
Novel Hybrid Benzoazacrown Ligand as a Chelator for Copper and Lead Cations: What Difference Does Pyridine Make

 , , ,
, , , 

Abstract
:1. Introduction
2. Results
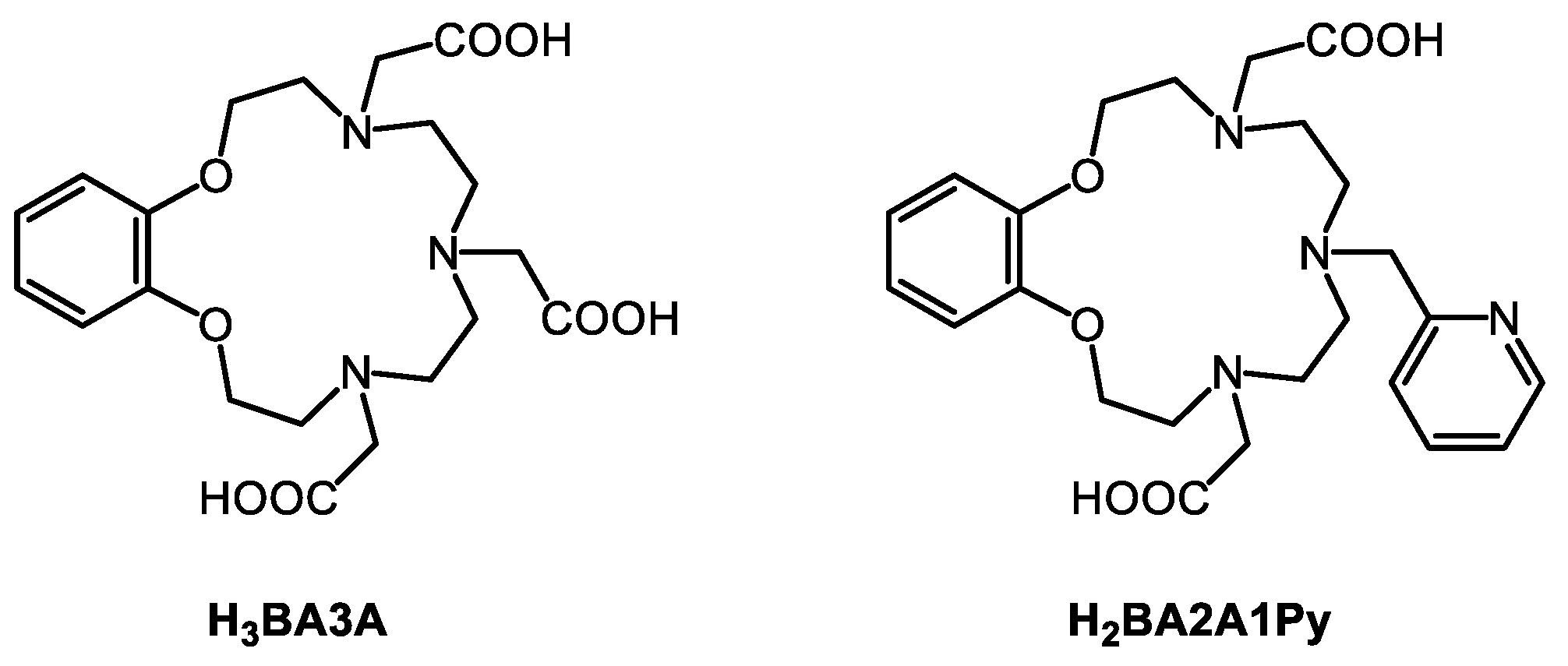
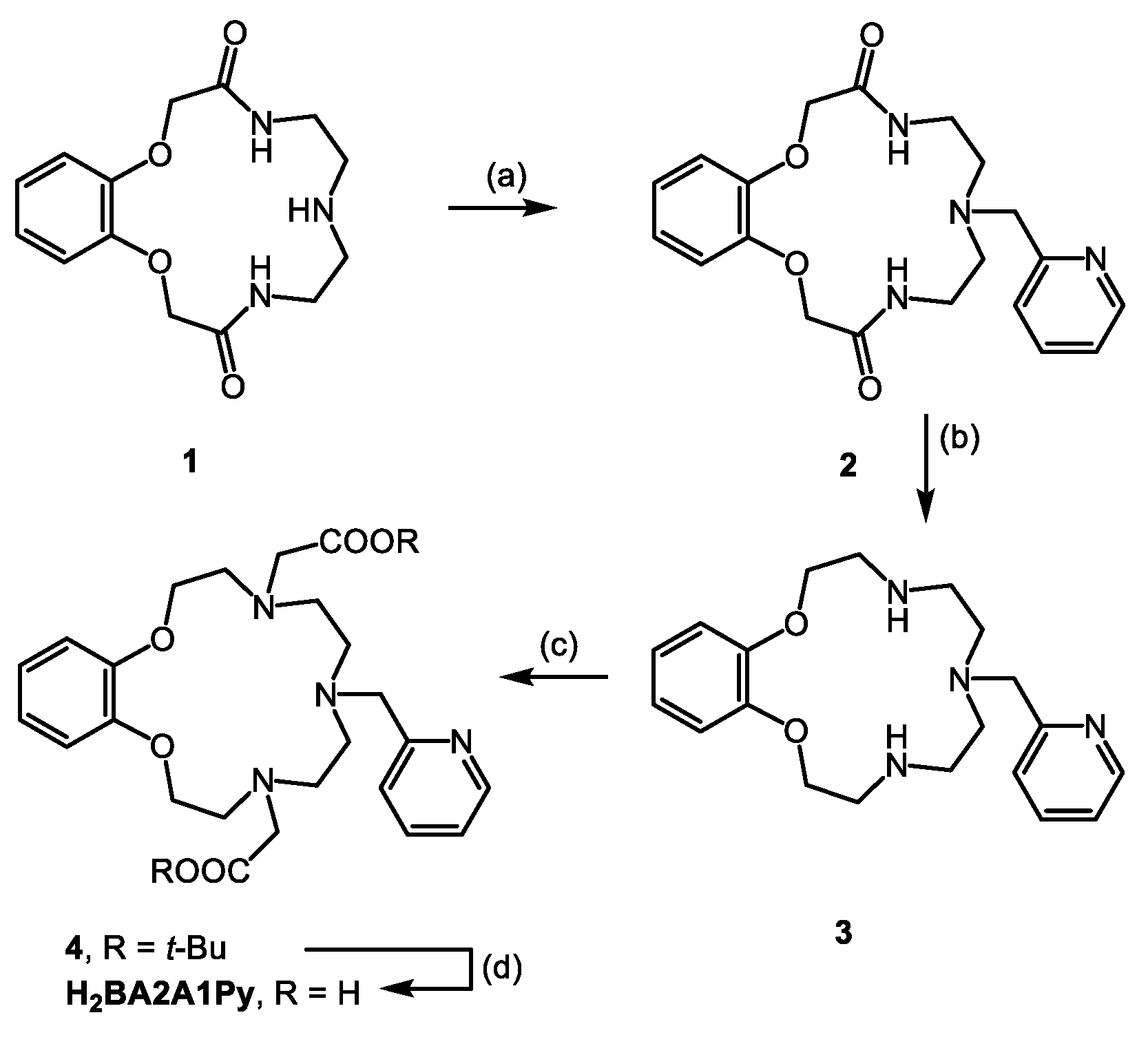
2.1. Synthesis
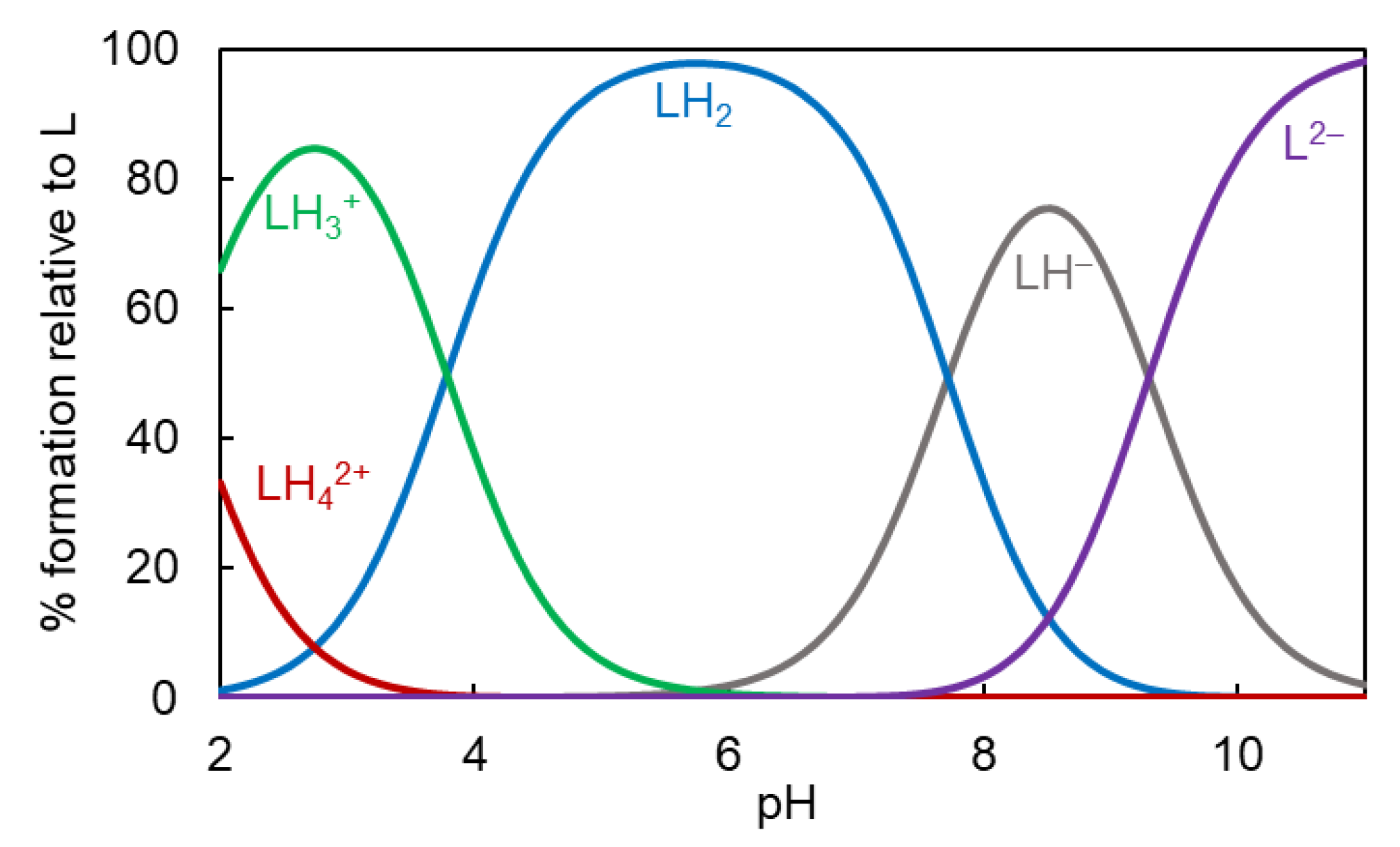
2.2. Ligand Protonation
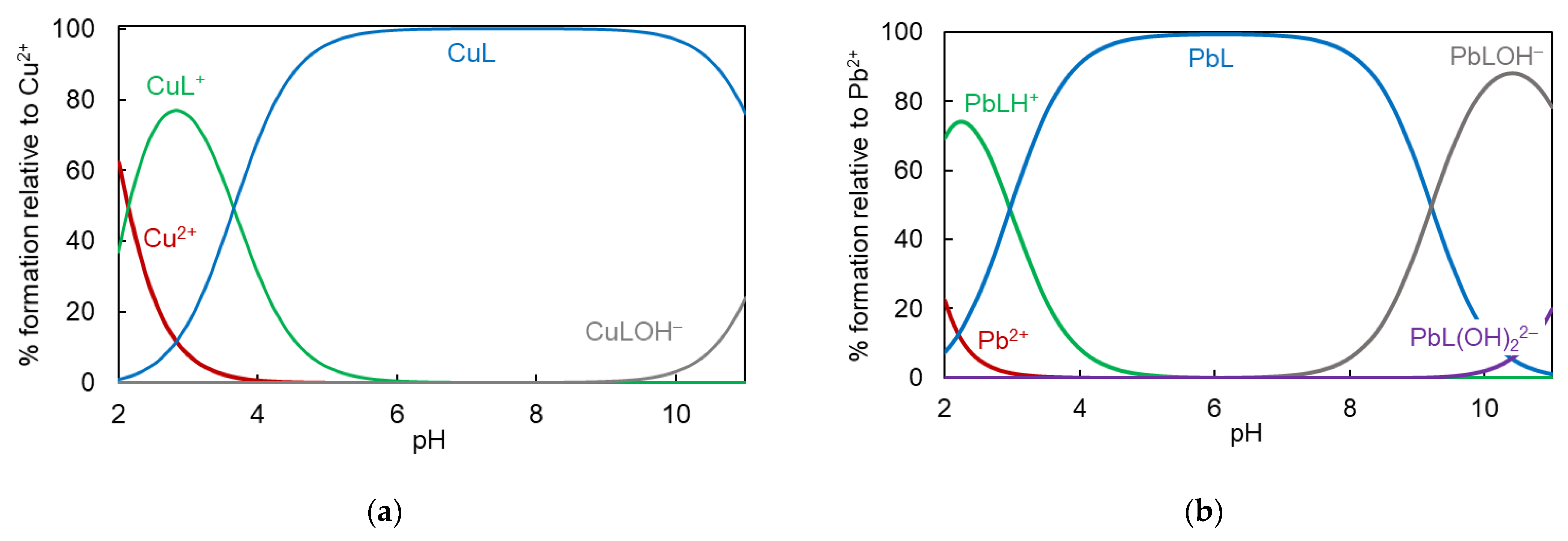
2.3. Complexation Study
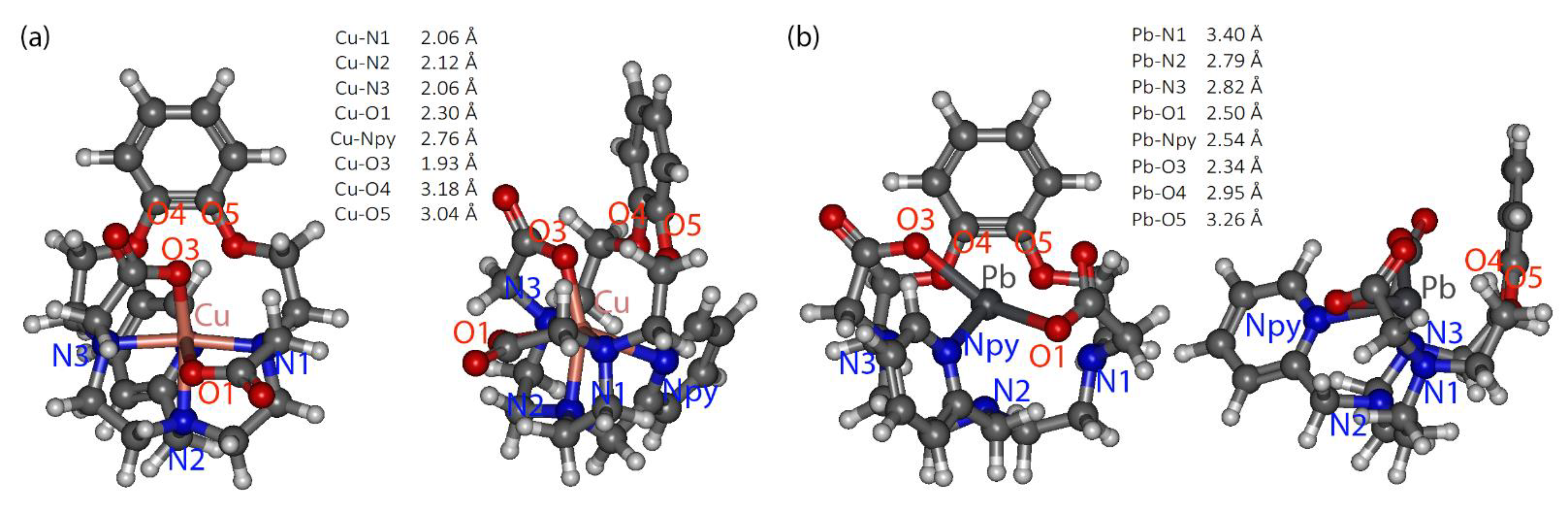
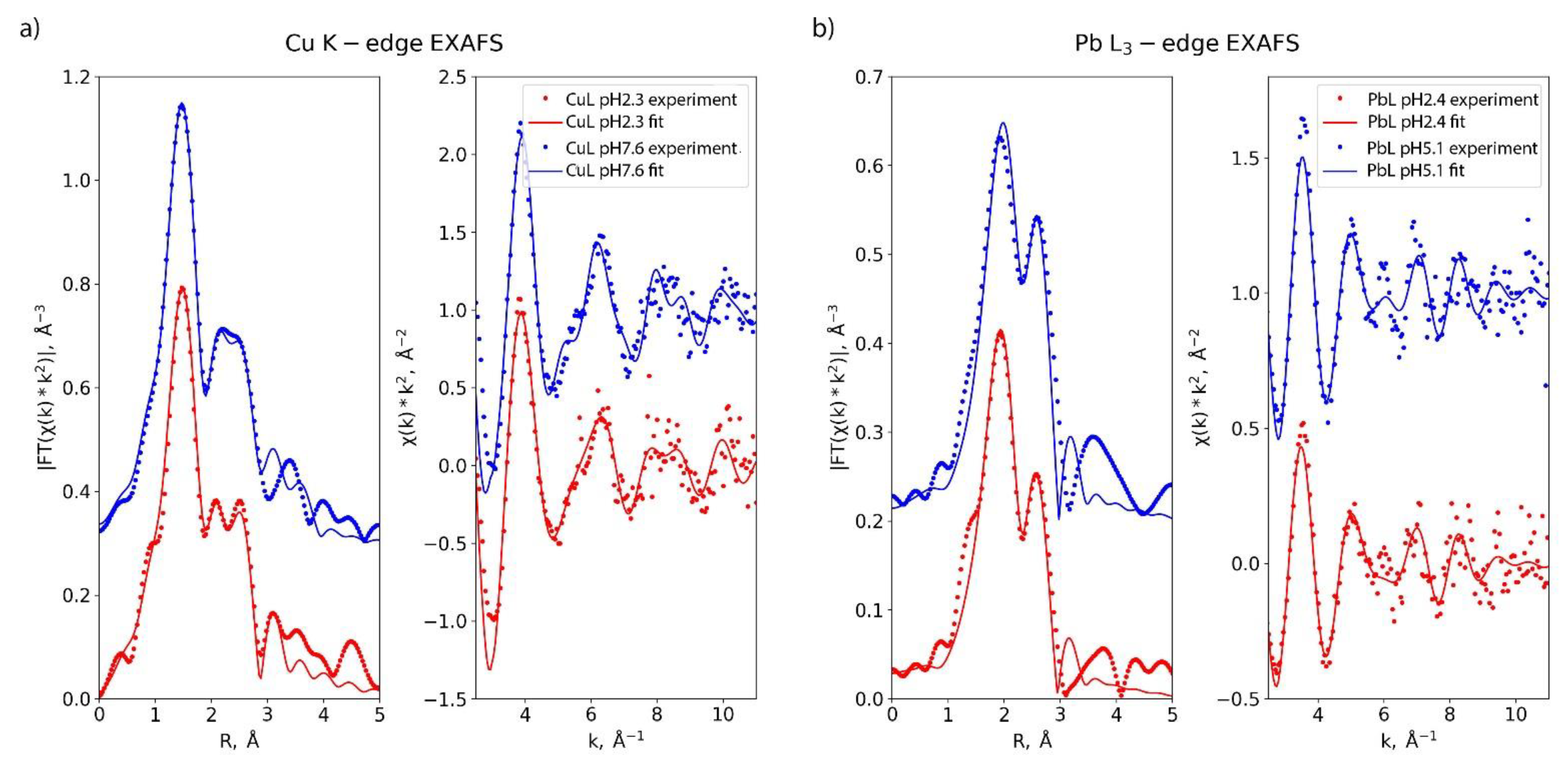
2.4. Complexes’ Structure Studies
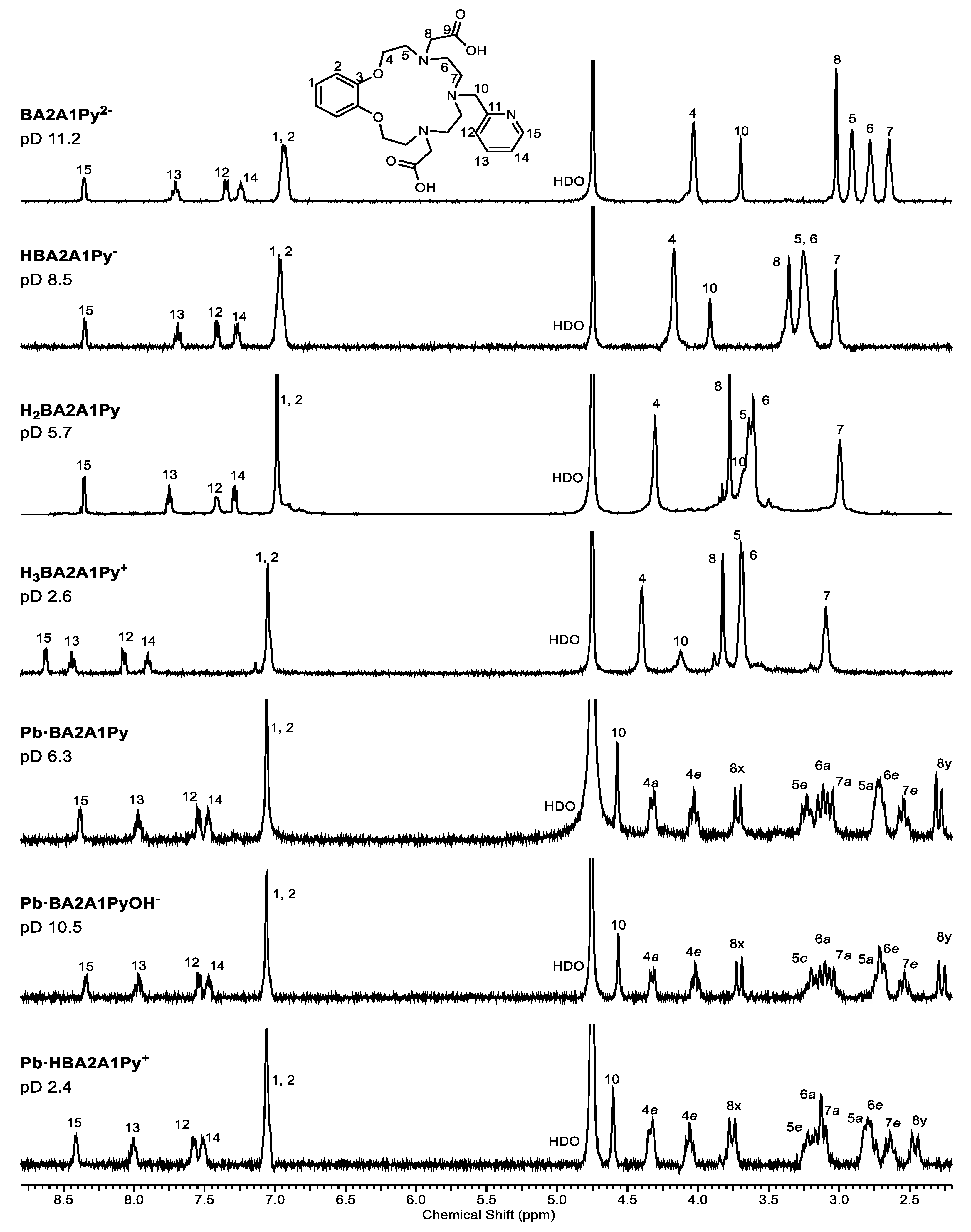
2.5. NMR Spectroscopy of PbBA2A1Py
2.6. Ligand Lipophilicity
2.7. Radiolabeling of Complexes
2.8. Stability of Radiolabeled Complexes in Biologically Relevant Media
3. Discussion
4. Materials and Methods
4.1. Synthesis and Analysis of Compounds
- Compound 3
- Compound 4
- Compound H2BA2A1Py
4.2. Potentiometric Titration
4.3. NMR Study of Ligand H2BA2A1Py Protonation and Complex Formation with Pb2+ Ion
4.4. Determination of logP
4.5. High Performance Liquid Chromatography (HPLC)
4.6. DFT Calculations
4.7. Radiochemical Separation of 210Pb
4.8. Production and Radiochemical Separation of 64Cu
4.9. Radiolabeling Studies
4.10. Stability of Complexes in Biologically Relevant Media
4.11. EXAFS Measurement and Data Treatment
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sinenko, I.L.; Kalmykova, T.P.; Likhosherstova, D.V.; Egorova, B.V.; Zubenko, A.D.; Vasiliev, A.N.; Ermolaev, S.V.; Lapshina, E.V.; Ostapenko, V.S.; Fedorova, O.A.; et al. 213Bi production and complexation with new picolinate containing ligands. J. Radioanal. Nucl. Chem. 2019, 321, 531–540. [Google Scholar] [CrossRef]
- Egorova, B.V.; Kalmykova, T.P.; Zubenko, A.D.; Shchukina, A.A.; Karnoukhova, V.A.; Likhosherstova, D.V.; Priselkova, A.B.; Fedorov, Y.V.; Fedorova, O.A.; Kalmykov, S.N. Comparative Study of Macrocyclic and Acyclic Picolinate Derivatives for Chelation of Copper Cations. Eur. J. Inorg. Chem. 2021, 2021, 4700–4709. [Google Scholar] [CrossRef]
- Fedorov, Y.V.; Fedorova, O.A.; Kalmykov, S.N.; Oshchepkov, M.S.; Nelubina, Y.V.; Arkhipov, D.E.; Egorova, B.V.; Zubenko, A.D. Potentiometric studies of complex formation of amidopyridine macrocycles bearing pendant arms with proton and heavy metal ions in aqueous solution. Polyhedron 2017, 124, 229–236. [Google Scholar] [CrossRef]
- Matazova, E.V.; Egorova, B.V.; Konopkina, E.A.; Aleshin, G.Y.; Zubenko, A.D.; Mitrofanov, A.A.; Karpov, K.V.; Fedorova, O.A.; Fedorov, Y.V.; Kalmykov, S.N. Benzoazacrown compound: A highly effective chelator for therapeutic bismuth radioisotopes. Medchemcomm 2019, 10, 1641–1645. [Google Scholar] [CrossRef] [PubMed]
- Egorova, B.V.; Matazova, E.V.; Aleshin, G.Y.; Zubenko, A.D.; Pashanova, A.V.; Konopkina, E.A.; Mitrofanov, A.A.; Smirnova, A.A.; Trigub, A.L.; Karnoukhova, V.A.; et al. Investigating the Bismuth Complexes with Benzoazacrown Tri- and Tetra-Acetates. Eur. J. Inorg. Chem. 2021, 2021, 3344–3354. [Google Scholar] [CrossRef]
- Zubenko, A.D.; Egorova, B.V.; Kalmykov, S.N.; Shepel, N.E.; Karnoukhova, V.A.; Fedyanin, I.V.; Fedorov, Y.V.; Fedorova, O.A. Out-cage metal ion coordination by novel benzoazacrown bisamides with carboxyl, pyridyl and picolinate pendant arms. Tetrahedron 2019, 75, 2848–2859. [Google Scholar] [CrossRef]
- Egorova, B.V.; Zamurueva, L.S.; Zubenko, A.D.; Pashanova, A.V.; Pillai, Z.S.; Fedorova, O.A.; Kalmykov, S.N. Triacetate of benzoazacrown compound as a chelator for lead cations promising for targeted radiopharmaceuticals. Macroheterocycles 2021, 14, 157–163. [Google Scholar] [CrossRef]
- Aleshin, G.Y.; Egorova, B.V.; Priselkova, A.B.; Zamurueva, L.S.; Khabirova, S.Y.; Zubenko, A.D.; Karnoukhova, V.A.; Fedorova, O.A.; Kalmykov, S.N. Zinc and copper complexes with azacrown ethers and their comparative stabilityin vitroandin vivo. Dalton Trans. 2020, 49, 6249–6258. [Google Scholar] [CrossRef]
- Yang, H.; Gao, F.; McNeil, B.; Zhang, C.; Yuan, Z.; Zeisler, S.; Kumlin, J.; Zeisler, J.; Bénard, F.; Ramogida, C.; et al. Synthesis of DOTA-pyridine chelates for 64Cu coordination and radiolabeling of αMSH peptide. EJNMMI Radiopharm. Chem. 2021, 6, 3. [Google Scholar] [CrossRef]
- Egorova, B.V.; Oshchepkov, M.S.; Fedorov, Y.V.; Fedorova, O.A.; Budylin, G.S.; Shirshin, E.A.; Kalmykov, S.N. Complexation of Bi3+, Ac3+, Y3+, Lu3+, La3+and Eu3+with benzo-diaza-crown ether with carboxylic pendant arms. Radiochim. Acta 2016, 104, 555–565. [Google Scholar] [CrossRef]
- Panchenko, P.A.; Zubenko, A.D.; Chernikova, E.Y.; Fedorov, Y.V.; Pashanova, A.V.; Karnoukhova, V.A.; Fedyanin, I.V.; Fedorova, O.A. Synthesis, structure and metal ion coordination of novel benzodiazamacrocyclic ligands bearing pyridyl and picolinate pendant side-arms. New J. Chem. 2019, 43, 15072–15086. [Google Scholar] [CrossRef]
- Gasser, G.; Tjioe, L.; Graham, B.; Belousoff, M.J.; Juran, S.; Walther, M.; Künstler, J.U.; Bergmann, R.; Stephan, H.; Spiccia, L. Synthesis, copper(II) complexation, 64Cu-labeling, and bioconjugation of a new bis(2-pyridylmethyl) derivative of 1,4,7- triazacyclononane. Bioconjug. Chem. 2008, 19, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.J.; Ferrier, M.; Radchenko, V.; Maassen, J.R.; Engle, J.W.; Batista, E.R.; Martin, R.L.; Nortier, F.M.; Fassbender, M.E.; John, K.D.; et al. Evaluation of nitrogen-rich macrocyclic ligands for the chelation of therapeutic bismuth radioisotopes. Nucl. Med. Biol. 2015, 42, 428–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zubenko, A.D.; Egorova, B.V.; Zamurueva, L.S.; Kalmykov, S.N.; Fedorova, O.A. Synthesis of benzoaza-15(18)-crown-5(6) ethers and study of their complexes with lead(II). Mendeleev Commun. 2021, 31, 194–196. [Google Scholar] [CrossRef]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- Jang, Y.H.; Blanco, M.; Dasgupta, S.; Keire, D.A.; Shively, J.E.; Goddard, W.A. Mechanism and Energetics for Complexation of 90 Y with Model for Cancer Radioimmunotherapy. J. Am. Chem. Soc. 1999, 121, 6142–6151. [Google Scholar] [CrossRef]
- Moreau, J.; Guillon, E.; Pierrard, J.C.; Rimbault, J.; Port, M.; Aplincourt, M. Complexing mechanism of the lanthanide cations Eu3+, Gd 3+, and Tb3+ with 1,4,7,10-tetrakis (carboxymethyl)-1,4,7, 10-tetraazacyclododecane (dota)-characterization of three successive complexing phases: Study of the thermodynamic and structural properti. Chem.-Eur. J. 2004, 10, 5218–5232. [Google Scholar] [CrossRef]
- Sharghi, H.; Khalifeh, R.; Salimi Beni, A.R. Synthesis of new lariat ethers containing polycyclic phenols and heterocyclic aromatic compound on graphite surface via mannich reaction. J. Iran. Chem. Soc. 2010, 7, 275–288. [Google Scholar] [CrossRef]
- Gans, P.; O’Sullivan, B. GLEE, a new computer program for glass electrode calibration. Talanta 2000, 51, 33–37. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Caldeweyher, E.; Bannwarth, C.; Grimme, S. Extension of the D3 dispersion coefficient model. J. Chem. Phys. 2017, 147, 034112. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef]
- Metz, B.; Stoll, H.; Dolg, M. Small-core multiconfiguration-Dirac–Hartree–Fock-adjusted pseudopotentials for post- d main group elements: Application to PbH and PbO. J. Chem. Phys. 2000, 113, 2563–2569. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef]
- Analytical Procedures, Lead-210 and Polonium-210 in Water. 2014. Available online: https://www.eichrom.com/wp-content/uploads/2018/02/pbw01-21_pb-po-water-vbs.pdf (accessed on 24 December 2021).
- JANIS Nuclear Database. Available online: http://www.oecd-nea.org/janisweb (accessed on 24 December 2021).
- Aliev, R.A.; Belyshev, S.S.; Kuznetsov, A.A.; Dzhilavyan, L.Z.; Khankin, V.V.; Aleshin, G.Y.; Kazakov, A.G.; Priselkova, A.B.; Kalmykov, S.N.; Ishkhanov, B.S. Photonuclear production and radiochemical separation of medically relevant radionuclides: 67Cu. J. Radioanal. Nucl. Chem. 2019, 321, 125–132. [Google Scholar] [CrossRef]
- Chernyshov, A.A.; Veligzhanin, A.A.; Zubavichus, Y.V. Structural Materials Science end-station at the Kurchatov Synchrotron Radiation Source: Recent instrumentation upgrades and experimental results. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrom. Detect. Assoc. Equip. 2009, 603, 95–98. [Google Scholar] [CrossRef]
- Newville, M. EXAFS analysis using FEFF and FEFFIT. J. Synchrotron Radiat. 2001, 8, 96–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zabinsky, S.I.; Rehr, J.J.; Ankudinov, A.; Albers, R.C.; Eller, M.J. Multiple-scattering calculations of X-ray-absorption spectra. Phys. Rev. B 1995, 52, 2995–3009. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cation | Complex a | BA2A1Py2− | BA3A3− [7,8] | |
|---|---|---|---|---|
| 0.1 M NaClO4 | 0.1 M KNO3 | 0.1 M KNO3 | ||
| H+ | HL | 9.0 (1) | 9.3 (1)/9.3 | 9.9 (1)/9.9 |
| H2L | 16.7 (1) | 17.0 (1)/7.7 | 17.7 (1)/7.8 | |
| H3L | 21.2 (1) | 20.8 (1)/3.8 | 21.2 (1)/3.5 | |
| H4L | 22.9 (1) | 22.5 (1)/1.7 | 23.8 (2)/2.6 | |
| Cu2+ | HLCu | 21.2 (1) | 20.0 (1) | 20.4 (1) |
| LCu | 16.2 (2) | 16.3 (1) | 16.8 (1) | |
| LCuOH | 18.8 (2) | 18.6 (1) | ||
| Pb2+ | HLPb | - | 18.6 (1) | 20.1 ± 0.1 |
| LPb | - | 15.6 (1) | 17.2 ± 0.1 | |
| LPbOH | - | 20.2 (2) | 23.1 ± 0.1 | |
| LPb(OH)2 | - | 22.4 (2) | 26.3 ± 0.1 | |
| Sample | N/O | C/N/O | C/N/O | C/N/O | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | R, Å | σ, Å | N | R, Å | σ, Å | N | R, Å | σ, Å | N | R, Å | σ, Å | |
| CuBA2A1Py_pH2.3 | 4.0 | 2.00 | 0.011 | 4.6 | 2.72 | 0.005 | 6.9 | 2.94 | 0.005 | 1.6 | 3.29 | 0.005 |
| CuBA2A1Py_pH7.6 | 4.0 | 1.99 | 0.010 | 3.4 | 2.73 | 0.005 | 5.7 | 2.92 | 0.005 | 2.2 | 3.41 | 0.005 |
| PbBA2A1Py_pH2.4 | 5.5 | 2.54 | 0.014 | 6.6 | 3.31 | 0.009 | - | |||||
| PbBA2A1Py_pH5.1 | 6.1 | 2.55 | 0.016 | 8.6 | 3.31 | 0.009 | ||||||
| c(H2BA2A1Py), μM | PbL, %, 0.15 M Ac (No Ac) | CuL, %, 0.15 M Ac |
|---|---|---|
| 1000 | 100 ± 7 | 97 ± 9 |
| 500 | 100 ± 9 | 92 ± 8 |
| 200 | 99 ± 9 | – |
| 100 | 100 ± 8 (99%) | 99 ± 2 |
| 80 | 99 ± 7 | – |
| 50 | 95 ± 9 (99%) | 96 ± 3 |
| 20 | 73 ± 7 | – |
| 10 | 62 ± 3 | 34 ± 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Egorova, B.V.; Zamurueva, L.S.; Zubenko, A.D.; Pashanova, A.V.; Mitrofanov, A.A.; Priselkova, A.B.; Fedorov, Y.V.; Trigub, A.L.; Fedorova, O.A.; Kalmykov, S.N. Novel Hybrid Benzoazacrown Ligand as a Chelator for Copper and Lead Cations: What Difference Does Pyridine Make. Molecules 2022, 27, 3115. https://doi.org/10.3390/molecules27103115
Egorova BV, Zamurueva LS, Zubenko AD, Pashanova AV, Mitrofanov AA, Priselkova AB, Fedorov YV, Trigub AL, Fedorova OA, Kalmykov SN. Novel Hybrid Benzoazacrown Ligand as a Chelator for Copper and Lead Cations: What Difference Does Pyridine Make. Molecules. 2022; 27(10):3115. https://doi.org/10.3390/molecules27103115
Chicago/Turabian StyleEgorova, Bayirta V., Lyubov S. Zamurueva, Anastasia D. Zubenko, Anna V. Pashanova, Artem A. Mitrofanov, Anna B. Priselkova, Yuri V. Fedorov, Alexander L. Trigub, Olga A. Fedorova, and Stepan N. Kalmykov. 2022. "Novel Hybrid Benzoazacrown Ligand as a Chelator for Copper and Lead Cations: What Difference Does Pyridine Make" Molecules 27, no. 10: 3115. https://doi.org/10.3390/molecules27103115
APA StyleEgorova, B. V., Zamurueva, L. S., Zubenko, A. D., Pashanova, A. V., Mitrofanov, A. A., Priselkova, A. B., Fedorov, Y. V., Trigub, A. L., Fedorova, O. A., & Kalmykov, S. N. (2022). Novel Hybrid Benzoazacrown Ligand as a Chelator for Copper and Lead Cations: What Difference Does Pyridine Make. Molecules, 27(10), 3115. https://doi.org/10.3390/molecules27103115