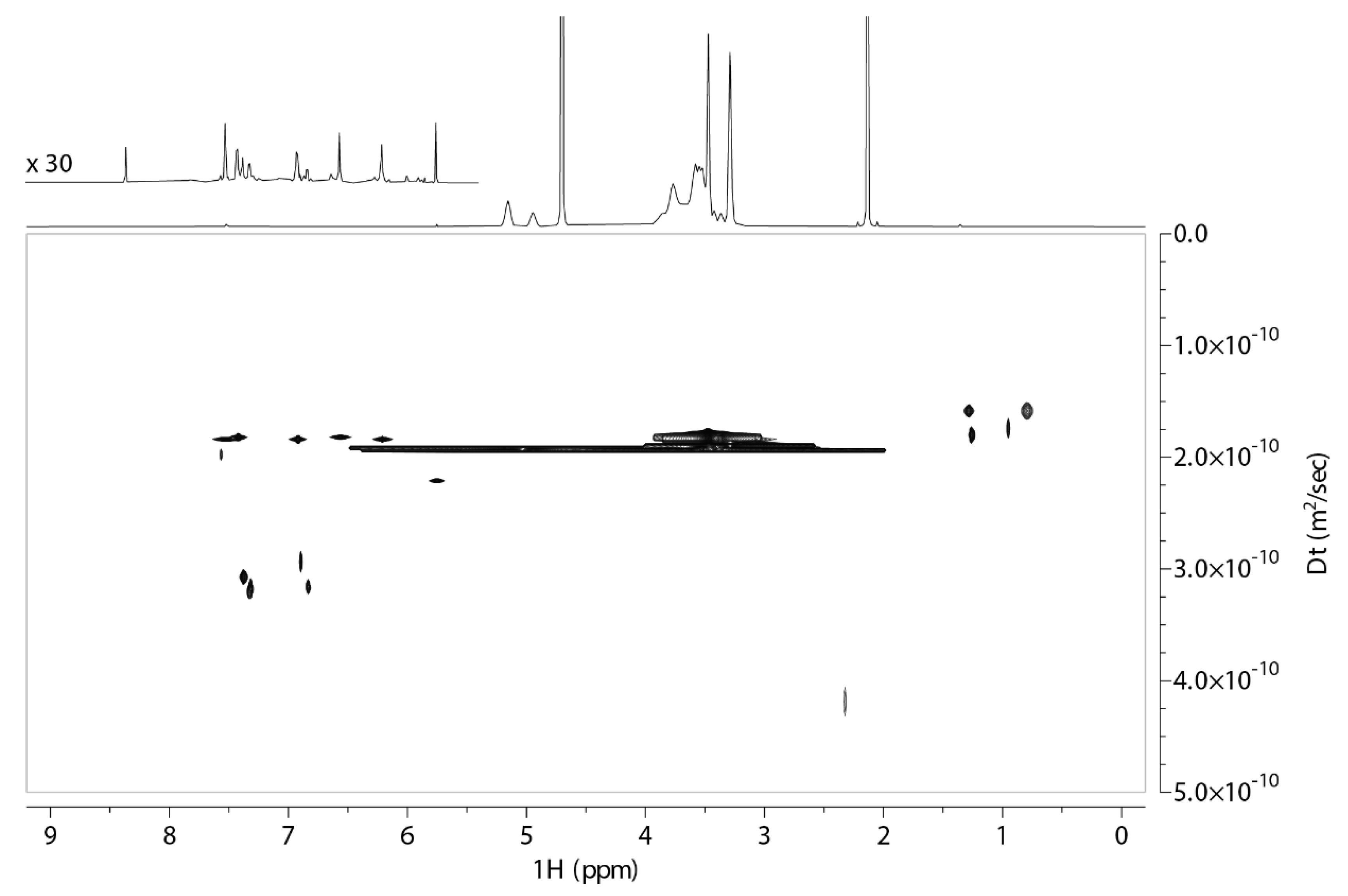
2.1. 2D DOSY Experiment
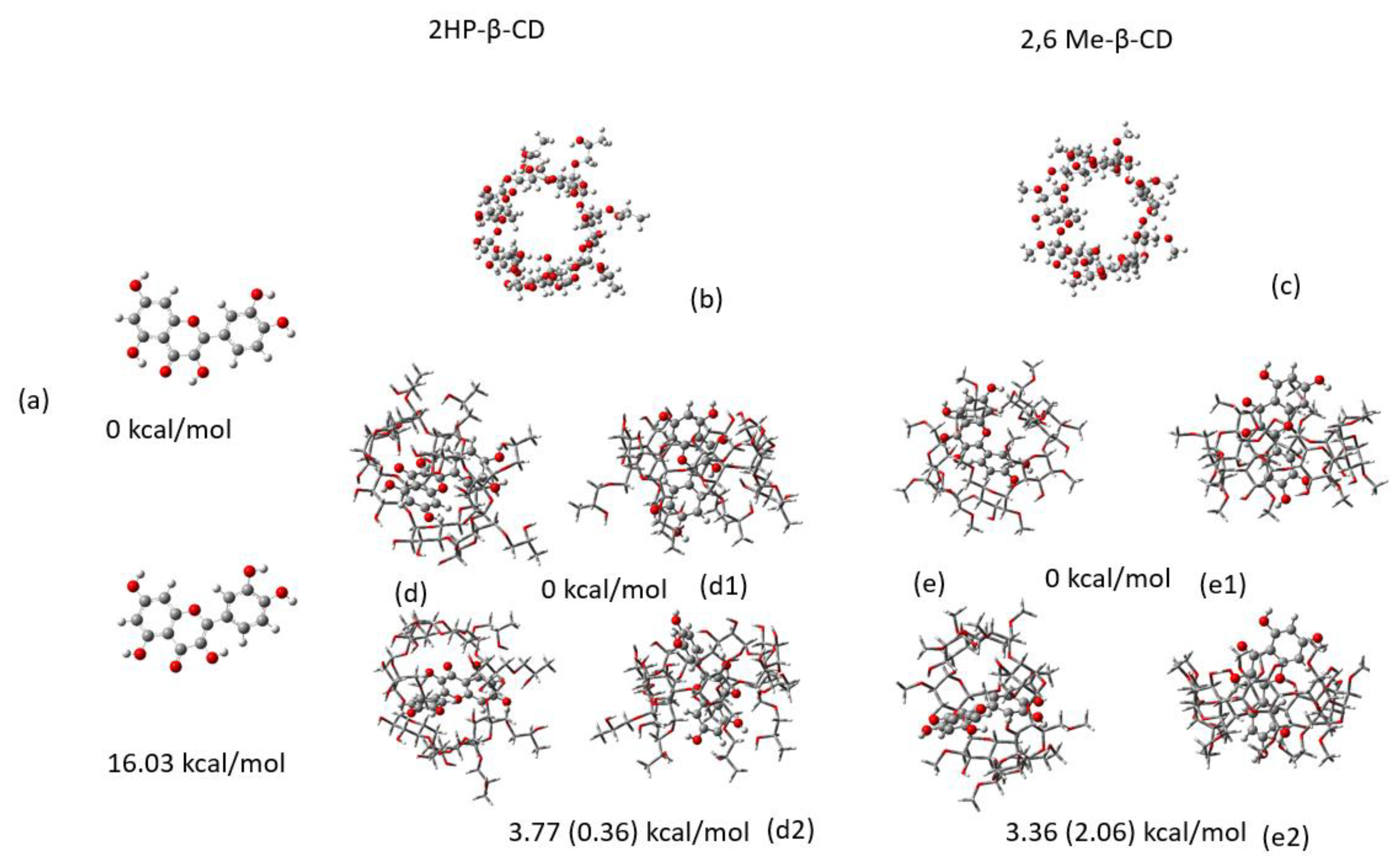
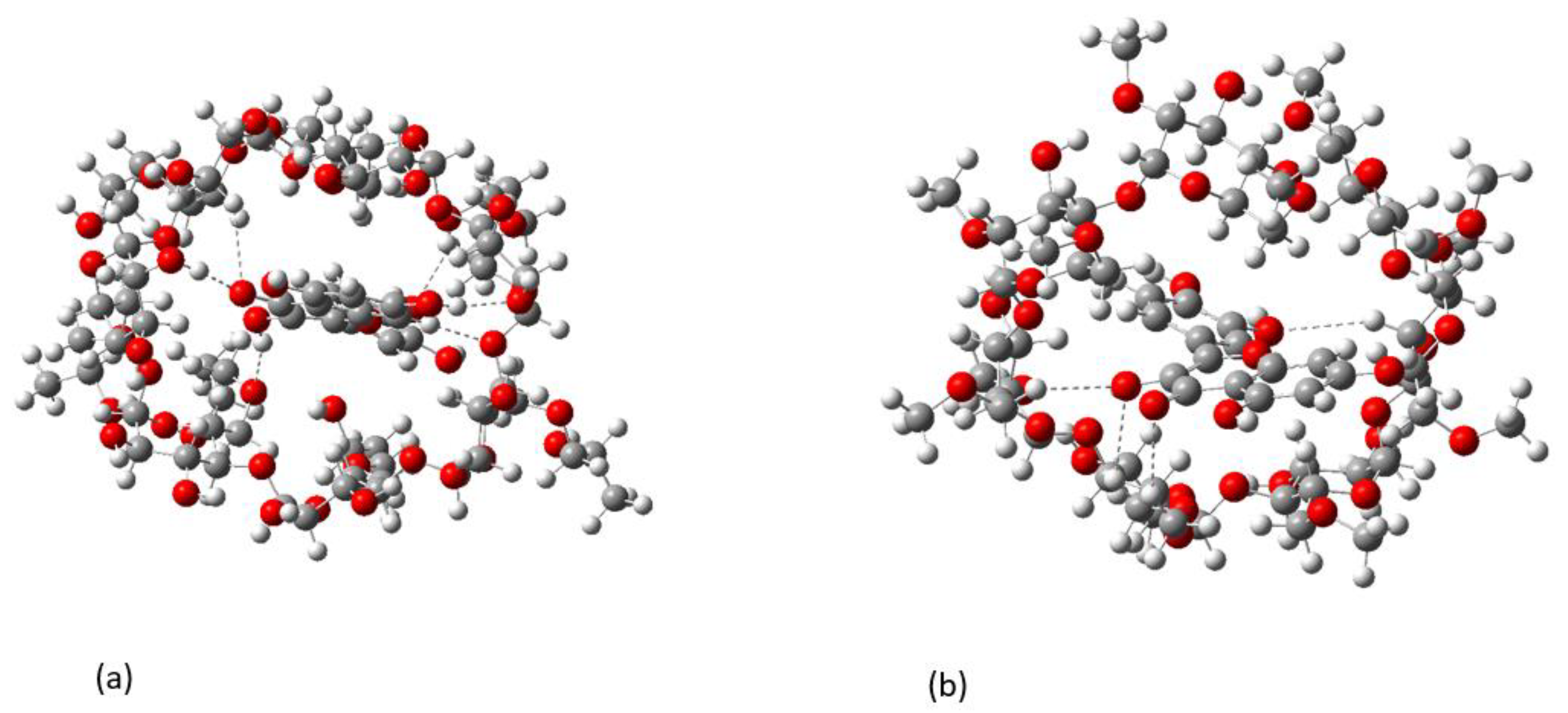
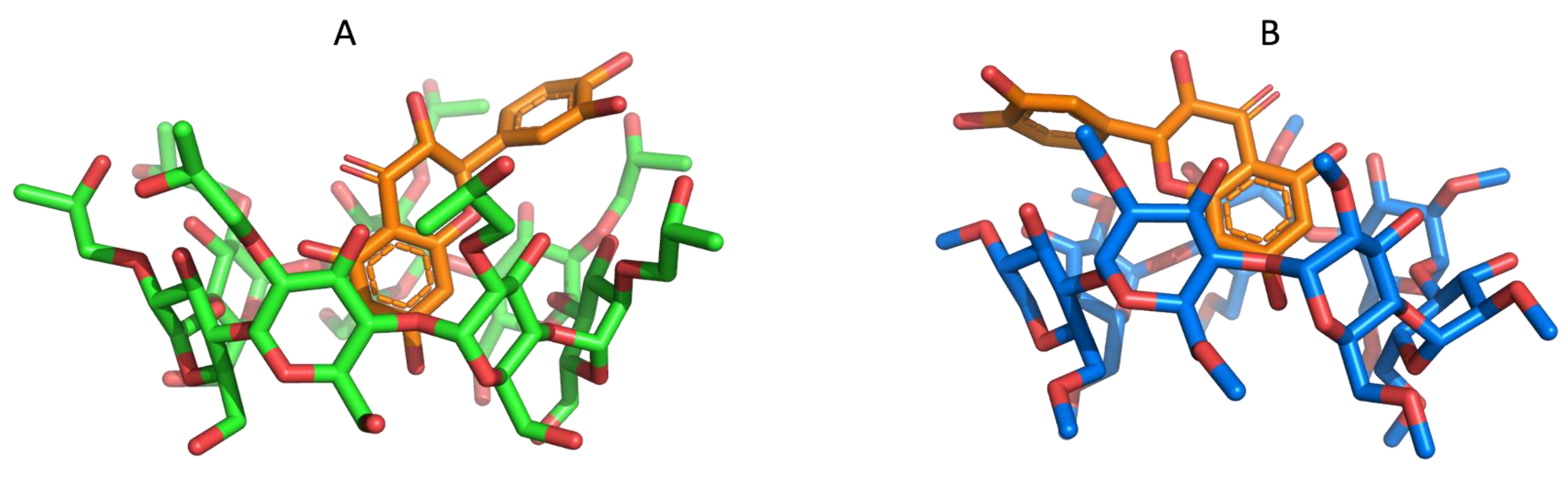
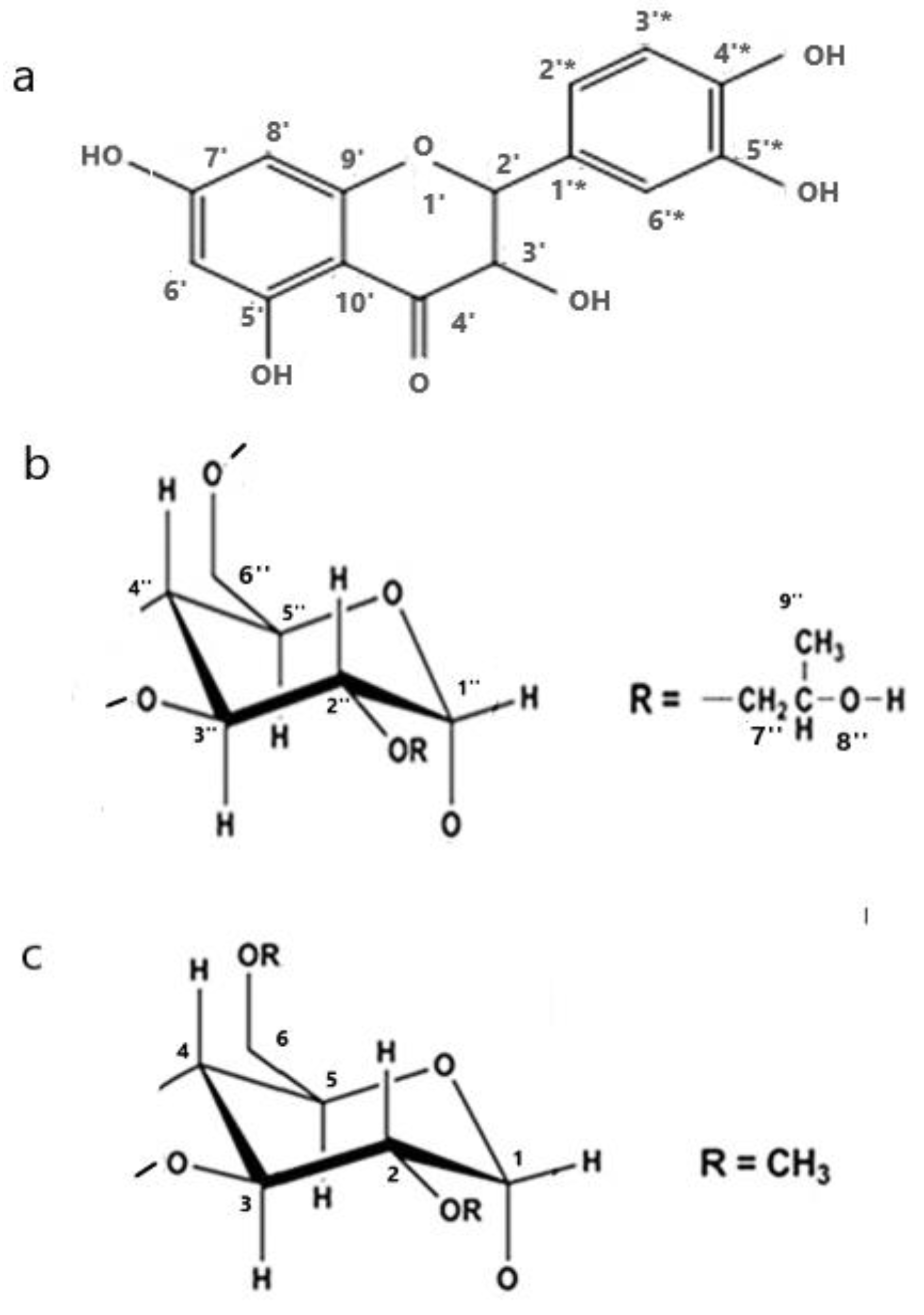
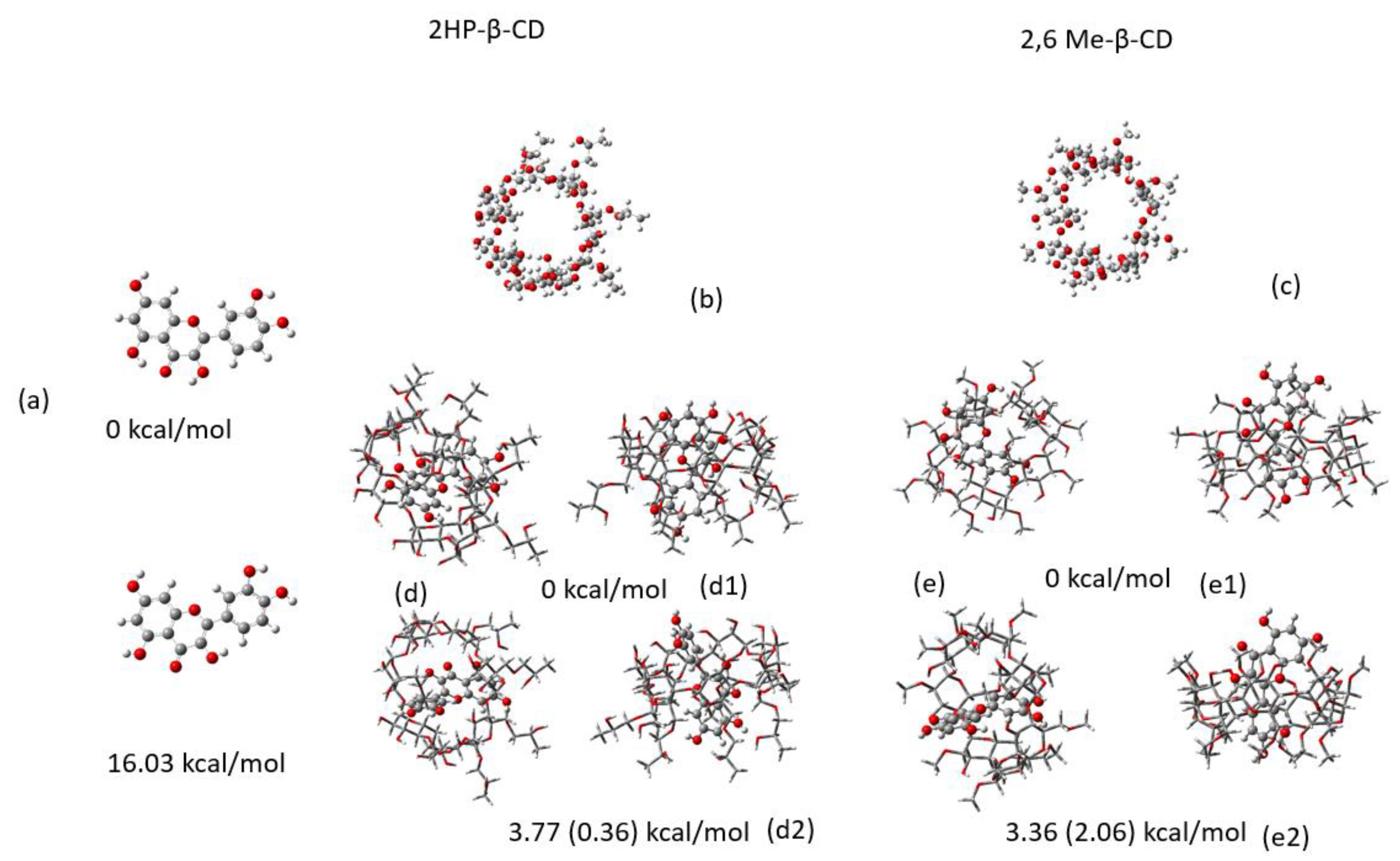
2.2. Conformational Analysis
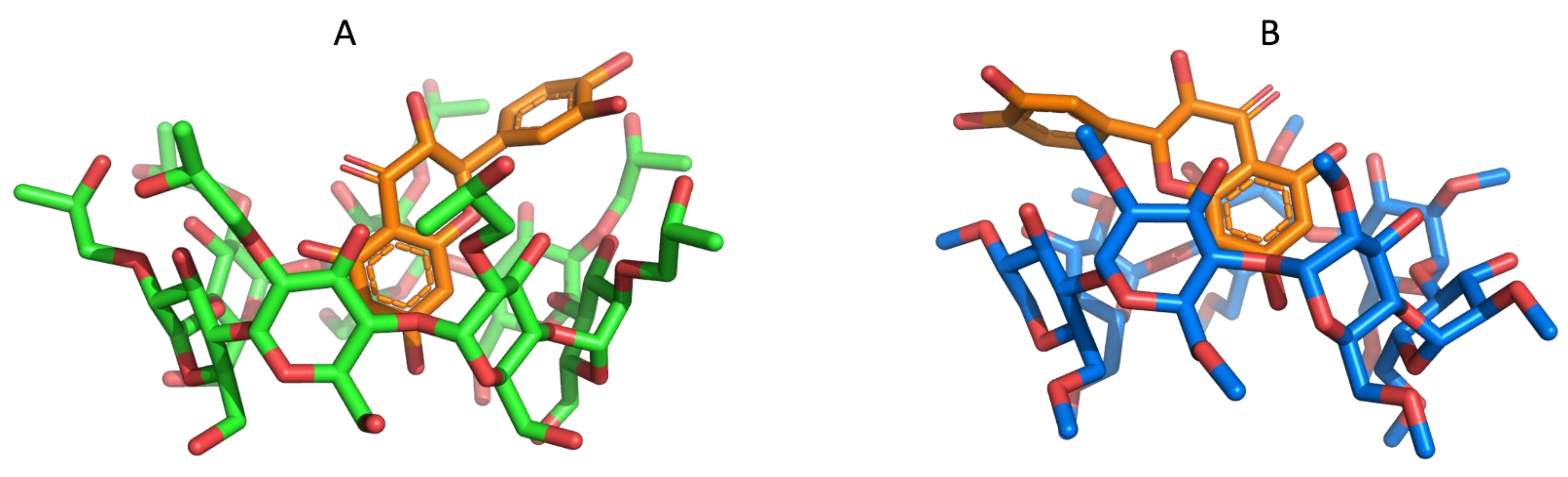
2.3. MM/GBSA Estimates of the Free Energy of Binding Using MD Simulations
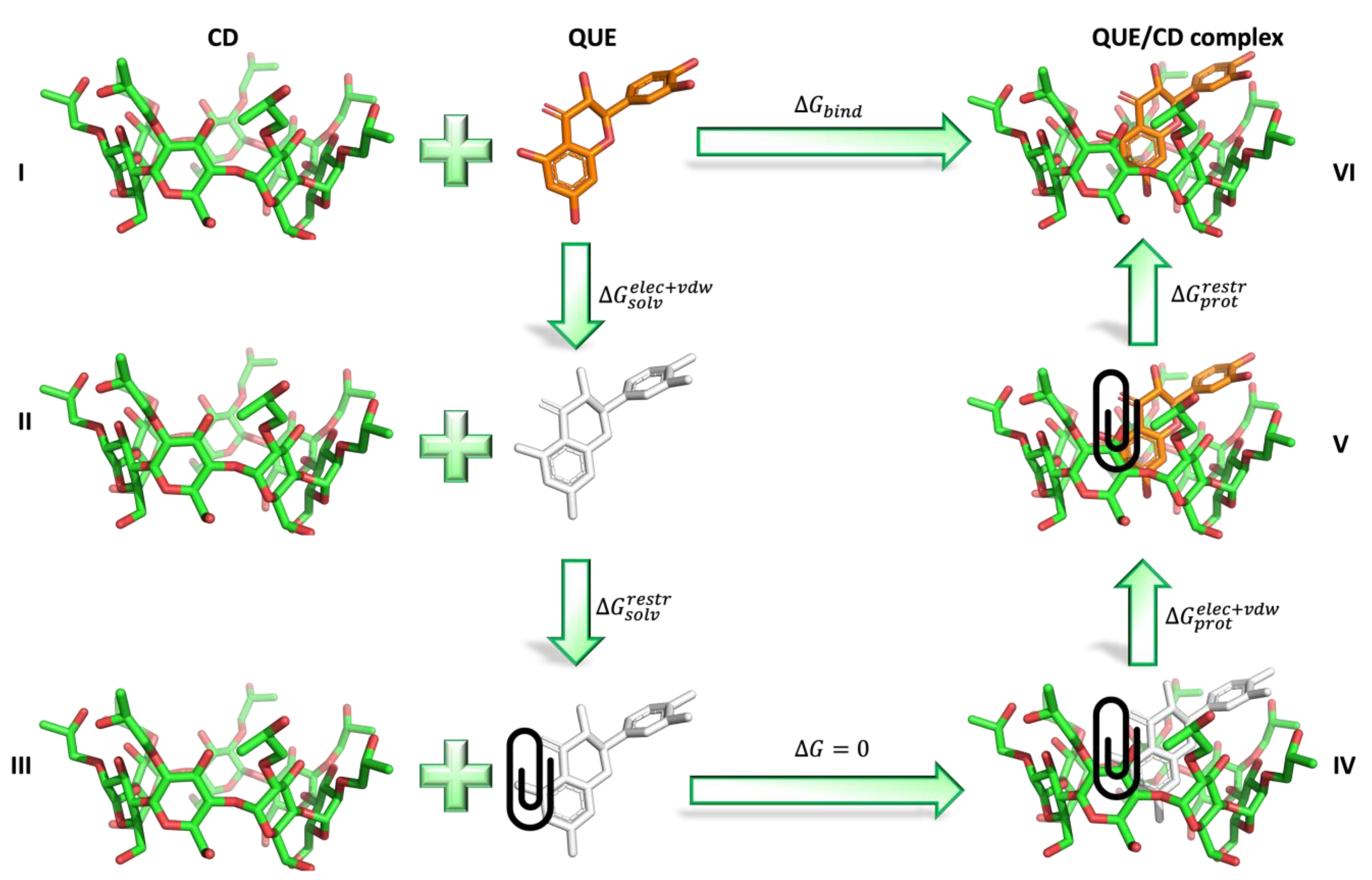
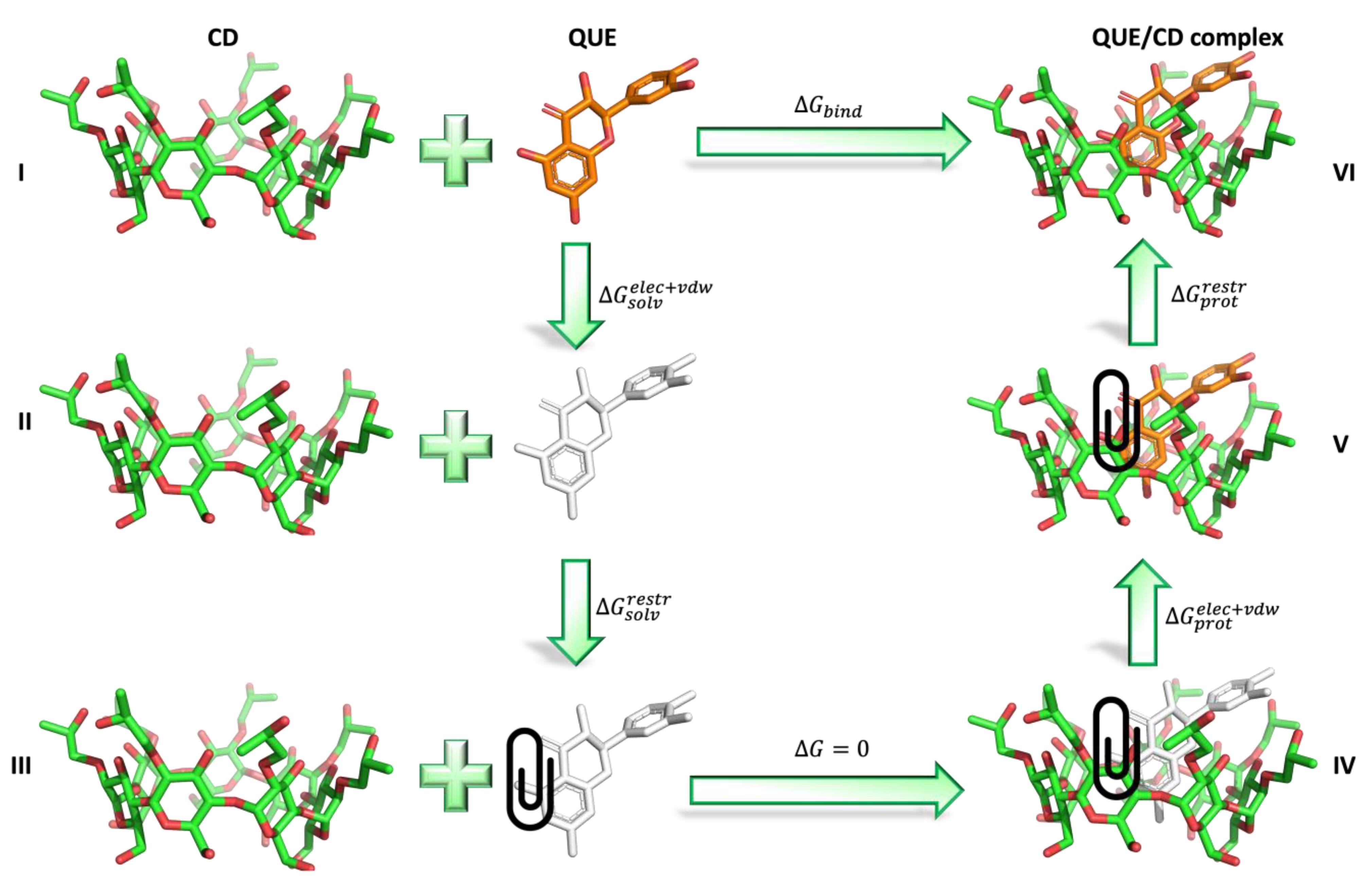
2.4. Absolute Binding Free Energy Calculations
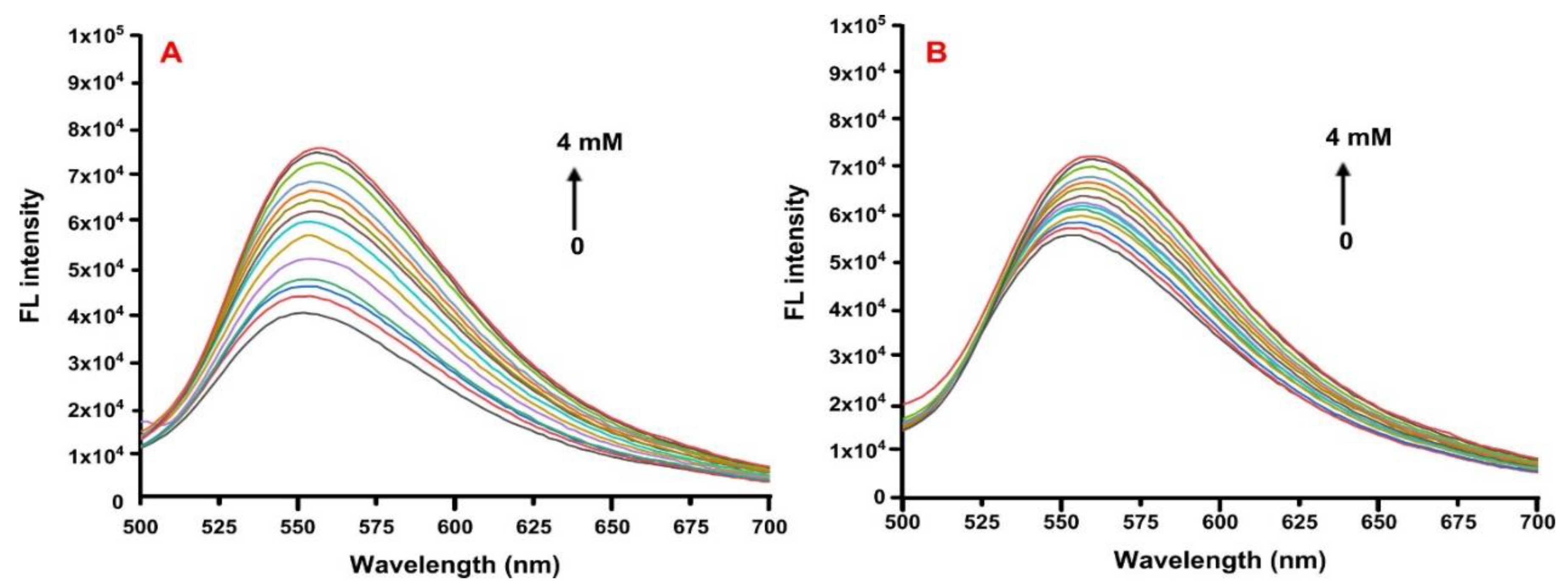
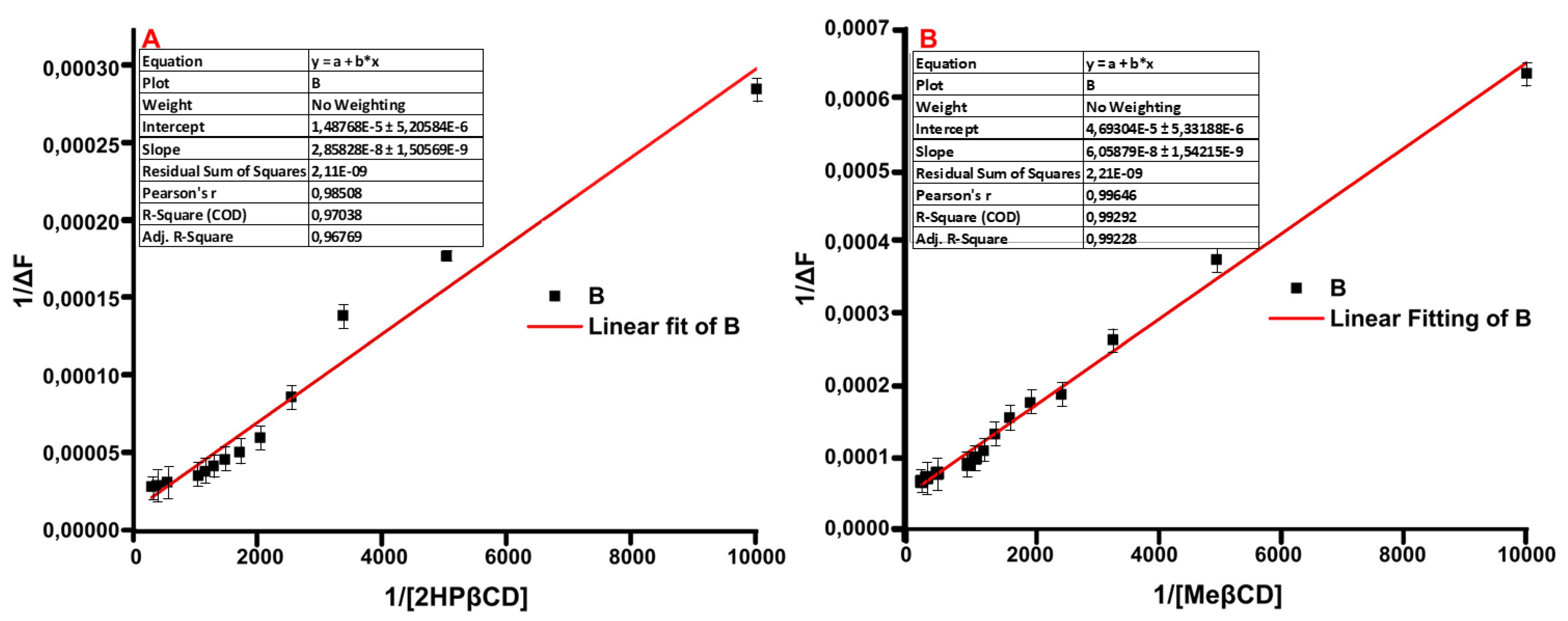
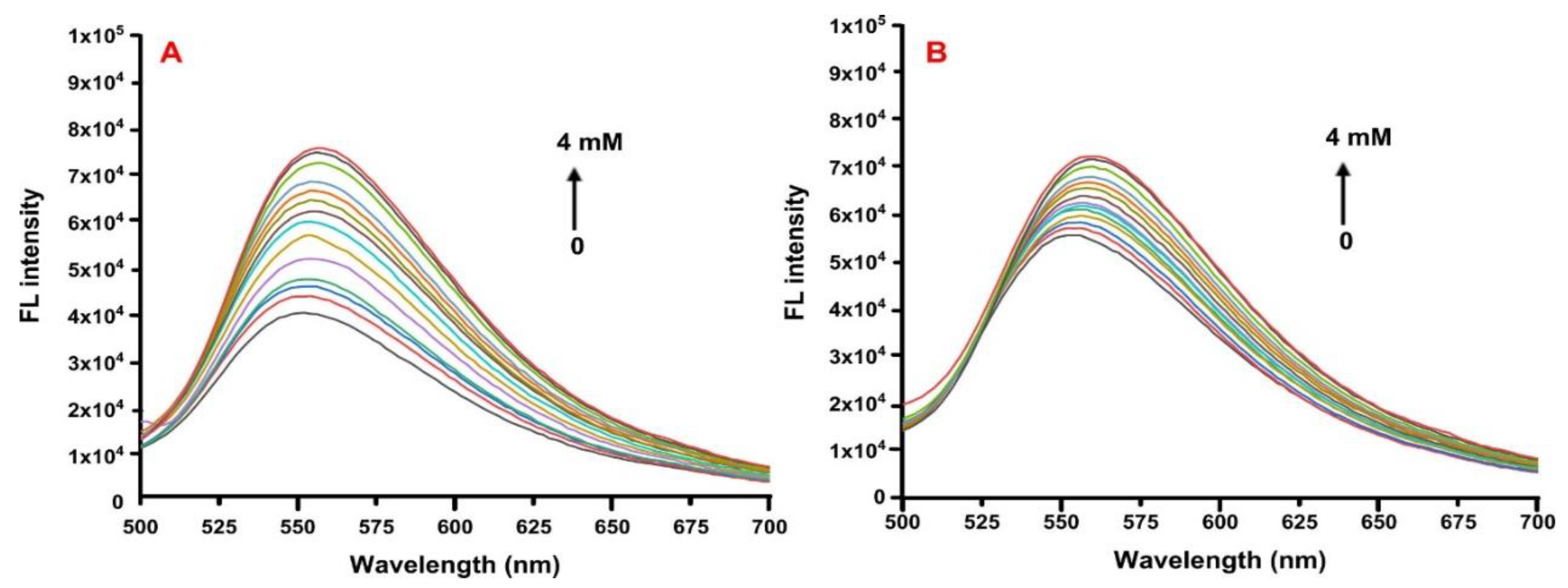
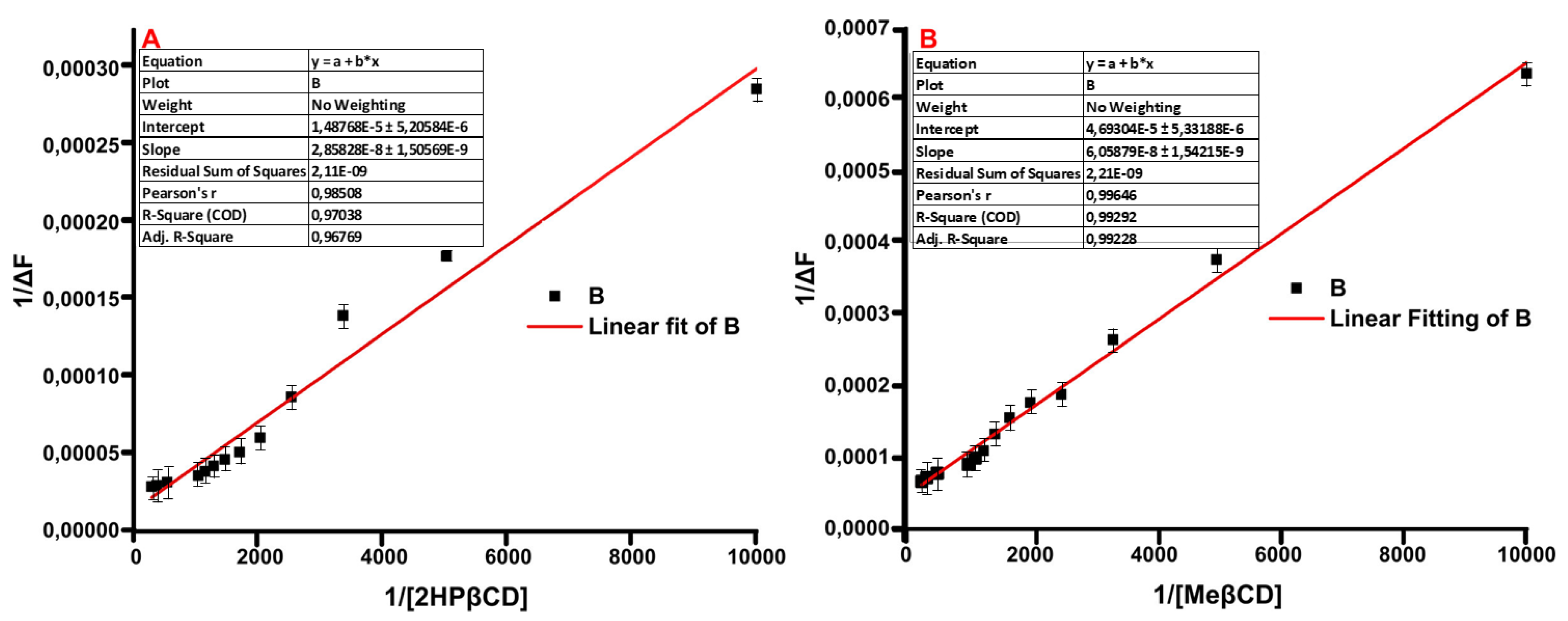
2.5. Fluorescence Spectroscopy
3. Materials and Methods
3.1. Chemicals
3.2. Preparation of the Complex
3.3. High Resolution 1H NMR Spectroscopy
3.4. Conformational Analysis (Quantum Mechanics Calculations)
3.5. MD Simulations
3.6. Absolute Binding Free Energy Calculations
3.7. Fluorescence Spectroscopy Studies
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Ilyich, T.V.; Kovalenia, T.A.; Lapshina, E.A.; Stępniak, A.; Palecz, B.; Zavodnik, I.B. Thermodynamic parameters and mitochondrial effects of supramolecular complexes of quercetin with β-cyclodextrins. J. Mol. Liq. 2021, 325, 115184. [Google Scholar] [CrossRef]
- Havsteen, B.H. The biochemistry and medical significance of the flavonoids. Pharmacol. Ther. 2002, 96, 67–202. [Google Scholar] [CrossRef]
- Andersen, Ø.M.; Markham, K.R. (Eds.) Flavonoids: Chemistry, Biochemistry, and Applications; CRC, Taylor & Francis: Boca Raton, FL, USA, 2006. [Google Scholar]
- Wang, W.; Sun, C.; Mao, L.; Ma, P.; Liu, F.; Yang, J.; Gao, Y. The biological activities, chemical stability, metabolism and delivery systems of quercetin: A review. Trends Food Sci. Technol. 2016, 56, 21–38. [Google Scholar] [CrossRef]
- Mallya, R.; Patil, K.; Patil, K.; Patil, K. Recent Developments in Formulation Design of a Multifunctional Phytochemical Quercetin: A Review. Pharmacogn. Rev. 2021, 15, 32–46. [Google Scholar] [CrossRef]
- Dueñas, M.; González-Manzano, S.; González-Paramás, A.; Santos-Buelga, C. Antioxidant evaluation of O-methylated metabolites of catechin, epicatechin and quercetin. J. Pharm. Biomed. Anal. 2010, 51, 443–449. [Google Scholar] [CrossRef]
- Ji, L.; Ma, Y.; Wang, Z.; Cai, Z.; Pang, C.; Wang, Z. Quercetin Prevents Pyrrolizidine Alkaloid Clivorine-Induced Liver Injury in Mice by Elevating Body Defense Capacity. PLoS ONE 2014, 9, e98970. [Google Scholar] [CrossRef]
- Suganthy, N.; Devi, K.P.; Nabavi, S.F.; Braidy, N.; Nabavi, S.M. Bioactive effects of quercetin in the central nervous system: Focusing on the mechanisms of actions. Biomed. Pharmacother. 2016, 84, 892–908. [Google Scholar] [CrossRef]
- Kumari, A.; Yadav, S.K.; Pakade, Y.B.; Singh, B.; Yadav, S.C. Development of biodegradable nanoparticles for delivery of quercetin. Colloids Surf. B Biointerfaces 2010, 80, 184–192. [Google Scholar] [CrossRef]
- Kleemann, R.; Verschuren, L.; Morrison, M.; Zadelaar, S.; van Erk, M.J.; Wielinga, P.Y.; Kooistra, T. Anti-inflammatory, anti-proliferative and anti-atherosclerotic effects of quercetin in human in vitro and in vivo models. Atherosclerosis 2011, 218, 44–52. [Google Scholar] [CrossRef]
- Phachonpai, W.; Wattanathorn, J.; Muchimapura, S.; Tong-Un, T.; Preechagoon, D. Neuroprotective Effect of Quercetin Encapsulated Liposomes: A Novel Therapeutic Strategy against Alzheimer’s Disease. Am. J. Appl. Sci. 2010, 7, 480–485. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Tang, Y.; Gao, C.; Li, Y.; Chen, S.; Xiong, T.; Li, J.; Du, M.; Gong, Z.; Chen, H.; et al. Characterization and biodistribution in vivo of quercetin-loaded cationic nanostructured lipid carriers. Colloids Surf. B Biointerfaces 2014, 115, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Gang, W.; Jie, W.J.; Ping, Z.L.; Ming, D.S.; Ying, L.J.; Lei, W.; Fang, Y. Liposomal quercetin: Evaluating drug delivery in vitro and biodistribution in vivo. Expert Opin. Drug Deliv. 2012, 9, 599–613. [Google Scholar] [CrossRef] [PubMed]
- Vicentini, F.T.M.C.; Vaz, M.M.O.L.L.; Fonseca, Y.M.; Bentley, M.V.L.B.; Fonseca, M.J.V. Characterization and stability study of a water-in-oil microemulsion incorporating quercetin. Drug Dev. Ind. Pharm. 2011, 37, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Rawat, M.S.M.; Semalty, A.; Semalty, M. Quercetin-Phospholipid Complex: An Amorphous Pharmaceutical System in Herbal Drug Delivery. Curr. Drug Discov. Technol. 2012, 9, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Landi-Librandi, A.P.; Chrysostomo, T.N.; Azzolini, A.E.C.S.; Marzocchi-Machado, C.M.; de Oliveira, C.A.; Lucisano-Valim, Y.M. Study of quercetin-loaded liposomes as potential drug carriers: In vitro evaluation of human complement activation. J. Liposome Res. 2012, 22, 89–99. [Google Scholar] [CrossRef]
- Dhawan, S.; Kapil, R.; Singh, B. Formulation development and systematic optimization of solid lipid nanoparticles of quercetin for improved brain delivery. J. Pharm. Pharmacol. 2011, 63, 342–351. [Google Scholar] [CrossRef]
- Varshosaz, J.; Jafarian, A.; Salehi, G.; Zolfaghari, B. Comparing different sterol containing solid lipid nanoparticles for targeted delivery of quercetin in hepatocellular carcinoma. J. Liposome Res. 2014, 24, 191–203. [Google Scholar] [CrossRef]
- Sun, M.; Nie, S.; Pan, X.; Zhang, R.; Fan, Z.; Wang, S. Quercetin-nanostructured lipid carriers: Characteristics and anti-breast cancer activities in vitro. Colloids Surf. B Biointerfaces 2014, 113, 15–24. [Google Scholar] [CrossRef]
- Arbain, N.H.A.N.H.; Ngan, C.L.; Salim, N.; Wui, W.T.; Rahman, M.B.A. Aerosolized nanoemulsion system encapsulating quercertin for lung cancer treatment. eProc. Chem. 2017, 3, 1. Available online: http://161.139.21.153/index.php/FYP/article/view/192 (accessed on 27 December 2021).
- Priprem, A.; Sutthiparinyanont, S.; Na, H.-K.; Surh, Y.-J.; Chulasiri, M. Effect of Formulations of Nanosized Quercetin Liposomes on COX-2 and NF-kB in MCF-10A Cells. Pharm. Nanotechnol. 2013, 1, 26–34. [Google Scholar] [CrossRef]
- Tablet, C.; Hillebrand, M. Theoretical and experimental study of the inclusion complexes of the 3-carboxy-5,6-benzocoumarinic acid with cyclodextrins. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2008, 70, 740–748. [Google Scholar] [CrossRef]
- Yousuf, S.; Radhika, D.; Enoch, I.V.M.V.; Easwaran, M. The influence of β-cyclodextrin encapsulation on the binding of 2′-hydroxyflavanone with calf thymus DNA. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2012, 98, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Shafi, A.A.; Al-Shihry, S.S. Fluorescence enhancement of 1-napthol-5-sulfonate by forming inclusion complex with β-cyclodextrin in aqueous solution. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2009, 72, 533–537. [Google Scholar] [CrossRef]
- Kosar, B.; Albayrak, C. Spectroscopic investigations and quantum chemical computational study of (E)-4-methoxy-2- [(p-tolylimino)methyl]phenol. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2011, 78, 160–167. [Google Scholar] [CrossRef]
- Abdel-Shafi, A.A. Inclusion complex of 2-naphthylamine-6-sulfonate with β-cyclodextrin: Intramolecular charge transfer versus hydrogen bonding effects. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2007, 66, 1228–1236. [Google Scholar] [CrossRef]
- Bilensoy, E. Cyclodextrins in Pharmaceutics, Cosmetics, and Biomedicine: Current and Future Industrial Applications; Wiley: Hoboken, NJ, USA, 2011. [Google Scholar]
- Carneiro, S.B.; Costa Duarte, F.Í.; Heimfarth, L.; Siqueira Quintans, J.D.S.; Quintans-Júnior, L.J.; Veiga Júnior, V.F.D.; De Lima, A.N. Cyclodextrin–Drug Inclusion Complexes: In Vivo and In Vitro Approaches. Int. J. Mol. Sci. 2019, 20, 642. [Google Scholar] [CrossRef]
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef]
- Topuz, F.; Uyar, T. Antioxidant, antibacterial and antifungal electrospun nanofibers for food packaging applications. Food Res. Int. 2020, 130, 108927. [Google Scholar] [CrossRef]
- Loftsson, T.; Brewster, M.E. Pharmaceutical Applications of Cyclodextrins. 1. Drug Solubilization and Stabilization. J. Pharm. Sci. 1996, 85, 1017–1025. [Google Scholar] [CrossRef]
- Roux, M.; Perly, B.; Djedaini-Pilard, F. Self-assemblies of amphiphilic cyclodextrins. Eur. Biophys. J. 2007, 36, 861–867. [Google Scholar] [CrossRef]
- Marques, H.M.C. A review on cyclodextrin encapsulation of essential oils and volatiles. Flavour Fragr. J. 2010, 25, 313–326. [Google Scholar] [CrossRef]
- Špehar, T.K.; Pocrnić, M.; Klarić, D.; Bertoša, B.; Čikoš, A.; Jug, M.; Padovan, J.; Dragojević, S.; Galić, N. Investigation of Praziquantel/Cyclodextrin Inclusion Complexation by NMR and LC-HRMS/MS: Mechanism, Solubility, Chemical Stability, and Degradation Products. Mol. Pharm. 2021, 18, 4210–4223. [Google Scholar] [CrossRef]
- Rajendrakumar, K.; Madhusudan, S.; Pralhad, T. Cyclodextrin complexes of valdecoxib: Properties and anti-inflammatory activity in rat. Eur. J. Pharm. Biopharm. 2005, 60, 39–46. [Google Scholar] [CrossRef]
- Zhang, L.; Man, S.; Qiu, H.; Liu, Z.; Zhang, M.; Ma, L.; Gao, W. Curcumin-cyclodextrin complexes enhanced the anti-cancer effects of curcumin. Environ. Toxicol. Pharmacol. 2016, 48, 31–38. [Google Scholar] [CrossRef]
- Yee, E.M.; Hook, J.M.; Bhadbhade, M.M.; Vittorio, O.; Kuchel, R.P.; Brandl, M.B.; Tilley, R.D.; Black, D.S.; Kumar, N. Preparation, characterization and in vitro biological evaluation of (1:2) phenoxodiol-β-cyclodextrin complex. Carbohydr. Polym. 2017, 165, 444–454. [Google Scholar] [CrossRef]
- Kellici, T.F.; Ntountaniotis, D.; Leonis, G.; Chatziathanasiadou, M.; Chatzikonstantinou, A.V.; Becker-Baldus, J.; Glaubitz, C.; Tzakos, A.G.; Viras, K.; Chatzigeorgiou, P.; et al. Investigation of the Interactions of Silibinin with 2-Hydroxypropyl-β-cyclodextrin through Biophysical Techniques and Computational Methods. Mol. Pharm. 2015, 12, 954–965. [Google Scholar] [CrossRef]
- Kellici, T.; Chatziathanasiadou, M.V.; Diamantis, D.; Chatzikonstantinou, A.V.; Andreadelis, I.; Christodoulou, E.; Valsami, G.; Mavromoustakos, T.; Tzakos, A.G. Mapping the interactions and bioactivity of quercetin (2-hydroxypropyl)-β-cyclodextrin complex. Int. J. Pharm. 2016, 511, 303–311. [Google Scholar] [CrossRef]
- Sfihi, H.; Legrand, A.; Doussot, J.; Guy, A. Solid-state 13C NMR study of β-cyclodextrin/substituted aromatic ketone complexes: Evidence for two kinds of complexation of the guest molecules. Colloids Surf. A Physicochem. Eng. Asp. 1996, 115, 115–126. [Google Scholar] [CrossRef]
- Figueiras, A.; Carvalho, R.A.; Ribeiro, L.; Torres-Labandeira, J.J.; Veiga, F.J.B. Solid-state characterization and dissolution profiles of the inclusion complexes of omeprazole with native and chemically modified β-cyclodextrin. Eur. J. Pharm. Biopharm. 2007, 67, 531–539. [Google Scholar] [CrossRef]
- Mendes, C.; Buttchevitz, A.; Kruger, J.H.; Bernardi, L.S.; Oliveira, P.R.; Silva, M.A.S. Quantitative Analysis of Norfloxacin in β-Cyclodextrin Inclusion Complexes—Development and Validation of a Stability-indicating HPLC Method. Anal. Sci. 2015, 31, 1083–10895. [Google Scholar] [CrossRef]
- Cravotto, G.; Binello, A.; Baranelli, E.; Carraro, P.; Trotta, F. Cyclodextrins as Food Additives and in Food Processing. Curr. Nutr. Food Sci. 2006, 2, 343–350. [Google Scholar] [CrossRef]
- Chattah, A.K.; Mroue, K.H.; Pfund, L.Y.; Ramamoorthy, A.; Longhi, M.R.; Garnero, C. Insights into Novel Supramolecular Complexes of Two Solid Forms of Norfloxacin and β-Cyclodextrin. J. Pharm. Sci. 2013, 102, 3717–3724. [Google Scholar] [CrossRef]
- Malanga, M.; Szemán, J.; Fenyvesi, E.; Puskás, I.; Csabai, K.; Gyémánt, G.; Fenyvesi, F.; Szente, L. “Back to the Future”: A New Look at Hydroxypropyl Beta-Cyclodextrins. J. Pharm. Sci. 2016, 105, 2921–2931. [Google Scholar] [CrossRef]
- Pitha, J.; Milecki, J.; Fales, H.; Pannell, L.; Uekama, K. Hydroxypropyl-β-cyclodextrin: Preparation and characterization; effects on solubility of drugs. Int. J. Pharm. 1986, 29, 73–82. [Google Scholar] [CrossRef]
- Trinadha, C.; Pitha, J.; Lindberg, B.; Lindberg, J. Distribution of substituents in O-(2-hydroxypropyl) derivatives of cyclomalto-oligosaccharides (cyclodextrins: Influence of increasing substitution of the base used in the preparation, and of macrocyclic size. Carbohydr. Res. 1992, 223, 99–107. [Google Scholar] [CrossRef]
- Laquintana, V.; Asim, M.H.; Lopedota, A.; Cutrignelli, A.; Lopalco, A.; Franco, M.; Bernkop-Schnürch, A.; Denora, N. Thiolated hydroxypropyl-β-cyclodextrin as mucoadhesive excipient for oral delivery of budesonide in liquid paediatric formulation. Int. J. Pharm. 2019, 572, 118820. [Google Scholar] [CrossRef]
- Nicoletti, C.D.; Queiroz, M.d.H.; Lima, C.G.d.; da Silva, F.d.; Futuro, D.O.; Ferreira, V.F. An improved method for the preparation of β-lapachone:2-hydroxypropyl-β-cyclodextrin inclusion complexes. J. Drug Deliv. Sci. Technol. 2020, 58, 101777. [Google Scholar] [CrossRef]
- Başaran, E.; Öztürk, A.A.; Şenel, B.; Demïrel, M.; Sarica, Ş. Quercetin, Rutin and Quercetin-Rutin Incorporated Hydroxypropyl β-Cyclodextrin Inclusion Complexes. Eur. J. Pharm. Sci. 2022, 172, 106153. [Google Scholar] [CrossRef]
- Tzeli, D.; Petsalakis, I.D.; Theodorakopoulos, G. Compression in encapsulated carboxylic acid homodimers. Chem. Phys. Lett. 2013, 573, 48–55. [Google Scholar] [CrossRef]
- Tzeli, D.; Tsoungas, P.G.; Petsalakis, I.D.; Kozielewicz, P.; Zloh, M. Intramolecular cyclization of β-nitroso-o-quinone methides. A theoretical endoscopy of a potentially useful innate “reclusive” reaction. Tetrahedron 2015, 71, 359–369. [Google Scholar] [CrossRef]
- Gapsys, V.; Yildirim, A.; Aldeghi, M.; Khalak, Y.; van der Spoel, D.; de Groot, B.L. Accurate absolute free energies for ligand–protein binding based on non-equilibrium approaches. Commun. Chem. 2021, 4, 1. [Google Scholar] [CrossRef]
- Khalak, Y.; Tresadern, G.; Aldeghi, M.; Baumann, H.M.; Mobley, D.L.; de Groot, B.L.; Gapsys, V. Alchemical absolute protein–ligand binding free energies for drug design. Chem. Sci. 2021, 12, 13958–13971. [Google Scholar] [CrossRef] [PubMed]
- Alexandrino, G.L.; Calderini, A.; Morgon, N.H.; Pessine, F.B.T. Spectroscopic (fluorescence, 1D-ROESY) and theoretical studies of the thiabendazole and β-cyclodextrin inclusion complex. J. Incl. Phenom. Macrocycl. Chem. 2013, 75, 93–99. [Google Scholar] [CrossRef]
- Mercader-Ros, M.T.; Lucas-Abellán, C.; Gabaldón, J.A.; Fortea, M.I.; Martínez-Cachá, A.; Núñez-Delicado, E. Kaempferol Complexation in Cyclodextrins at Basic pH. J. Agric. Food Chem. 2010, 58, 4675–4680. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, G.; Ghosh, H.N. Efficient Electron Injection from Twisted Intramolecular Charge Transfer (TICT) State of 7-Diethyl amino coumarin 3-carboxylic Acid (D-1421) Dye to TiO2 Nanoparticle. J. Phys. Chem. A 2002, 106, 2545–2553. [Google Scholar] [CrossRef]
- Manta, K.; Papakyriakopoulou, P.; Chountoulesi, M.; Diamantis, D.A.; Spaneas, D.; Vakali, V.; Naziris, N.; Chatziathanasiadou, M.V.; Andreadelis, I.; Moschovou, K.; et al. Preparation and Biophysical Characterization of Quercetin Inclusion Complexes with β-Cyclodextrin Derivatives to be Formulated as Possible Nose-to-Brain Quercetin Delivery Systems. Mol. Pharm. 2020, 17, 4241–4255. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Blaudeau, J.-P.; McGrath, M.P.; Curtiss, L.A.; Radom, L. Extension of Gaussian-2 (G2) theory to molecules containing third-row atoms K and Ca. J. Chem. Phys. 1997, 107, 5016–5021. [Google Scholar] [CrossRef]
- Cossi, M.; Scalmani, G.; Rega, N.; Barone, V. New developments in the polarizable continuum model for quantum mechanical and classical calculations on molecules in solution. J. Chem. Phys. 2002, 117, 43–54. [Google Scholar] [CrossRef]
- Tzeli, D.; Mavridis, A.; Xantheas, S.S. First Principles Examination of the Acetylene−Water Clusters, HCCH−(H2O)x, x = 2, 3, and 4. J. Phys. Chem. A 2002, 106, 11327–11337. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Betzel, C.; Saenger, W.; Hingerty, B.E.; Brown, G.M. Topography of cyclodextrin inclusion complexes, part 20. Circular and flip-flop hydrogen bonding in. beta.-cyclodextrin undecahydrate: A neutron diffraction study. J. Am. Chem. Soc. 1984, 106, 7545–7557. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-Like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Arsiccio, A.; Rospiccio, M.; Shea, J.-E.; Pisano, R. Force Field Parameterization for the Description of the Interactions between Hydroxypropyl-β-Cyclodextrin and Proteins. J. Phys. Chem. B 2021, 125, 7397–7405. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Ermak, D.L.; McCammon, J.A. Brownian dynamics with hydrodynamic interactions. J. Chem. Phys. 1978, 69, 1352–1360. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; van Gunsteren, W.F.; Mark, A.E. Peptide Folding: When Simulation Meets Experiment. Angew. Chem. Int. Ed. 1999, 38, 236–240. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinform. 2004, 55, 351–367. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of the Crystal Environment in Determining Protein Side-chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins Struct. Funct. Bioinform. 2011, 79, 2794–2812. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Buckner, J.K.; Boudon, S.; Tirado-Rives, J. Efficient computation of absolute free energies of binding by computer simulations. Application to the methane dimer in water. J. Chem. Phys. 1988, 89, 3742–3746. [Google Scholar] [CrossRef]
- Gilson, M.K.; Given, J.A.; Bush, B.L.; McCammon, J.A. The statistical-thermodynamic basis for computation of binding affinities: A critical review. Biophys. J. 1997, 72, 1047–1069. [Google Scholar] [CrossRef]
- Boresch, S.; Tettinger, F.; Leitgeb, M.; Karplus, M. Absolute Binding Free Energies: A Quantitative Approach for Their Calculation. J. Phys. Chem. B 2003, 107, 9535–9551. [Google Scholar] [CrossRef]
- Shirts, M.R.; Bair, E.; Hooker, G.; Pande, V.S. Equilibrium free energies from nonequilibrium measurements using maximum-likelihood methods. Phys. Rev. Lett. 2003, 91, 140601. [Google Scholar] [CrossRef]
- Crooks, G.E. Entropy production fluctuation theorem and the nonequilibrium work relation for free energy differences. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Topics 1999, 60, 2721–2726. [Google Scholar] [CrossRef]
- Aldeghi, M.; Heifetz, A.; Bodkin, M.J.; Knapp, S.; Biggin, P.C. Accurate calculation of the absolute free energy of binding for drug molecules. Chem. Sci. 2015, 7, 207–218. [Google Scholar] [CrossRef]
- Lindahl, E.; Abraham, M.J.; Hess, B.; van der Spoel, D. GROMACS 2021.4 Manual; GROMACS Development Team: Stockholm, Sweden, 2021. [Google Scholar] [CrossRef]
- Alibay, I.; Magarkar, A.; Seeliger, D.; Biggin, P.C. Evaluating the use of Absolute Binding Free Energy in the Fragment Optimization Process. p. 23. Available online: https://chemrxiv.org/engage/chemrxiv/article-details/61f802834b9500e1881041b5 (accessed on 9 June 2022).
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Gapsys, V.; Michielssens, S.; Seeliger, D.; de Groot, B.L. pmx: Automated protein structure and topology generation for alchemical perturbations. J. Comput. Chem. 2015, 36, 348–354. [Google Scholar] [CrossRef] [Green Version]
- GitLab. Solvation Free Energy with Couple-Intramol Set to No. Available online: https://gitlab.com/gromacs/gromacs/-/issues/3403 (accessed on 9 June 2022).
- GitLab. Remove False Positives from FE Excluded Pair Check. Available online: https://gitlab.com/gromacs/gromacs/-/merge_requests/2626 (accessed on 9 June 2022).
- GitLab. Add Fep Exclusion Check at Pairlist Generation. Available online: https://gitlab.com/gromacs/gromacs/-/merge_requests/2639 (accessed on 9 June 2022).
- Zhang, H.; Yin, C.; Yan, H.; van der Spoel, D. Evaluation of generalized born models for large scale affinity prediction of cyclodextrin host–guest complexes. J. Chem. Inf. Model. 2016, 56, 2080–2092. [Google Scholar] [CrossRef] [PubMed]
- Di, P.; Chen, J.; Liu, L.; Li, W.; Tang, Y.; Liu, G. In silico prediction of binding capacity and interaction forces of organic compounds with α- and β-cyclodextrins. J. Mol. Liquids 2020, 302, 112585. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Cournia, Z.; Allen, B.K.; Beuming, T.; Pearlman, D.A.; Radak, B.K.; Sherman, W. Rigorous Free Energy Simulations in Virtual Screening. J. Chem. Inf. Model. 2020, 60, 4153–4169. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, A.; Jensen, T.; Slochower, D.R.; Aldeghi, M.; Gapsys, V.; Ntekoumes, D.; Bosisio, S.; Papadourakis, M.; Henriksen, N.M.; de Groot, B.L.; et al. The SAMPL6 SAMPLing challenge: Assessing the reliability and efficiency of binding free energy calculations. J. Comput. Aided Mol. Des. 2020, 34, 601–633. [Google Scholar] [CrossRef] [PubMed]
- Cournia, Z.; Allen, B.; Sherman, W. Relative Binding Free Energy Calculations in Drug Discovery: Recent Advances and Practical Considerations. J. Chem. Inf. Model. 2017, 57, 2911–2937. [Google Scholar] [CrossRef]
- Armacost, K.A.; Riniker, S.; Cournia, Z. Novel Directions in Free Energy Methods and Applications. J. Chem. Inf. Model. 2020, 60, 1–5. [Google Scholar] [CrossRef]
- Singh, N.; Li, W. Absolute Binding Free Energy Calculations for Highly Flexible Protein MDM2 and Its Inhibitors. Int. J. Mol. Sci. 2020, 21, 4765. [Google Scholar] [CrossRef]
- Berishvili, V.P.; Kuimov, A.N.; Voronkov, A.E.; Radchenko, E.V.; Kumar, P.; Choonara, Y.E.; Pillay, V.; Kamal, A.; Palyulin, V.A. Discovery of Novel Tankyrase Inhibitors through Molecular Docking-Based Virtual Screening and Molecular Dynamics Simulation Studies. Molecules 2020, 25, 3171. [Google Scholar] [CrossRef]
- Mendoza-Martinez, C.; Papadourakis, M.; Llabrés, S.; Gupta, A.A.; Barlow, P.N.; Michel, J. Energetics of a protein disorder–order transition in small molecule recognition. Chem. Sci. 2022, 13, 5220–5229. [Google Scholar] [CrossRef]
- Li, Z.; Huang, Y.Y.; Wu, Y.N.; Chen, J.Y.; Wu, D.Y.; Zhan, C.G.; Luo, H.B. Absolute Binding Free Energy Calculation and Design of a Subnanomolar Inhibitor of Phosphodiesterase-10. J. Med. Chem. 2019, 62, 2099–2111. [Google Scholar] [CrossRef] [PubMed]
- Gkeka, P.; Eleftheratos, S.; Kolocouris, A.; Cournia, Z. Free Energy Calculations Reveal the Origin of Binding Preference for Aminoadamantane Blockers of Influenza A/M2TM Pore. J. Chem. Theory Comput. 2013, 9, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, H.; Drakopoulos, A.; Tzitzoglaki, C.; Homeyer, N.; Kolarov, F.; Gkeka, P.; Freudenberger, K.; Liolios, C.; Gauglitz, G.; Cournia, Z.; et al. Alchemical Free Energy Calculations and Isothermal Titration Calorimetry Measurements of Aminoadamantanes Bound to the Closed State of Influenza A/M2TM. J. Chem. Inf. Model. 2016, 56, 862–876. [Google Scholar] [CrossRef] [PubMed]
- Amezcua, M.; El Khoury, L.; Mobley, D.L. SAMPL7 Host–Guest Challenge Overview: Assessing the reliability of polarizable and non-polarizable methods for binding free energy calculations. J. Comput. Aided Mol. Des. 2021, 35, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Cournia, Z.; Chipot, C.; Roux, B.; York, D.M.; Sherman, W. Free Energy Methods in Drug Discovery—Introduction. In Free Energy Methods in Drug Discovery: Current State and Future Directions; American Chemical Society: Washington, DC, USA, 2021; pp. 1–38. [Google Scholar] [CrossRef]
- Baumann, H.M.; Gapsys, V.; de Groot, B.L.; Mobley, D.L. Challenges Encountered Applying Equilibrium and Nonequilibrium Binding Free Energy Calculations. J. Phys. Chem. B 2021, 125, 4241–4261. [Google Scholar] [CrossRef] [PubMed]
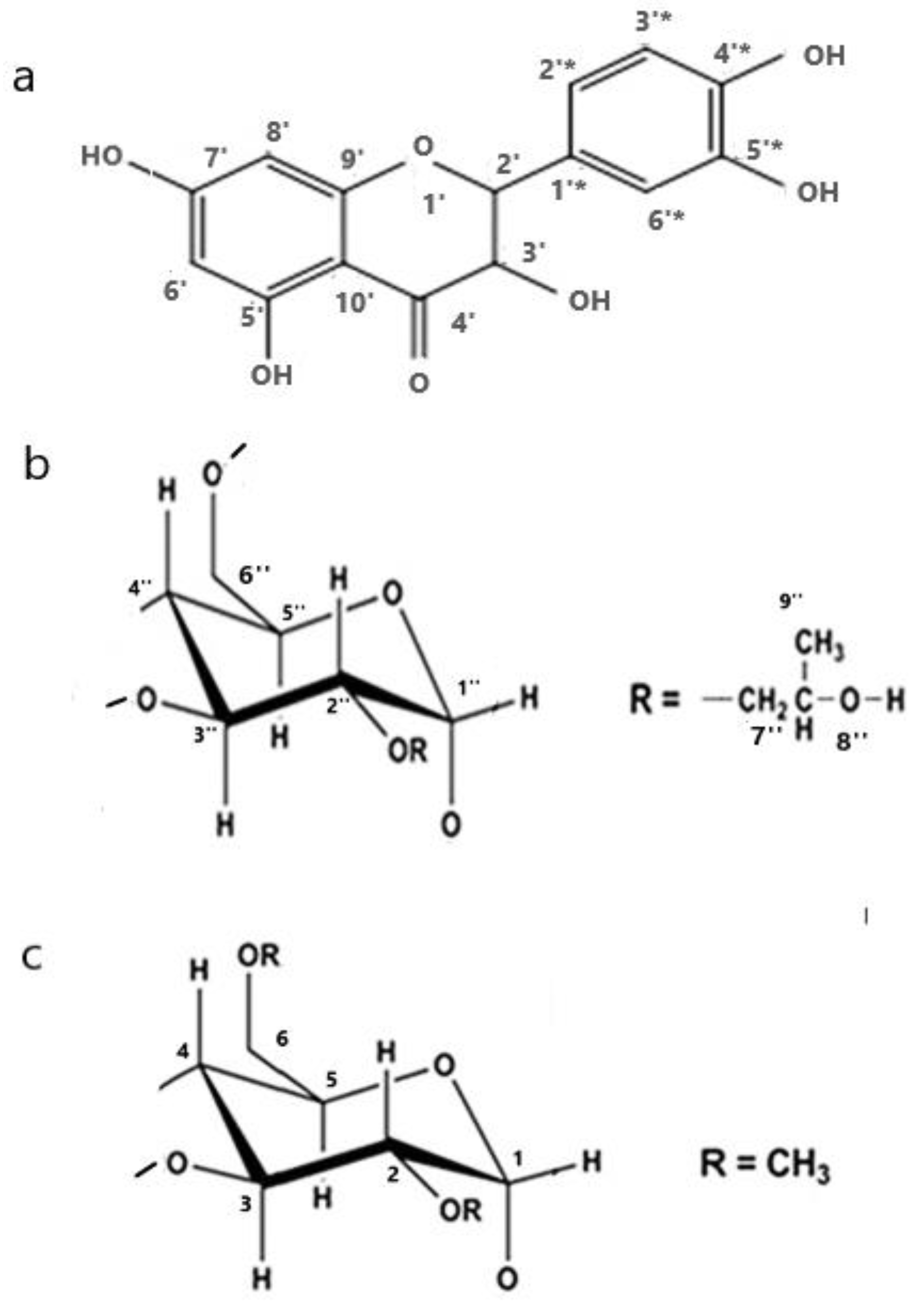
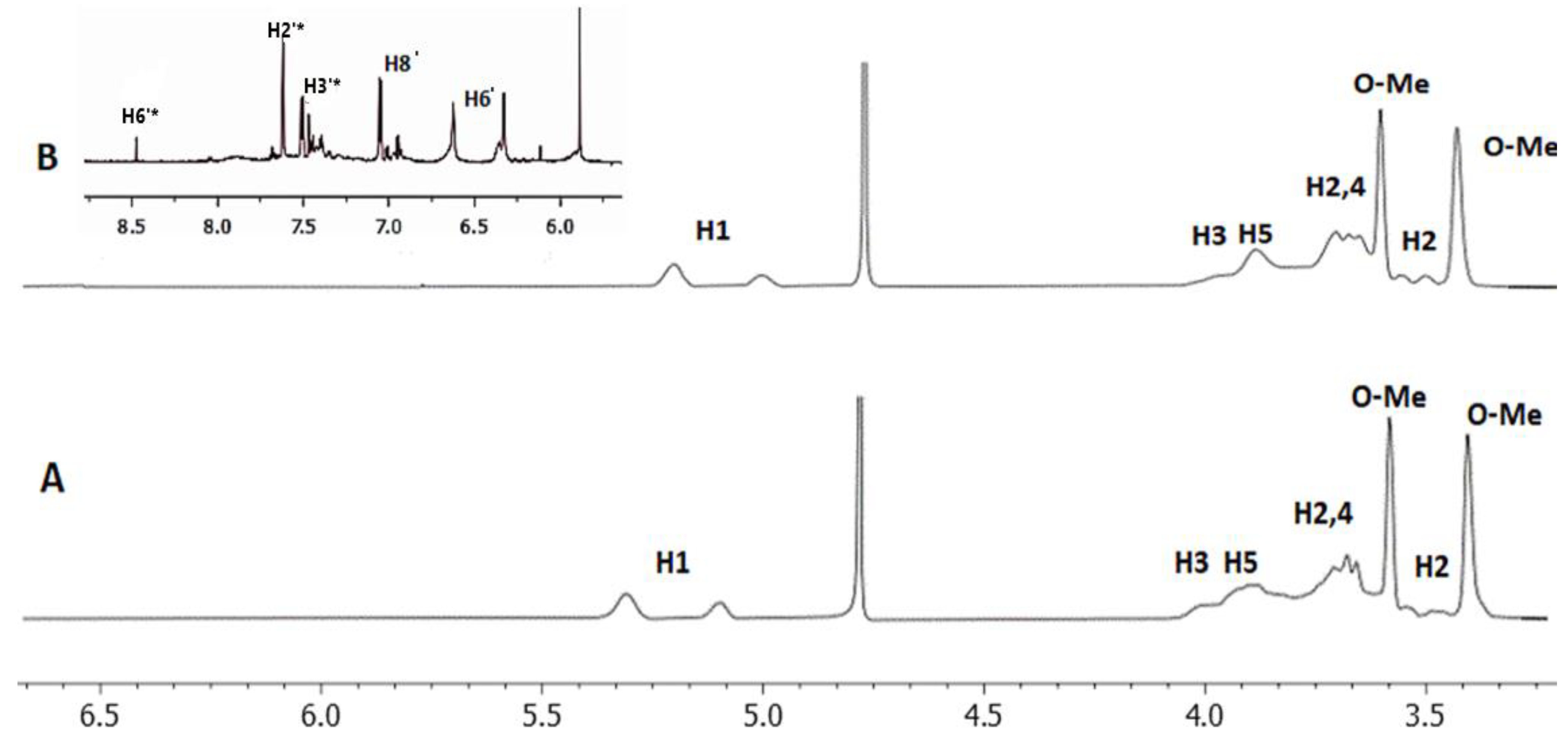
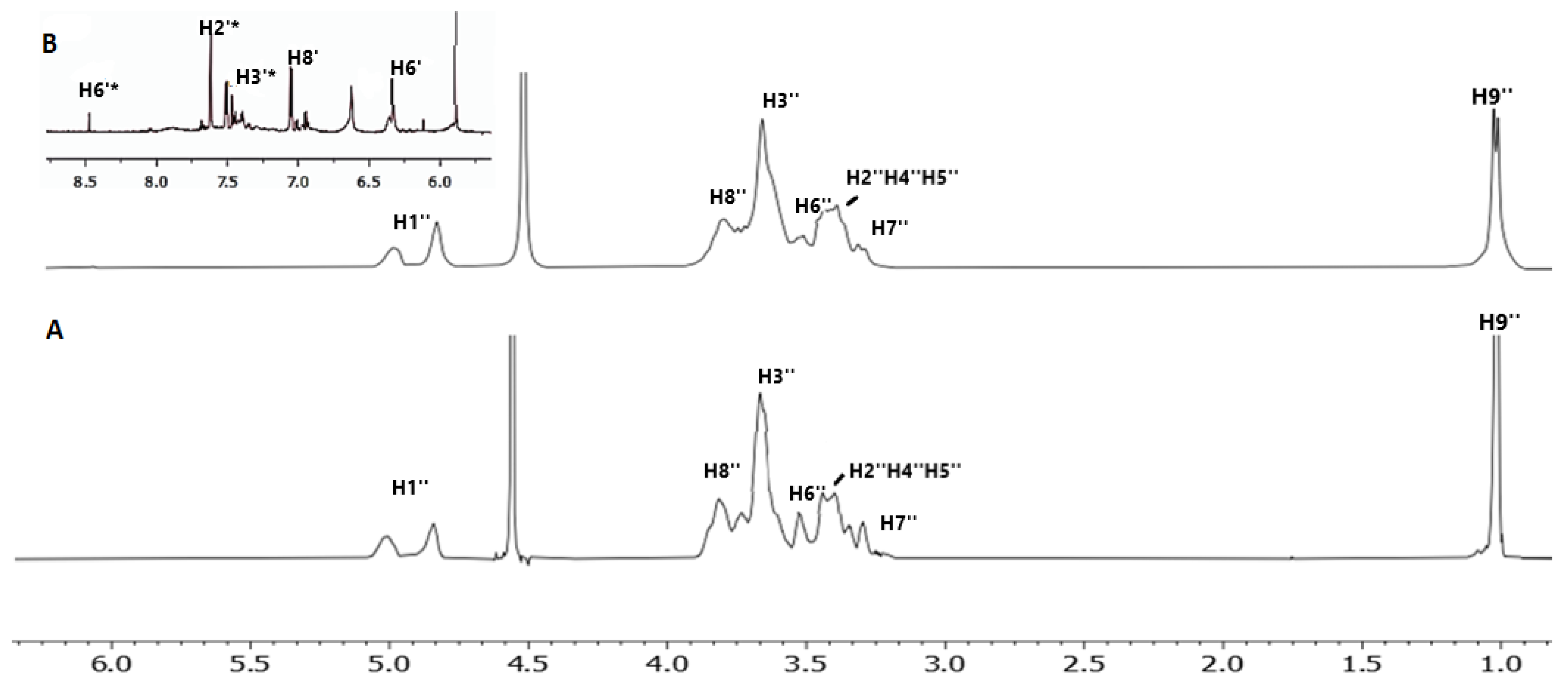
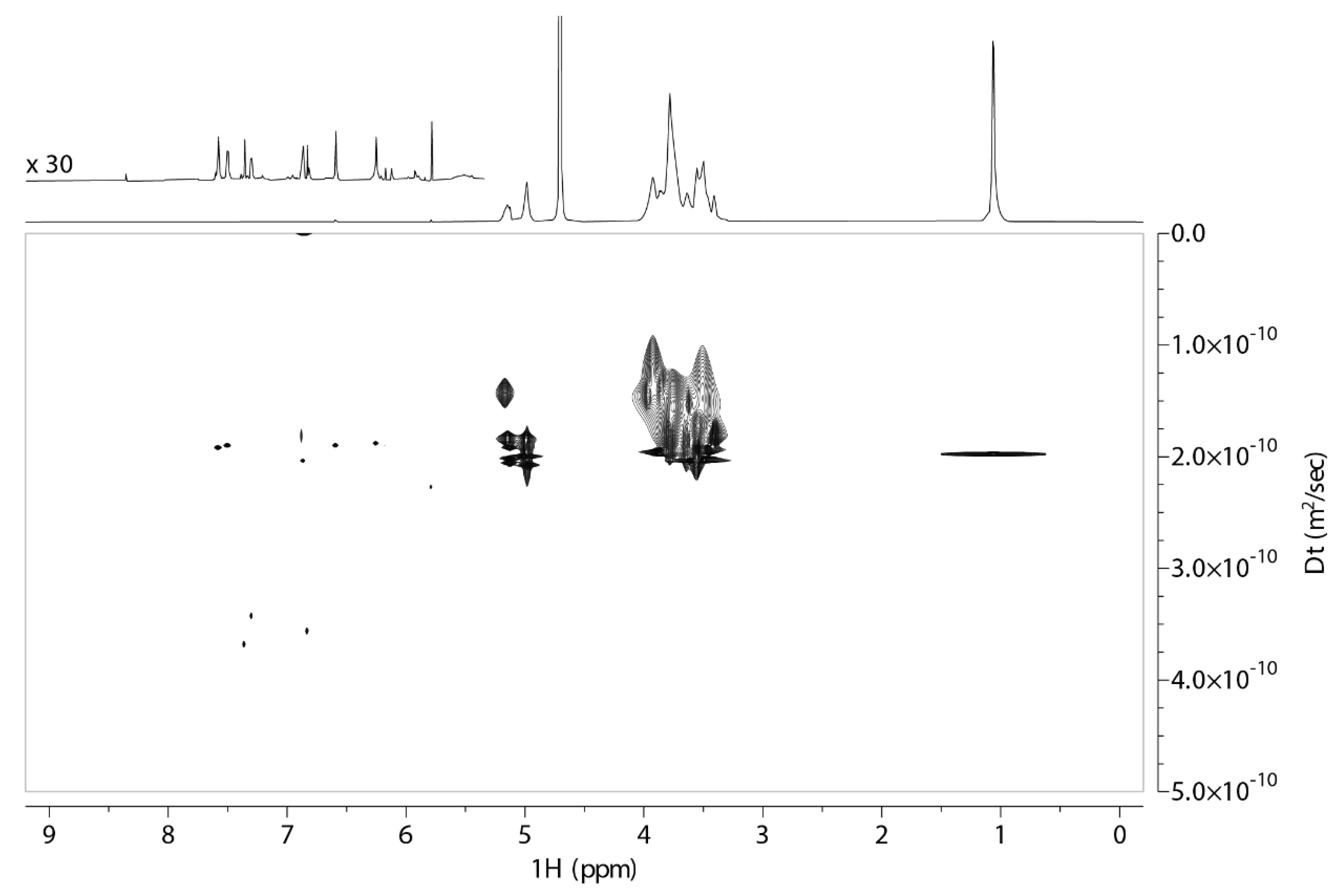

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protons of 2.6 Μe-β-CD | Chemical Shifts | Chemical Shifts of Complex | Δδ (ppm) | Multiplicity |
|---|---|---|---|---|
| H1 | 5.35−5.09 | 5.29−5.03 | 0.02 | singlet |
| H3 | 4.01 | 4.00 | 0.01 | singlet |
| H5 | 3.94 | 3.90 | 0.04 | doublet |
| H6 | 3.91 | 3.88 | 0.03 | singlet |
| H2 | 3.72 | 3.68 | 0.04 | singlet |
| H4 | 3.68 | 3.67 | 0.01 | doublet |
| H6-O-Me | 3.59 | 3.59 | 0 | singlet |
| H2 | 3.49 | 3.44 | 0.05 | singlet |
| H2-O-Me | 3.41 | 3.42 | 0.01 | singlet |
| Protons of 2,6Μe-β-CD | Chemical Shifts of Complex Experimental Results | Chemical Shifts of Complex SCF GIAO Method |
|---|---|---|
| H1 | 5.29−5.03 | 5.30−5.07 |
| H3 | 4.00 | 4.01 |
| H5 | 3.90 | 3.90 |
| H6 | 3.88 | 3.89 |
| H2 | 3.68 | 3.68 |
| H4 | 3.67 | 3.67 |
| H6-O-Me | 3.59 | 3.55 |
| 2 | 3.44 | 3.43 |
| H2-O-Me | 3.42 | 3.40 |
| Protons of 2HP-β-CD | Chemical Shifts | Δδ (ppm) | Multiplicity |
|---|---|---|---|
| H1″ | 5.03−4.92 | 0.01 | singlet |
| H8″ | 3.87 | 0 | singlet |
| H3″ | 3.75 | 0 | singlet |
| H6″ | 3.58 | 0.04 | singlet |
| H2″-H4″-H5″ | 3.47 | 0.04 | multiple peaks |
| H7″ | 3.37 | 0.01 | singlet |
| H9″ | 1.01 | 0 | singlet |
| Protons of 2HP-β-CD | Chemical Shifts of Complex Experimental Results | Chemical Shifts of Complex SCF GIAO Method |
|---|---|---|
| H1″ | 5.03−4.92 | 5.08−4.90 |
| H8″ | 3.87 | 3.84 |
| H3″ | 3.75 | 3.75 |
| H6″ | 3.54 | 3.57 |
| H2″-H4″-H5″ | 3.43 | 3.40 |
| H7″ | 3.36 | 3.36 |
| H9″ | 1.01 | 1.00 |
| Protons of QUE | Chemical Shifts | Complex with 2,6 Me-β-CD Experimental | Complex with Me-β-CD Using SCF GIAO Method | Complex with 2HΡ-β-CD Experimental | Complex with 2HΡ-β-CD Using SCF GIAO Method | Multiplicity |
|---|---|---|---|---|---|---|
| H6′ | 7.87 | 7.65 | 7.73 | 7.70 | 7.70 | singlet |
| H2′* | 7.65 | 7.57 | 7.50 | 7.60 | 7.30 | singlet |
| H3′* | 7.15 | 7.05 | 7.35 | 7.02 | 7.00 | doublet |
| H8′ | 6.65 | 6.59 | 6.27 | 6.69 | 6.44 | doublet |
| H6′* | 5.90–5.93 | 6.06–6.09 | 5.99–6.01 | 5.86–5.89 | 5.80–5.90 | broad singlet |
| Peak Name | Τ1 2HΡ-β-CD | T1 2HP-β-CD with QUE | Percentage Change |
|---|---|---|---|
| H1″ | 1.40 s−1.54 s | 1.39 s−1.52 s | 2% |
| H8″ | 1.72 s | 1.73 s | 1% |
| H3″ | 1.23 s | 1.23 s | 0% |
| H6″ | 1.44 s | 1.28 s | 16% |
| H2″-H4″-H5″ | 1.42 s | 1.20 s | 22% |
| H7″ | 1.55 s | 1.55 s | 0% |
| H9″ | 1.19 s | 1.20 s | 1% |
| Peak Name | Τ1 2,6Μe-β-CD | Τ1 2,6Μe-β-CD with QUE | Percentage Change |
|---|---|---|---|
| H1 | 1.36 s−1.4 s | 1.35 s−1.4 s | 1% |
| H3 | 1.49 s | 1.51 s | 2% |
| H5 | 1.41 s | 1.18 s | 23% |
| H6 | 1.34 s | 1.12 s | 22% |
| H2 | 1.36 s | 1.12 s | 24% |
| H4 | 1.35 s | 1.28 s | 7% |
| H6-O-Me | 1.76 s | 1.79 s | 3% |
| H2 | 1.51 s | 1.39 s | 12% |
| H2-O-Me | 1.64 s | 1.62 s | 2% |
| 2HP-β-CD (d1) a | 2HP-β-CD (d2) a | 2,6Μe-β-CD (e1) a | 2,6Μe-β-CD (e2) a | |
|---|---|---|---|---|
| Deformation_QUE | 3.89 (19.91) b | 3.42 (19.44) b | 0.59 (16.62) b | 7.31 (23.34) b |
| Def_CD | 20.15 | 18.99 | 11.75 | 20.75 |
| BE | −8.91 (7.12) b | −10.56 (5.46) b | 0.83 (16.85) b | 3.85 (19.87) b |
| BE(BSSE) | 10.73 (26.76) b | 10.90 (26.93) b | 11.68 (27.71) b | 20.03 (36.06) b |
| BE_raw | −32.95 | −32.97 | −11.52 | −24.21 |
| BE(BSSE)_fcp | −13.31 | −11.50 | −0.66 | −8.03 |
| Energy Component (kcal/mol) | 2HP-β-CD | 2,6Me-β-CD |
|---|---|---|
| MM/GBSA ΔGvdW | −27.35 ± 1.10 | −27.90 ± 0.92 |
| MM/GBSA ΔGCoulomb | −2.08 ± 1.36 | −5.37 ± 1.90 |
| MM/GBSA ΔGSolvGB | +9.13 ± 0.85 | +9.99 ± 1.65 |
| MM/GBSA ΔGLipo | −12.70 ± 1.81 | −6.26 ± 1.54 |
| MM/GBSA ΔGHbond | −0.48 ± 0.16 | −0.54 ± 0.13 |
| MM/GBSA ΔGCovalent | +1.69 ± 0.79 | +1.63 ± 0.73 |
| MM/GBSA ΔGbind | −31.73 ± 3.21 | −26.77 ± 2.37 |
| Systems under Study | Replica1 | Replica2 | Replica3 | Replica4 | |
|---|---|---|---|---|---|
| 2HP-β-CD | −1.45 ± 0.46 | −1.39 ± 0.28 | −1.55 ± 0.40 | −1.21 ± 0.36 | −1.40 ± 0.76 |
| 2,6Me-β-CD | −5.21 ± 0.37 | −5.14 ± 0.28 | −4.53 ± 0.45 | −5.47 ± 0.39 | −5.09 ± 0.76 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vakali, V.; Papadourakis, M.; Georgiou, N.; Zoupanou, N.; Diamantis, D.A.; Javornik, U.; Papakyriakopoulou, P.; Plavec, J.; Valsami, G.; Tzakos, A.G.; et al. Comparative Interaction Studies of Quercetin with 2-Hydroxyl-propyl-β-cyclodextrin and 2,6-Methylated-β-cyclodextrin. Molecules 2022, 27, 5490. https://doi.org/10.3390/molecules27175490
Vakali V, Papadourakis M, Georgiou N, Zoupanou N, Diamantis DA, Javornik U, Papakyriakopoulou P, Plavec J, Valsami G, Tzakos AG, et al. Comparative Interaction Studies of Quercetin with 2-Hydroxyl-propyl-β-cyclodextrin and 2,6-Methylated-β-cyclodextrin. Molecules. 2022; 27(17):5490. https://doi.org/10.3390/molecules27175490
Chicago/Turabian StyleVakali, Vasiliki, Michail Papadourakis, Nikitas Georgiou, Nikoletta Zoupanou, Dimitrios A. Diamantis, Uroš Javornik, Paraskevi Papakyriakopoulou, Janez Plavec, Georgia Valsami, Andreas G. Tzakos, and et al. 2022. "Comparative Interaction Studies of Quercetin with 2-Hydroxyl-propyl-β-cyclodextrin and 2,6-Methylated-β-cyclodextrin" Molecules 27, no. 17: 5490. https://doi.org/10.3390/molecules27175490
APA StyleVakali, V., Papadourakis, M., Georgiou, N., Zoupanou, N., Diamantis, D. A., Javornik, U., Papakyriakopoulou, P., Plavec, J., Valsami, G., Tzakos, A. G., Tzeli, D., Cournia, Z., & Mauromoustakos, T. (2022). Comparative Interaction Studies of Quercetin with 2-Hydroxyl-propyl-β-cyclodextrin and 2,6-Methylated-β-cyclodextrin. Molecules, 27(17), 5490. https://doi.org/10.3390/molecules27175490