
Synthesis, Characterization, and Preliminary In Vitro Cytotoxic Evaluation of a Series of 2-Substituted Benzo [d] [1,3] Azoles

 , , , , and
, , , , and 
Abstract
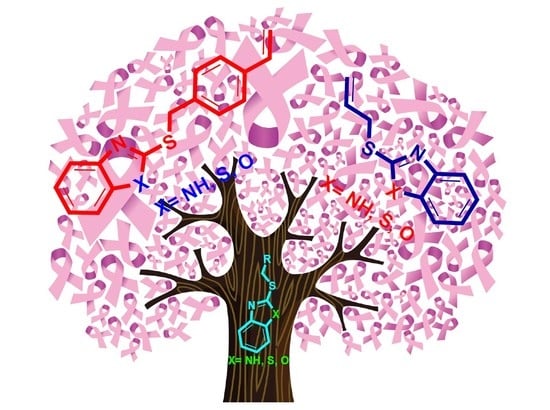
1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization
2.2. Preliminary In Vitro Antiproliferative Screening
2.3. Molecular Docking Studies
3. Materials and Methods
3.1. Reagents and Apparatus
3.2. Synthesis of 2-Substituted Benzo[d] [1,3] Azole Heterocycle Derivatives (BTA-1, BZM-2, BOX-3, BTA-4, BZM-5, and BOX-6)
3.3. Biological Evaluation
3.4. Methodology and Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Makowski, S.J.; Lacher, M.; Lermer, C.; Schnick, W. Supramolecular hydrogen-bonded structures between melamine and N-heterocycles. J. Mol. Struct. 2012, 1013, 19–25. [Google Scholar] [CrossRef]
- Didehban, K.; Vessally, E.; Salary, M.; Edjlali, L.; Babazadeh, M. Synthesis of a variety of key medicinal heterocyclic compounds via chemical fixation of CO2 onto o-alkynylaniline derivatives. J. CO2 Util. 2018, 23, 42–50. [Google Scholar] [CrossRef]
- Kotian, S.Y.; Mohan, C.D.; Merlo, A.A.; Rangappa, S.; Nayak, S.C.; Rai, K.M.L.; Rangappa, K.S. Small molecule based five-membered heterocycles: A view of liquid crystalline properties beyond the biological applications. J. Mol. Liq. 2020, 297, 111686. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Ivchenko, P.V.; Vinogradov, A.A. Heterocycle-fused cyclopentadienyl metal complexes: Heterocene synthesis, structure and catalytic applications. Coord. Chem. Rev. 2021, 426, 213515. [Google Scholar] [CrossRef]
- Bansal, Y.; Silakari, O. The therapeutic journey of benzimidazoles: A review. Bioorg. Med. Chem. 2012, 20, 6208–6236. [Google Scholar] [CrossRef]
- Akhtar, W.; Khan, M.F.; Verma, G.; Shaquiquzzaman, M.; Rizvi, M.A.; Mehdi, S.H.; Akhter, M.; Alam, M.M. Therapeutic evolution of benzimidazole derivatives in the last quinquennial period. Eur. J. Med. Chem. 2017, 126, 705–753. [Google Scholar] [CrossRef]
- Salahuddin Shaharyar, M.; Mazumder, A. Benzimidazoles: A biologically active compounds. Arab. J. Chem. 2017, 10, S157–S173. [Google Scholar] [CrossRef]
- Beltran-Hortelano, I.; Alcolea, V.; Font, M.; Pérez-Silanes, S. The role of imidazole and benzimidazole heterocycles in Chagas disease: A review. Eur. J. Med. Chem. 2020, 206, 112692. [Google Scholar] [CrossRef]
- Bondock, S.; Fadaly, W.; Metwally, M.A. Synthesis and antimicrobial activity of some new thiazole, thiophene and pyrazole derivatives containing benzothiazole moiety. Eur. J. Med. Chem. 2010, 45, 3692–3701. [Google Scholar] [CrossRef]
- Keri, R.S.; Patil, M.R.; Patil, S.A.; Budagupi, S. comprehensive review in current developments of benzothiazole-based molecules in medicinal chemistry. Eur. J. Med. Chem. 2015, 89, 207–251. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, M.V.; Bloomer, W.D.; Rosenzweig, H.S.; Kaiser, M.; Chatelain, E.; Ioset, J.-R. Novel 3-nitro-1H-1,2,4-triazole-based piperazines and 2-amino-1,3-benzothiazoles as antichagasic agents. Bioorg. Med. Chem. 2013, 21, 6600–6607. [Google Scholar] [CrossRef]
- Djuidje, E.N.; Sciabica, S.; Buzzi, R.; Dissette, V.; Balzarini, J.; Liekens, S.; Serra, E.; Andreotti, E.; Manfredini, S.; Vertuani, S.; et al. Design, synthesis and evaluation of benzothiazole derivatives as multifunctional agents. Bioorg. Chem. 2020, 101, 103960. [Google Scholar] [CrossRef] [PubMed]
- Demmer, C.S.; Bunch, L. Benzoxazoles and oxazolopyridines in medicinal chemistry studies. Eur. J. Med. Chem. 2015, 97, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Pathak, D.P.; Sharma, V.; Wakode, S. Synthesis, biological evaluation and docking study of a new series of di-substituted benzoxazole derivatives as selective COX-2 inhibitors and anti-inflammatory agents. Bioorg. Med. Chem. 2018, 26, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Desai, S.; Desai, V.; Shingade, S. In-vitro anti-cancer assay and apoptotic cell pathway of newly synthesized benzoxazole-N-heterocyclic hybrids as potent tyrosine kinase inhibitors. Bioorg. Chem. 2020, 94, 103382. [Google Scholar] [CrossRef] [PubMed]
- World and Health Organization. Cancer. Available online: https://www.who.int/health-topics/cancer#tab=tab_1 (accessed on 1 October 2020).
- Mccormack, V.A.; Boffetta, P. Today’s lifestyles, tomorrow’s cancers: Trends in lifestyle risk factors for cancer in low- and middle-income countries. Ann. Oncol. 2011, 22, 2349–2357. [Google Scholar] [CrossRef] [PubMed]
- Fishman, J.; Osborne, M.P.; Telang, N.T. The role of estrogen in mammary carcinogenesis. Ann. N. Y. Acad. Sci. 1995, 768, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.; Palmieri, C.; Tilley, W.D. Renewed interest in the progesterone receptor in breast cancer. Br. J. Cancer 2016, 115, 909–911. [Google Scholar] [CrossRef]
- Pulliam, N.; Tang, J.; Nephew, K.P. Estrogen receptor regulation of microRNAs in breast cancer. In Estrogen Receptor and Breast Cancer: Celebrating the 60th Anniversary of the Discovery of ERZ; Xiaoting, Z., Ed.; Springer Nature: Cham, Switzerland, 2019; pp. 129–143. [Google Scholar] [CrossRef]
- Saha, T.; Makar, S.; Swetha, R.; Gutti, G.; Singh, S.K. Estrogen signaling: An emanating therapeutic target for breast cancer treatment. Eur. J. Med. Chem. 2019, 177, 116–143. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhou, S.; Gustafsson, J.-Å. Nuclear receptors: Recent drug discovery for cancer therapies. Endocr. Rev. 2019, 40, 1207–1249. [Google Scholar] [CrossRef]
- Hewitt, S.C.; Korach, K.S. Estrogen receptors: New directions in the new millennium. Endocr. Rev. 2018, 39, 664–675. [Google Scholar] [CrossRef]
- Ali, S.; Coombes, R.C. Estrogen receptor alpha in human breast cancer: Occurrence and significance. J. Mammary Gland Biol. Neoplasia 2000, 5, 271–281. [Google Scholar] [CrossRef]
- Hagrass, H.A.; Pasha, H.F.; Ali, A.M. Estrogen receptor alpha (ERα) promoter methylation status in tumor and serum DNA in Egyptian breast cancer patients. Gene 2014, 552, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Chen, M.; Lyu, W.; Zhao, R.; Xu, Q.; You, Q.; Xiang, H. Design, synthesis, biological evaluation and molecular docking studies of novel 3-aryl-4-anilino-2H-chromen-2-one derivatives targeting ERα as anti-breast cancer agents. Bioorg. Med. Chem. Lett. 2017, 27, 2668–2673. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Berezov, A.; Wang, Q.; Zhang, G.; Drebin, J.; Murali, R.; Greene, M.I. ErbB receptors: From oncogenes to targeted cancer treatment. J. Clin. Investig. 2007, 117, 2051–2058. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Quinn, J.A.; Sabo, E. Association of the membrane estrogen receptor, GPR30, with breast tumor metastasis and transactivation of the epidermal growth factor receptor. Steroids 2008, 73, 870–873. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.F.; Wu, T.-T.; Yang, J.-Y.; Dong, C.-R.; Wang, N.; Liu, X.-H.; Liua, Z.-M. 17β-Estradiol promotes the invasion and migration of nuclear estrogen receptor-negative breast cancer cells through cross-talk between GPER1 and CXCR1. J. Steroid Biochem. Mol. Biol. 2013, 138, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R., Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef]
- Ignatov, A.; Ignatov, T.; Roessner, A.; Costa, S.D.; Kalinski, T. Role of GPR30 in the mechanisms of tamoxifen resistance in breast cancer MCF-7 cells. Breast Cancer. Breast Cancer Res. Treat. 2010, 123, 87–96. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Arterburn, J.B.; Smith, H.O.; Oprea, T.I.; Sklar, L.A.; Hathaway, H.J. Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Annu. Rev. Physiol. 2008, 70, 165–190. [Google Scholar] [CrossRef]
- Wagland, R.; Richardson, A.; Ewings, S.; Armes, J.; Lennan, E.; Hankins, M.; Griffiths, P. Prevalence of cancer chemotherapy-related problems, their relation to health-related quality of life and associated supportive care: A cross-sectional survey. Support. Care Cancer 2016, 24, 4901–4911. [Google Scholar] [CrossRef] [PubMed]
- Breen, S.; Kofoed, S.; Ritchie, D.; Dryden, T.; Maguire, R.; Kearney, N.; Aranda, S. Remote real-time monitoring for chemotherapy side-effects in patients with blood cancers. Collegian 2017, 24, 541–549. [Google Scholar] [CrossRef]
- Liu, Y.-Q.; Wang, X.-L.; He, D.-H.; Cheng, Y.-X. Protection against chemotherapy- and radiotherapy-induced side effects: A review based on the mechanisms and therapeutic opportunities of phytochemicals. Phytomedicine 2021, 80, 153402. [Google Scholar] [CrossRef]
- Refaat, H.M. Synthesis and anticancer activity of some novel 2-substituted benzimidazole derivatives. Eur. J. Med. Chem. 2010, 45, 2949–2956. [Google Scholar] [CrossRef] [PubMed]
- Ammazzalorso, A.; Carradori, S.; Amoroso, R.; Fernández, I.F. 2-substituted benzothiazoles as antiproliferative agents: Novel insights on structure-activity relationships. Eur. J. Med. Chem. 2020, 207, 112762. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.A. Estrogen receptor-β: Recent lessons from in vivo studies. Mol. Endocrinol. 2007, 21, 1–13. [Google Scholar] [CrossRef]
- Noolvi, M.N.; Patel, H.M.; Kaur, M. Benzothiazoles: Search for anticancer agents. Eur. J. Med. Chem. 2012, 54, 447–462. [Google Scholar] [CrossRef]
- Rezazadeh, S.; Navidpour, L.; Shafie, A. Synthesis of substituted 2-heteroarylbenzazol-5-ol derivatives as potential ligands for estrogen receptors. Tetrahedron 2013, 69, 6076–6082. [Google Scholar] [CrossRef]
- Payton-Stewart, F.; Tilghman, S.L.; Williams, L.G.; Winfield, L.L. Benzimidazoles diminish ERE transcriptional activity and cell growth in breast cancer cells. Eur. J. Med. Chem. 2014, 450, 1358–1362. [Google Scholar] [CrossRef]
- Singla, R.; Gupta, K.B.; Upadhyay, S.; Dhiman, M.; Jaitak, V. Design, synthesis and biological evaluation of novel indole-benzimidazole hybrids targeting estrogen receptor alpha (ER-α). Eur. J. Med. Chem. 2018, 146, 206–219. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.C.; Sharma, D.; Sharma, A.; Bansal, K.K.; Rajak, H.; Sharma, S.; Thakur, V.K. New horizons in benzothiazole scaffold for cancer therapy: Advances in bioactivity, functionality, and chemistry. Appl. Mater. Today 2020, 20, 100783. [Google Scholar] [CrossRef]
- Omar, A.-M.M.E.; AboulWafa, O.M.; El-Shoukrofy, M.S.; Amr, M.E. Benzoxazole derivatives as new generation of anti-breast cancer agents. Bioorg. Chem. 2020, 96, 103593. [Google Scholar] [CrossRef]
- Avila-Sorrosa, A.; Tapia-Alvarado, J.D.; Nogueda-Torres, B.; Chacón-Vargas, K.F.; Díaz-Cedillo, F.; Vargas-Díaz, M.E.; Morales-Morales, D. Facile synthesis of a series of non-symmetric thioethers including a benzothiazole moiety and their use as efficient in vitro anti-trypanosoma cruzi agents. Molecules 2019, 17, 3077. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. MTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Acharya, R.; Chacko, S.; Bose, P.; Lapenna, A.; Pattanayak, S.P. Structure based multitargeted molecular docking analysis of selected furanocoumarins against breast cancer. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Crippen, G.M. Atomic physicochemical parameters for three-dimensional-structure-directed quantitative structure-activity relationships. 2. Modeling dispersive and hydrophobic interactions. J. Chem. Inf. Comput. Sci. 1987, 27, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-B.; Liu, X.; Gao, M.-Y.; Dong, Z.-B. One-pot synthesis of 2-benzyl/2-allyl-substituted thiobenzoazoles using transition-metal-free conditions in water. J. Org. Chem. 2018, 83, 14933–14941. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, C.-Y.; Li, Y.-S.; Dong, Z.-B. Three-component synthesis of 2-substituted thiobenzoazoles using tetramethyl thiuram monosulfide (TMTM) as thiocarbonyl surrogate. Eur. J. Org. Chem. 2020, 2020, 6770–6775. [Google Scholar] [CrossRef]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Nat. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeerschd, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform, J. Cheminform. 2012, 4, 1–17. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation modle based on solute electron density and a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Palacio-Rodríguez, K.; Lans, I.; Cavasotto, C.N.; Cossio, P. Exponential consensus ranking improves the outcome in docking and receptor ensemble docking. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Morris, G.; Huey, R. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef] [PubMed]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998, 95, 927–937. [Google Scholar] [CrossRef]
- Petit-Topin, I.; Fay, M.; Resche-Rigon, M.; Ulmann, A.; Gainer, E.; Rafestin-Oblin, M.E.; Fagart, J. Molecular determinants of the recognition of ulipristal acetate by oxo-steroid receptors. J. Steroid Biochem. Mol. Biol. 2014, 144, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007, 11, 217–227. [Google Scholar] [CrossRef]
- Marz, A.M.; Fabian, A.-K.; Kozany, C.; Bracher, A.; Hausch, F. Large FK506-binding proteins shape the pharmacology of rapamycin. Mol. Cell. Biol. 2013, 33, 1357–1367. [Google Scholar] [CrossRef]
- Herrera-Calderon, O.; Yepes-Pérez, A.F.; Quintero-Saumeth, J.; Rojas-Armas, J.P.; Palomino-Pacheco, M.; Ortiz-Sánchez, J.M.; Cieza-Macedo, E.C.; Arroyo-Acevedo, J.L.; Figueroa-Salvador, L.; Peña-Rojas, G.; et al. Carvacrol: An in silico approach of a candidate drug on HER2, PI3Kα, mTOR, HER-α, PR, and EGFR receptors in the breast cancer, evidence-based complement. Altern. Med. 2020, 2020. [Google Scholar] [CrossRef]
- Guda, R.; Kumar, G.; Korra, R.; Balaji, S.; Dayakar, G.; Palabindela, R.; Myadaraveni, P.; Yellu, N.R.; Kasula, M. EGFR, HER2 target based molecular docking analysis, in vitro screening of 2, 4, 5-trisubstituted imidazole derivatives as potential anti-oxidant and cytotoxic agents. J. Photochem. Photobiol. B Biol. 2017, 176, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Landeros-Martinez, L.L.; Glossman-Mitnik, D.; Orrantia-Borunda, E.; Flores-Holguin, N. A combined molecular docking and electronic structure study for a breast cancer drug design. In Molecular Docking; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand à protein interaction diagrams for drug discovery. J. Cheminform. 2011, 3, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, LLC. The PyMOL Molecular Graphics System, Version~1.8; Schrödinger, LLC: New York, NY, USA, 2015. [Google Scholar]
- Schrödinger, LLC. Maestro; Schrödinger, LLC: New York, NY, USA, 2020. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | 2-Mercaptobenzo [d] Azole | Br-R | Product | Yield (%) a |
| 1 |  |  |  | 70 |
| 2 |  |  |  | 83 |
| 3 |  |  |  | 80 |
| 4 |  |  |  | 75 |
| 5 |  |  |  | 70 |
| 6 |  |  |  | 77 |
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Comp. | H4 | H5 | H6 | H7 | H8 | H10 | H11 | H13 | H15a | H15b |
| BTA-1 | 7.89 | 7.44–7.25a | 7.44–7.25a | 7.73 | 4.58 | 7.40–7.38 | 7.35–7.36 | 6.68 | 5.72 | 5.23 |
| BZM-2 | 7.43–7.40 | 7.12–7.08 | 7.12–7.08 | 7.43–7.40 | 4.38 | 7.12–7.08 | 7.12–7.08 | 6.51 | 5.57 | 5.10 |
| BOX-3 | 7.62 | 7.30–7.20 | 7.30–7.20 | 7.44–7.43 | 4.54 | 7.41–7.40 | 7.37–7.36 | 6.68 | 5.73 | 5.24 |
| BTA-4 | 7.68 | 7.36–7.31 | 7.24–7.19 | 7.67 | 3.92 | − | − | 6.01–5.88 | 5.30 | 5.13 |
| BZM-5 | 7.47–7.37 | 7.12–7.04 | 7.12–7.04 | 7.47–7.37 | 3.87 | − | − | 5.97–5.83 | 5.18 | 5.01 |
| BOX-6 | 7.60 | 7.29–7.21 | 7.29–7.21 | 7.43 | 3.95 | − | − | 6.08–6.00 | 5.39 | 5.21 |
| Compound | U-251 | PC-3 | K-562 | HTC-15 | MCF-7 | SKLU-1 | FGH |
|---|---|---|---|---|---|---|---|
 | 15.7 | 12.7 | 14.6 | 14.0 | 21.4 | 38.4 | NA |
 | 27.5 | 47.0 | 37.2 | 31.2 | 46.8 | 41 | 7.7 |
 | NA | NA | NA | 1.1 | 16.7 | 10.4 | NA |
 | NA | 2.5 | NA | 5.8 | 4.0 | 12.4 | NA |
 | NA | NA | NA | NA | 10.3 | 12.1 | 12.0 |
 | NA | NA | NA | NA | 2.5 | 3.2 | NA |
 | 80.6 | 36.3 | 54.3 | 58.8 | 71.3 | 43.3 | 100 |
| Compound | Receptor | Binding Energy (kcal/mol) AD4 | Binding Energy (kcal/mol) Vina | Binding Energy (kcal/mol) Smina | Mean RMSD (A°) |
|---|---|---|---|---|---|
| AEE788 | EGFR | −6.7 | −8.3 | −6.7 | 0.845 |
| TAM | Erα | −8.1 | −6.2 | −7.9 | 0.568 |
| Ulipristal acetate | Pr | −9.4 | −8.5 | −10.6 | 0.352 |
| RAP | mTOR | −15.0 | −9.1 | −6.9 | 0.158 |
| Ligand | EGFR | Erα | Pr | mTOR | LogP ± 0.47 |
|---|---|---|---|---|---|
| BTA-1 | 3 | 4 | 5 | 4 | 6.04 |
| BZM-2 | 4 | 2 | 4 | 3 | 4.60 |
| BOX-3 | 5 | 3 | 3 | 5 | 4.67 |
| BTA-4 | 6 | 5 | 6 | 6 | 4.25 |
| BZM-5 | 7 | 5 | 7 | 7 | 2.82 |
| BOX-6 | 8 | 6 | 7 | 7 | 2.86 |
| TAM | 1 | 1 * | 2 | 2 | 6.07 |
| Reference ligand (crystal structure) | 2 | 1 | 1 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linares-Anaya, O.; Avila-Sorrosa, A.; Díaz-Cedillo, F.; Gil-Ruiz, L.Á.; Correa-Basurto, J.; Salazar-Mendoza, D.; Orjuela, A.L.; Alí-Torres, J.; Ramírez-Apan, M.T.; Morales-Morales, D. Synthesis, Characterization, and Preliminary In Vitro Cytotoxic Evaluation of a Series of 2-Substituted Benzo [d] [1,3] Azoles. Molecules 2021, 26, 2780. https://doi.org/10.3390/molecules26092780
Linares-Anaya O, Avila-Sorrosa A, Díaz-Cedillo F, Gil-Ruiz LÁ, Correa-Basurto J, Salazar-Mendoza D, Orjuela AL, Alí-Torres J, Ramírez-Apan MT, Morales-Morales D. Synthesis, Characterization, and Preliminary In Vitro Cytotoxic Evaluation of a Series of 2-Substituted Benzo [d] [1,3] Azoles. Molecules. 2021; 26(9):2780. https://doi.org/10.3390/molecules26092780
Chicago/Turabian StyleLinares-Anaya, Ozvaldo, Alcives Avila-Sorrosa, Francisco Díaz-Cedillo, Luis Ángel Gil-Ruiz, José Correa-Basurto, Domingo Salazar-Mendoza, Adrian L. Orjuela, Jorge Alí-Torres, María Teresa Ramírez-Apan, and David Morales-Morales. 2021. "Synthesis, Characterization, and Preliminary In Vitro Cytotoxic Evaluation of a Series of 2-Substituted Benzo [d] [1,3] Azoles" Molecules 26, no. 9: 2780. https://doi.org/10.3390/molecules26092780
APA StyleLinares-Anaya, O., Avila-Sorrosa, A., Díaz-Cedillo, F., Gil-Ruiz, L. Á., Correa-Basurto, J., Salazar-Mendoza, D., Orjuela, A. L., Alí-Torres, J., Ramírez-Apan, M. T., & Morales-Morales, D. (2021). Synthesis, Characterization, and Preliminary In Vitro Cytotoxic Evaluation of a Series of 2-Substituted Benzo [d] [1,3] Azoles. Molecules, 26(9), 2780. https://doi.org/10.3390/molecules26092780