
Photoredox-Catalyzed Reduction of Halogenated Arenes in Water by Amphiphilic Polymeric Nanoparticles



Abstract
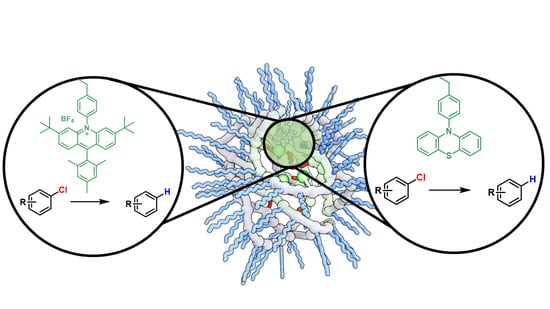
:
1. Introduction
2. Methodology and Results
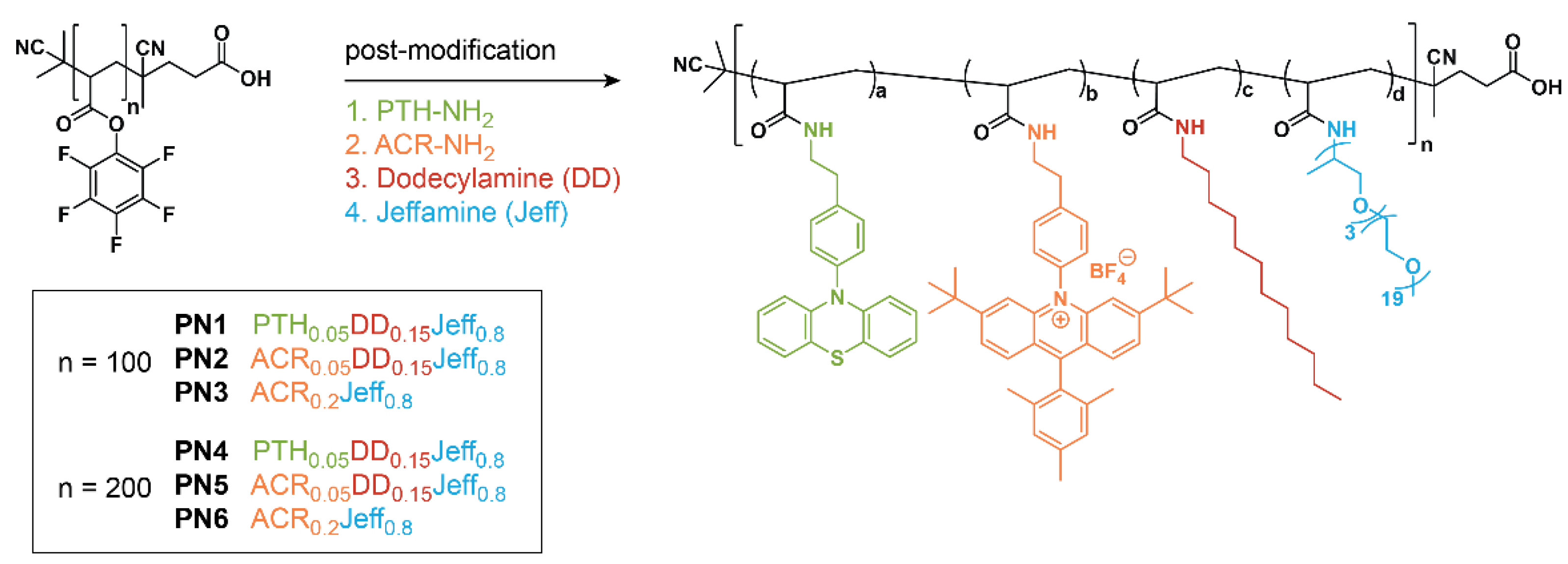
2.1. Molecular Design, Synthesis, and Characterization of Photocatalyst Loaded PNs
2.2. Reduction of Para-Substituted Aryl Chlorides in Water Using PNs
2.3. Investigating Selectivity of Dehalogenation with PTH and ACR
2.4. C-C Cross-Coupling Reaction with N-Methylpyrrole
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Methods
4.3. General Procedure for Photoredox Dehalogenation in Water
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Narayanam, J.M.R.; Stephenson, C.R.J. Visible light photoredox catalysis: Applications in organic synthesis. Chem. Soc. Rev. 2011, 40, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, N.; Shanmugam, S.; Xu, J.; Boyer, C. Photocatalysis in organic and polymer synthesis. Chem. Soc. Rev. 2016, 45, 6165–6212. [Google Scholar] [CrossRef] [PubMed]
- Glaser, F.; Wenger, O.S. Recent progress in the development of transition-metal based photoredox catalysts. Coord. Chem. Rev. 2020, 405, 213129. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannalire, R.; Pelliccia, S.; Sancineto, L.; Novellino, E.; Tron, G.C.; Giustiniano, M. Visible light photocatalysis in the late-stage functionalization of pharmaceutically relevant compounds. Chem. Soc. Rev. 2021, 50, 866–897. [Google Scholar] [CrossRef]
- Giedyk, M.; Narobe, R.; Weiß, S.; Touraud, D.; Kunz, W.; König, B. Photocatalytic activation of alkyl chlorides by assembly-promoted single electron transfer in microheterogeneous solutions. Nat. Catal. 2020, 3, 40–47. [Google Scholar] [CrossRef]
- Naumann, R.; Goez, M. A Green-LED Driven Source of Hydrated Electrons Characterized from Microseconds to Hours and Applied to Cross-Couplings. Chem. Eur. J. 2018, 24, 9833–9840. [Google Scholar] [CrossRef]
- Bu, M.; Cai, C.; Gallou, F.; Lipshutz, B.H. PQS-enabled visible-light iridium photoredox catalysis in water at room temperature. Green Chem. 2018, 20, 1233–1237. [Google Scholar] [CrossRef]
- Guerrero, I.; Kelemen, Z.; Viñas, C.; Romero, I.; Teixidor, F. Metallacarboranes as Photoredox Catalysts in Water. Chem. Eur. J. 2020, 26, 5027–5036. [Google Scholar] [CrossRef]
- Naumann, R.; Goez, M. How the sustainable solvent water unleashes the photoredox catalytic potential of ruthenium polypyridyl complexes for pinacol couplings. Green Chem. 2019, 21, 4470–4474. [Google Scholar] [CrossRef]
- Bobo, M.V.; Kuchta, J.J.; Vannucci, A.K. Recent advancements in the development of molecular organic photocatalysts. Org. Biomol. Chem. 2021, 19, 4816–4834. [Google Scholar] [CrossRef] [PubMed]
- Romero, N.A.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef]
- Lee, Y.; Kwon, M.S. Emerging Organic Photoredox Catalysts for Organic Transformations. Eur. J. Org. Chem. 2020, 2020, 6028–6043. [Google Scholar] [CrossRef]
- Discekici, E.H.; Treat, N.J.; Poelma, S.O.; Mattson, K.M.; Hudson, Z.M.; Luo, Y.; Hawker, C.J.; de Alaniz, J.R. A highly reducing metal-free photoredox catalyst: Design and application in radical dehalogenations. Chem. Commun. 2015, 51, 11705–11708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poelma, S.O.; Burnett, G.L.; Discekici, E.H.; Mattson, K.M.; Treat, N.J.; Luo, Y.; Hudson, Z.M.; Shankel, S.L.; Clark, P.G.; Kramer, J.W.; et al. Chemoselective Radical Dehalogenation and C–C Bond Formation on Aryl Halide Substrates Using Organic Photoredox Catalysts. J. Org. Chem. 2016, 81, 7155–7160. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, I.A.; Wang, L.; Onuska, N.P.R.; Williams, O.F.; Begam, K.; Moran, A.M.; Dunietz, B.D.; Nicewicz, D.A. Discovery and characterization of an acridine radical photoreductant. Nature 2020, 580, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Kerzig, C.; Goez, M. Combining energy and electron transfer in a supramolecular environment for the “green” generation and utilization of hydrated electrons through photoredox catalysis. Chem. Sci. 2016, 7, 3862–3868. [Google Scholar] [CrossRef] [Green Version]
- Naumann, R.; Lehmann, F.; Goez, M. Generating Hydrated Electrons for Chemical Syntheses by Using a Green Light-Emitting Diode (LED). Angew. Chem. Int. Ed. 2018, 57, 1078–1081. [Google Scholar] [CrossRef]
- Pallini, F.; Sangalli, E.; Sassi, M.; Roth, P.M.C.C.; Mattiello, S.; Beverina, L. Selective photoredox direct arylations of aryl bromides in water in a microfluidic reactor. Org. Biomol. Chem. 2021, 19, 3016–3023. [Google Scholar] [CrossRef]
- Bu, M.; Lu, G.; Jiang, J.; Cai, C. Merging visible-light photoredox and micellar catalysis: Arylation reactions with anilines nitrosated in situ. Catal. Sci. Technol. 2018, 8, 3728–3732. [Google Scholar] [CrossRef]
- Finck, L.; Brals, J.; Pavuluri, B.; Gallou, F.; Handa, S. Micelle-Enabled Photoassisted Selective Oxyhalogenation of Alkynes in Water under Mild Conditions. J. Org. Chem. 2018, 83, 7366–7372. [Google Scholar] [CrossRef]
- Xie, H.-Y.; Han, L.-S.; Huang, S.; Lei, X.; Cheng, Y.; Zhao, W.; Sun, H.; Wen, X.; Xu, Q.-L. N-Substituted 3(10 H)-Acridones as Visible-Light, Water-Soluble Photocatalysts: Aerobic Oxidative Hydroxylation of Arylboronic Acids. J. Org. Chem. 2017, 82, 5236–5241. [Google Scholar] [CrossRef]
- Eisenreich, F.; Meijer, E.W.; Palmans, A.R.A. Amphiphilic Polymeric Nanoparticles for Photoredox Catalysis in Water. Chem. Eur. J. 2020, 26, 10355–10361. [Google Scholar] [CrossRef]
- Lipshutz, B.H.; Ghorai, S.; Cortes-Clerget, M. The Hydrophobic Effect Applied to Organic Synthesis: Recent Synthetic Chemistry “in Water” . Chem. Eur. J. 2018, 1–25. [Google Scholar] [CrossRef]
- Lipshutz, B.H. Synthetic chemistry in a water world. New rules ripe for discovery. Curr. Opin. Green Sustain. Chem. 2018, 11, 1–8. [Google Scholar] [CrossRef]
- Bergbreiter, D.E.; Liu, Y.-S. Amphoteric, water-soluble polymer-bound hydrogenation catalysts. Tetrahedron Lett. 1997, 38, 3703–3706. [Google Scholar] [CrossRef]
- Liang, Y.; Bergbreiter, D.E. Recyclable polyisobutylene (PIB)-bound organic photoredox catalyst catalyzed polymerization reactions. Polym. Chem. 2016, 7, 2161–2165. [Google Scholar] [CrossRef]
- Bergbreiter, D.E. Soluble polymers as tools in catalysis. ACS Macro Lett. 2014, 3, 260–265. [Google Scholar] [CrossRef]
- Treat, N.J.; Sprafke, H.; Kramer, J.W.; Clark, P.G.; Barton, B.E.; Read De Alaniz, J.; Fors, B.P.; Hawker, C.J. Metal-Free Atom Transfer Radical Polymerization. J. Am. Chem. Soc. 2014, 136, 16096–16101. [Google Scholar] [CrossRef] [Green Version]
- Terashima, T.; Mes, T.; de Greef, T.F.A.; Gillissen, M.A.J.; Besenius, P.; Palmans, A.R.A.; Meijer, E.W. Single-chain folding of polymers for catalytic systems in water. J. Am. Chem. Soc. 2011, 133, 4742–4745. [Google Scholar] [CrossRef]
- Artar, M.; Souren, E.R.J.; Terashima, T.; Meijer, E.W.; Palmans, A.R.A. Single Chain Polymeric Nanoparticles as Selective Hydrophobic Reaction Spaces in Water. ACS Macro Lett. 2015, 4, 1099–1103. [Google Scholar] [CrossRef]
- Huerta, E.; Stals, P.J.M.; Meijer, E.W.; Palmans, A.R.A. Consequences of Folding a Water-Soluble Polymer around an Organocatalyst. Angew. Chem. Int. Ed. 2013, 52, 2906–2910. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Turunen, P.; de Waal, B.F.M.M.; Blank, K.G.; Rowan, A.E.; Palmans, A.R.A.; Meijer, E.W. Catalytic single-chain polymeric nanoparticles at work: From ensemble towards single-particle kinetics. Mol. Syst. Des. Eng. 2018, 3, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Pan, X.; Schmitt, M.; Wang, Z.; Bockstaller, M.R.; Matyjaszewski, K. Enhancing Initiation Efficiency in Metal-Free Surface-Initiated Atom Transfer Radical Polymerization (SI-ATRP). ACS Macro Lett. 2016, 5, 661–665. [Google Scholar] [CrossRef]
- White, A.; Wang, L.; Nicewicz, D. Synthesis and Characterization of Acridinium Dyes for Photoredox Catalysis. Synlett 2019, 30, 827–832. [Google Scholar] [CrossRef] [Green Version]
- Zilate, B.; Fischer, C.; Sparr, C. Design and application of aminoacridinium organophotoredox catalysts. Chem. Commun. 2020, 56, 1767–1775. [Google Scholar] [CrossRef] [Green Version]
- Timpe, H.-J.; Ulrich, S.; Decker, C.; Fouassier, J.-P. Acridinium salt mediated photopolymerization of acrylates and methacrylates with dihydropyridine-iodonium salt combinations. Eur. Polym. J. 1994, 30, 1301–1307. [Google Scholar] [CrossRef]
- Theato, P. Synthesis of well-defined polymeric activated esters. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 6677–6687. [Google Scholar] [CrossRef]
- Liu, Y.; Pauloehrl, T.; Presolski, S.I.; Albertazzi, L.; Palmans, A.R.A.; Meijer, E.W. Modular Synthetic Platform for the Construction of Functional Single-Chain Polymeric Nanoparticles: From Aqueous Catalysis to Photosensitization. J. Am. Chem. Soc. 2015, 137, 13096–13106. [Google Scholar] [CrossRef]
- ter Huurne, G.M.; de Windt, L.N.J.; Liu, Y.; Meijer, E.W.; Voets, I.K.; Palmans, A.R.A. Improving the Folding of Supramolecular Copolymers by Controlling the Assembly Pathway Complexity. Macromolecules 2017, 50, 8562–8569. [Google Scholar] [CrossRef]
- Yamamoto, H.; Morishima, Y. Effect of hydrophobe content on intra- and interpolymer self-associations of hydrophobically modified poly(sodium 2-(acrylamido)-2-methylpropanesulfonate) in water. Macromolecules 1999, 32, 7469–7475. [Google Scholar] [CrossRef]
- Morishima, Y.; Nomura, S.; Ikeda, T.; Seki, M.; Kamachi, M. Characterization of Unimolecular Micelles of Random Copolymers of Sodium 2-(Acrylamido)-2-methylpropanesulfonate and Methacrylamides Bearing Bulky Hydrophobic Substituents. Macromolecules 1995, 28, 2874–2881. [Google Scholar] [CrossRef]
- Steiner, A.; Williams, J.D.; Rincón, J.A.; de Frutos, O.; Mateos, C.; Kappe, C.O. Implementing Hydrogen Atom Transfer (HAT) Catalysis for Rapid and Selective Reductive Photoredox Transformations in Continuous Flow. Eur. J. Org. Chem. 2019, 2019, 5807–5811. [Google Scholar] [CrossRef]
- Connell, T.U.; Fraser, C.L.; Czyz, M.L.; Smith, Z.M.; Hayne, D.J.; Doeven, E.H.; Agugiaro, J.; Wilson, D.J.D.; Adcock, J.L.; Scully, A.D.; et al. The Tandem Photoredox Catalysis Mechanism of [Ir(ppy) 2 (dtb-bpy)] + Enabling Access to Energy Demanding Organic Substrates. J. Am. Chem. Soc. 2019, 141, 17646–17658. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; You, D.; Li, C.; Han, C.; Tang, N.; Yang, H.; Xue, X. Photolysis of Nitroaromatic Compounds under Sunlight: A Possible Daytime Photochemical Source of Nitrous Acid? Environ. Sci. Technol. Lett. 2021, 8, 747–752. [Google Scholar] [CrossRef]
- Cowper, N.G.W.; Chernowsky, C.P.; Williams, O.P.; Wickens, Z.K. Potent Reductants via Electron-Primed Photoredox Catalysis: Unlocking Aryl Chlorides for Radical Coupling. J. Am. Chem. Soc. 2020, 142, 2093–2099. [Google Scholar] [CrossRef] [PubMed]
- Fry, A.J.; Krieger, R.L. Electrolyte effects upon the polarographic reduction of alkyl halides in dimethyl sulfoxide. J. Org. Chem. 1976, 41, 54–57. [Google Scholar] [CrossRef]
- Takeda, N.; Poliakov, P.V.; Cook, A.R.; Miller, J.R. Faster Dissociation: Measured Rates and Computed Effects on Barriers in Aryl Halide Radical Anions. J. Am. Chem. Soc. 2004, 126, 4301–4309. [Google Scholar] [CrossRef]
- Cranwell, P.; Russell, A.; Smith, C. Methyl Hydrazinocarboxylate as a Practical Alternative to Hydrazine in the Wolff–Kishner Reaction. Synlett 2015, 27, 131–135. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}

| S1 | S2 | S3 | S4 | S5 |
|---|---|---|---|---|
| PN1 100% | PN1 2% | PN1 55% | PN1 0% | PN1 3% |
| PN2 100% | PN2 2% | PN2 23% | PN2 3% | PN2 10% |
| PN3 100% | PN3 18% | PN3 56% | PN3 13% | PN3 12% |
| PN4 100% | PN4 8% | PN4 45% | PN4 1% | PN4 2% |
| PN5 100% | PN5 2% | PN5 26% | PN5 6% | PN5 12% |
| PN6 100% | PN6 7% | PN6 47% | PN6 12% | PN6 22% |

| Reaction a | Reaction b | ||||
|---|---|---|---|---|---|
| R6 | R7 | R8 | R6 | R9 | R8 |
| PN1 65% | PN1 0% | PN1 2% | PN1 6% | PN1 6% | PN1 > 1% |
| PN2 68% | PN2 0% | PN2 4% | PN2 13% | PN2 13% | PN2 4% |
| PN3 51% | PN3 0% | PN3 6% | PN3 41% | PN3 41% | PN3 6% |
| PN4 63% | PN4 0% | PN4 1% | PN4 8% | PN4 8% | PN4 > 1% |
| PN5 68% | PN5 0% | PN5 7% | PN5 22% | PN5 22% | PN5 7% |
| PN6 38% | PN6 0% | PN6 8% | PN6 19% | PN6 19% | PN6 8% |
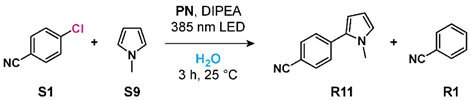
| PN | Conversion | Ratio R11:R1 |
|---|---|---|
| PN1 | 100% | 46:54 |
| PN2 | 100% | 37:63 |
| PN3 | 100% | 47:53 |
| PN4 | 100% | 48:52 |
| PN5 | 100% | 33:67 |
| PN6 | 100% | 48:52 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eisenreich, F.; Kuster, T.H.R.; van Krimpen, D.; Palmans, A.R.A. Photoredox-Catalyzed Reduction of Halogenated Arenes in Water by Amphiphilic Polymeric Nanoparticles. Molecules 2021, 26, 5882. https://doi.org/10.3390/molecules26195882
Eisenreich F, Kuster THR, van Krimpen D, Palmans ARA. Photoredox-Catalyzed Reduction of Halogenated Arenes in Water by Amphiphilic Polymeric Nanoparticles. Molecules. 2021; 26(19):5882. https://doi.org/10.3390/molecules26195882
Chicago/Turabian StyleEisenreich, Fabian, Tom H. R. Kuster, David van Krimpen, and Anja R. A. Palmans. 2021. "Photoredox-Catalyzed Reduction of Halogenated Arenes in Water by Amphiphilic Polymeric Nanoparticles" Molecules 26, no. 19: 5882. https://doi.org/10.3390/molecules26195882
APA StyleEisenreich, F., Kuster, T. H. R., van Krimpen, D., & Palmans, A. R. A. (2021). Photoredox-Catalyzed Reduction of Halogenated Arenes in Water by Amphiphilic Polymeric Nanoparticles. Molecules, 26(19), 5882. https://doi.org/10.3390/molecules26195882