
Analytical Methods for Determination of Phytic Acid and Other Inositol Phosphates: A Review



Abstract
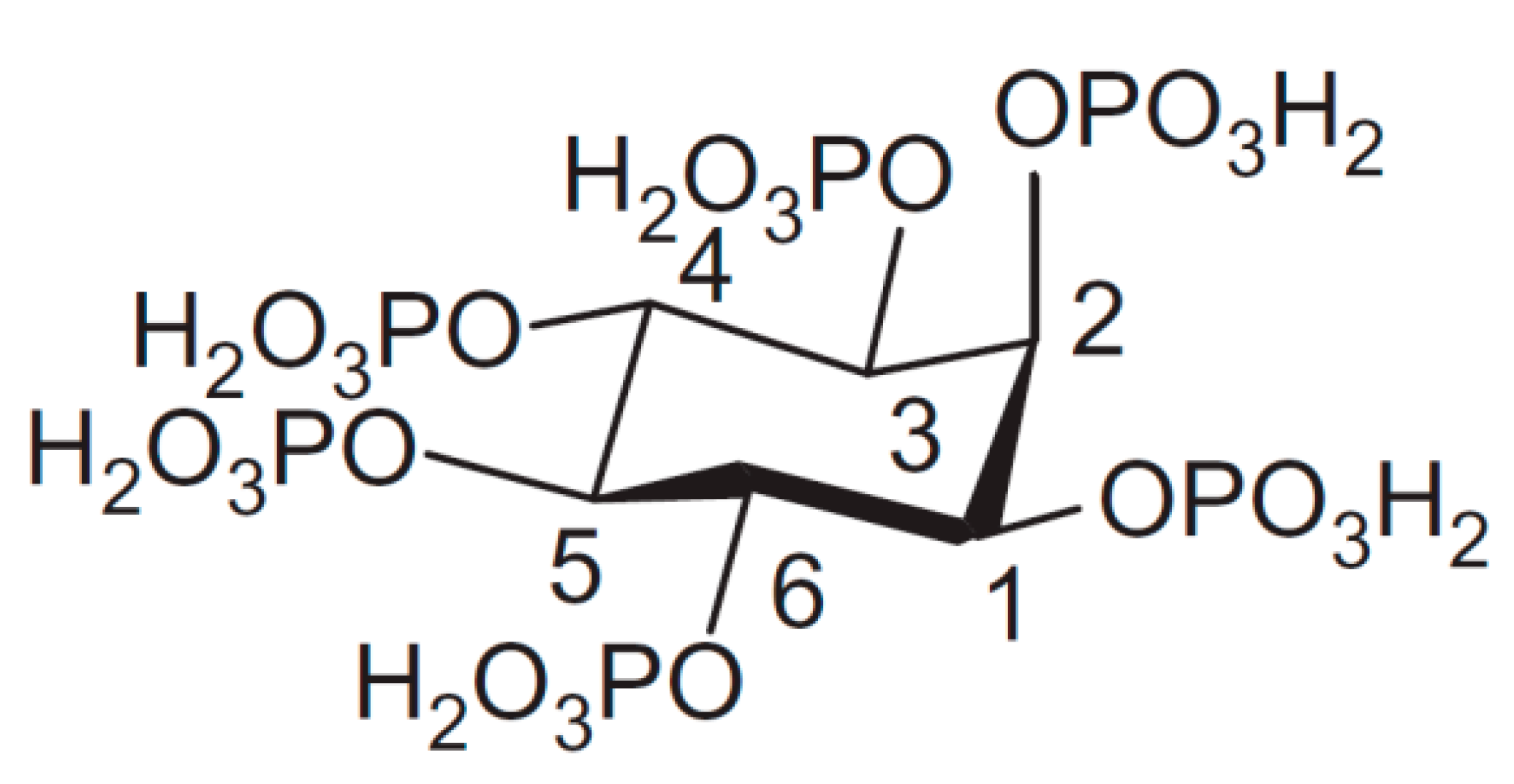
1. Introduction
2. Classical Analytical Methods
2.1. Precipitation Techniques
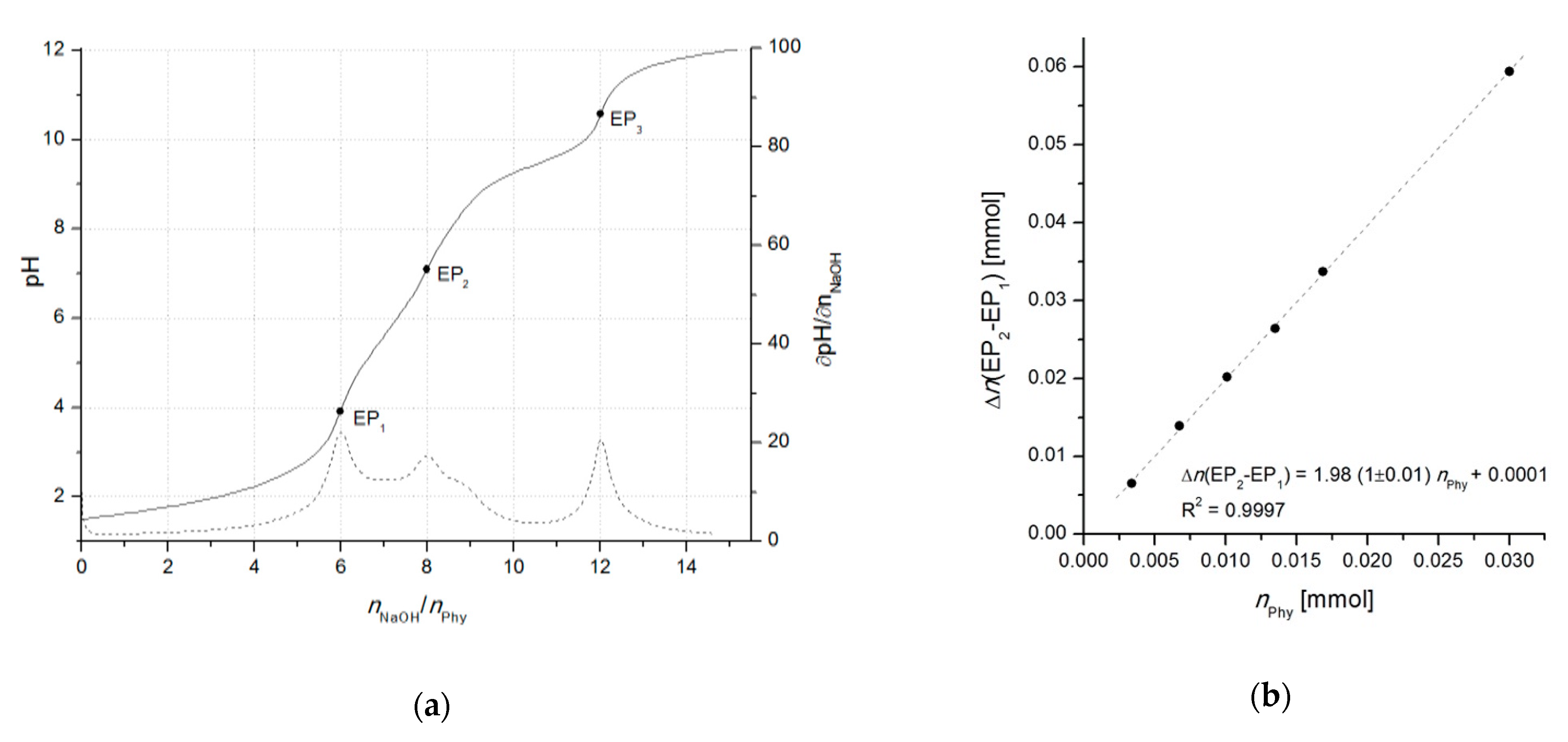
2.2. Potentiometric Titrations
3. Chromatography
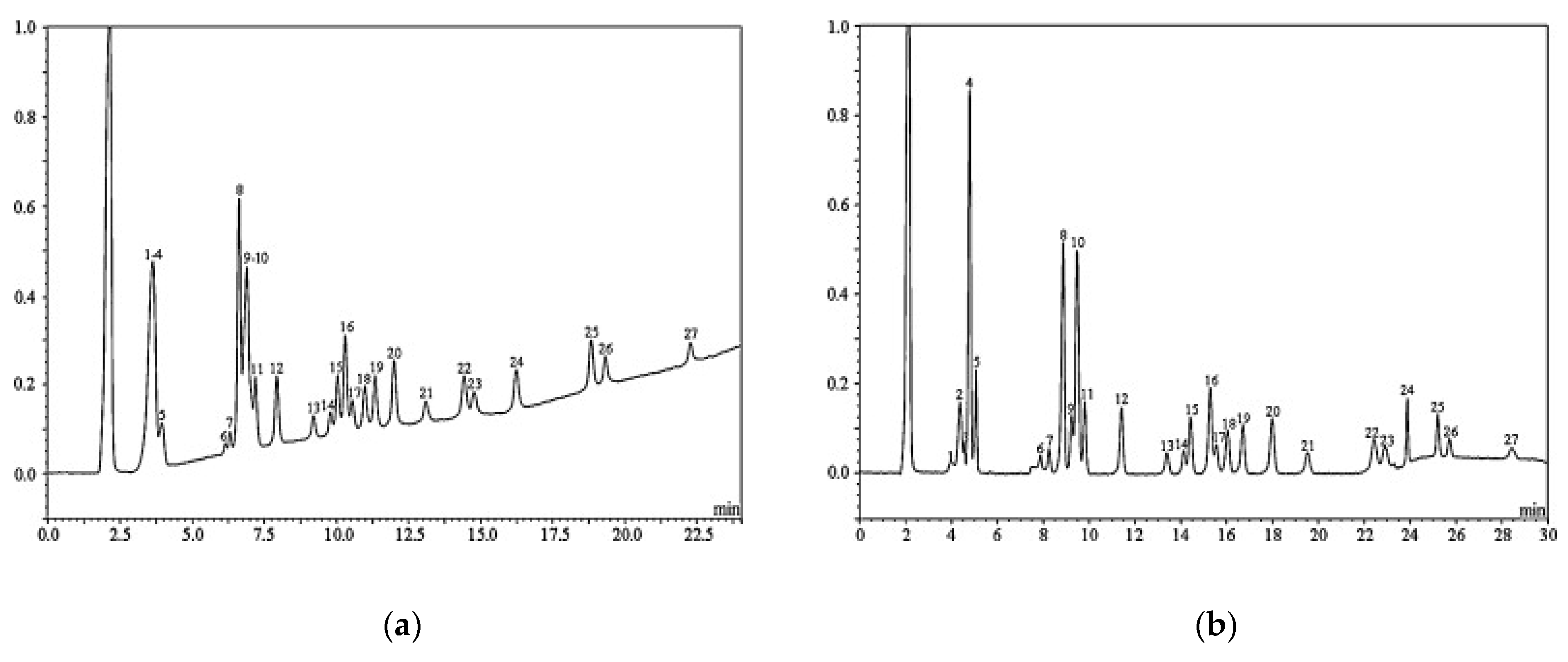
3.1. Liquid Chromatography (LC)
3.2. Ion-Exchange Chromatography (IC)
3.3. Gas Chromatography (GC)
3.4. Thin Layer Chromatography (TLC)
3.5. Electrophoresis
4. Spectroscopy
4.1. UV-Vis Spectrophotometry
4.2. Fluorescence Spectroscopy
4.3. Nuclear Magnetic Resonance (NMR)
4.4. Inductively Coupled Plasma (ICP)
5. Sensors
5.1. Electrochemical Biosensors
5.2. Fluorescence Nanoprobes
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Torres, J.; Veiga, N.; Gancheff, J.S.; Domínguez, S.; Mederos, A.; Sundberg, M.; Sánchez, A.; Castiglioni, J.; Díaz, A.; Kremer, C. Interaction of myo-inositol hexakisphosphate with alkali and alkaline earth metal ions: Spectroscopic, potentiometric and theoretical studies. J. Mol. Struct. 2008, 874, 77–88. [Google Scholar] [CrossRef]
- Brigando, C.; Mossoyan, J.C.; Favier, F.; Benlian, D. Conformational preferences and protonation sequence of myo-inositol hexaphosphate in aqueous solution; potentiometric and multinuclear magnetic resonance studies. J. Chem. Soc. Dalt. Trans. 1995, 575–578. [Google Scholar] [CrossRef]
- Veiga, N.; Torres, J.; MacHo, I.; Gómez, K.; González, G.; Kremer, C. Coordination, microprotonation equilibria and conformational changes of myo-inositol hexakisphosphate with pertinence to its biological function. Dalt. Trans. 2014, 43, 16238–16251. [Google Scholar] [CrossRef] [PubMed]
- Irvine, R.F.; Schell, M.J. Back in the water: The return of the inositol phosphates. Nat. Rev. Mol. Cell Biol. 2001, 2, 327–338. [Google Scholar] [CrossRef]
- Maga, J.A. Phytate—Its chemistry, occurrence, food interactions, nutritional significance, and methods of analysis. J. Agric. Food Chem. 1982, 30, 1–9. [Google Scholar] [CrossRef]
- Reddy, N.R.; Sathe, S.K.; Salunkhe, D.K. Phytates in legumes and cereals. Adv. Food Res. 1982, 28, 1–92. [Google Scholar] [CrossRef]
- Graf, E. Applications of phytic acid. J. Am. Oil Chem. Soc. 1983, 60, 1861–1867. [Google Scholar] [CrossRef]
- Chatree, S.; Thongmaen, N.; Tantivejkul, K.; Sitticharoon, C.; Vucenik, I. Role of inositols and inositol phosphates in energy metabolism. Molecules 2020, 25, 5079. [Google Scholar] [CrossRef]
- Graf, E. Antioxidant functions of phytic acid. Free Radic. Biol. Med. 1990, 8, 61–69. [Google Scholar] [CrossRef]
- Sasakawa, N.; Sharif, M.; Hanley, M.R. Metabolism and biological activities of inositol pentakisphosphate and inositol hexakisphosphate. Biochem. Pharmacol. 1995, 50, 137–146. [Google Scholar] [CrossRef]
- Maffucci, T.; Falasca, M. Signalling Properties of Inositol Polyphosphates. Molecules 2020, 25, 5281. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.; Domínguez, S.; Cerdá, M.F.; Obal, G.; Mederos, A.; Irvine, R.F.; Díaz, A.; Kremer, C. Solution behaviour of myo-inositol hexakisphosphate in the presence of multivalent cations. Prediction of a neutral pentamagnesium species under cytosolic/nuclear conditions. J. Inorg. Biochem. 2005, 99, 828–840. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Sinha, A.K.; Makkar, H.P.S.; Becker, K. Dietary roles of phytate and phytase in human nutrition: A review. Food Chem. 2010, 120, 945–959. [Google Scholar] [CrossRef]
- Marolt, G.; Gričar, E.; Pihlar, B.; Kolar, M. Complex Formation of Phytic Acid With Selected Monovalent and Divalent Metals. Front. Chem. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Lopez, H.W.; Leenhardt, F.; Coudray, C.; Remesy, C. Minerals and phytic acid interactions: Is it a real problem for human nutrition? Int. J. Food Sci. Technol. 2002, 37, 727–739. [Google Scholar] [CrossRef]
- Deshpande, S.S.; Damodaran, S. Effect of phytate onsolubility, activity andconformation oftrypsin andchymotrypsin. J. Food Sci. 1989, 54, 695–699. [Google Scholar] [CrossRef]
- Lee, S.-H.; Park, H.-J.; Chun, H.-K.; Cho, S.-Y.; Cho, S.-M.; Lillehoj, H.S. Dietary phytic acid lowers the blood glucose level in diabetic KK mice. Nutr. Res. 2006, 26, 474–479. [Google Scholar] [CrossRef]
- Matyka, S.; Korol, W.; Bogusz, G. The retention of phytin phosphorus from diets with fat supplements in broiler chicks. Anim. Feed Sci. Technol. 1990, 31, 223–230. [Google Scholar] [CrossRef]
- Noureddini, H.; Malik, M.; Byun, J.; Ankeny, A.J. Distribution of phosphorus compounds in corn processing. Bioresour. Technol. 2009, 100, 731–736. [Google Scholar] [CrossRef]
- Rao, D.E.C.S.; Rao, K.V.; Reddy, T.P.; Reddy, V.D. Molecular characterization, physicochemical properties, known and potential applications of phytases: An overview. Crit. Rev. Biotechnol. 2009, 29, 182–198. [Google Scholar] [CrossRef]
- Sanz-Penella, J.M.; Tamayo-Ramos, J.A.; Sanz, Y.; Haros, M. Phytate Reduction in Bran-Enriched Bread by Phytase-Producing Bifidobacteria. J. Agric. Food Chem. 2009, 57, 10239–10244. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, T.; Tange, T.; Anazawa, H. Dephosphorylation of Phytate by Using theAspergillus niger Phytase with a High Affinity for Phytate. Appl. Environ. Microbiol. 1999, 65, 4682–4684. [Google Scholar] [CrossRef] [PubMed]
- Reddy, N.R.; Sathe, S.K. Food Phytates; CRC Press: Boca Raton, FL, USA, 2001; ISBN 9781420014419. [Google Scholar]
- Menniti, F.S.; Miller, R.N.; Putney, J.W.; Shears, S.B. Turnover of inositol polyphosphate pyrophosphates in pancreatoma cells. J. Biol. Chem. 1993, 268, 3850–3856. [Google Scholar] [CrossRef] [PubMed]
- Oomah, B.D.; Blanchard, C.; Balasubramanian, P. Phytic acid, phytase, minerals, and antioxidant activity in Canadian dry bean (Phaseolus vulgaris L.) cultivars. J. Agric. Food Chem. 2008, 56, 11312–11319. [Google Scholar] [CrossRef]
- Vucenik, I.; Shamsuddin, A.K.M. Cancer Inhibition by Inositol Hexaphosphate (IP6) and Inositol: From Laboratory to Clinic. J. Nutr. 2003, 133, 3778S–3784S. [Google Scholar] [CrossRef]
- Vucenik, I.; Shamsuddin, A.M. Protection Against Cancer by Dietary IP 6 and Inositol. Nutr. Cancer 2006, 55, 109–125. [Google Scholar] [CrossRef]
- Marolt, G.; Šala, M.; Pihlar, B. Voltammetric Investigation of Iron(III) Interactions with Phytate. Electrochim. Acta 2015, 176, 1116–1125. [Google Scholar] [CrossRef]
- Graf, E.; Mahoney, J.R.; Bryant, R.G.; Eaton, J.W. Iron-catalyzed hydroxyl radical formation. Stringent requirement for free iron coordination site. J. Biol. Chem. 1984, 259, 3620–3624. [Google Scholar] [CrossRef]
- Grases, F.; Llobera, A. Experimental model to study sedimentary kidney stones. Micron 1998, 29, 105–111. [Google Scholar] [CrossRef]
- Jariwalla, R.J.; Sabin, R.; Lawson, S.; Herman, Z.S. Lowering of serum cholesterol and triglycerides and modulation of divalent cations by dietary phytate. J. Appl. Nutr. 1990, 42, 18–28. [Google Scholar]
- Graf, E. Phytic Acid: Chemistry & Applications; Pilatus Press: Stans, Switzerland, 1986; ISBN 9780961491505. [Google Scholar]
- Heubner, W.; Stadler, H. Über eine Titration-methode zur Bestimmung des Phytins. Biochem. Z. 1914, 64, 432–437. [Google Scholar]
- Rather, J.B. THE DETERMINATION OF PHYTIN PHOSPHORUS IN PLANT PRODUCTS. 1. J. Am. Chem. Soc. 1917, 39, 2506–2515. [Google Scholar] [CrossRef]
- Averill, H.P.; King, C.G. THE PHYTIN CONTENT OF FOODSTUFFS. J. Am. Chem. Soc. 1926, 48, 724–728. [Google Scholar] [CrossRef]
- Harris, R.S.; Mosher, L.M. Estimation of Phytin Phosphorus. Ind. Eng. Chem. Anal. Ed. 1934, 6, 320–321. [Google Scholar] [CrossRef]
- McCance, R.A.; Widdowson, E.M. Phytin in human nutrition. Biochem. J. 1935, 29, 2694–2699. [Google Scholar] [CrossRef]
- Padmanabhan, C.; Sundaram, S. A new method for the estimation of Nitrogen in Cellulose Nitrate. Curr. Sci. 1955, 24, 403–404. [Google Scholar]
- Briggs, A.P. A modification of the bell-doisy phosphate method. J. Biol. Chem. 1922, 53, 13–16. [Google Scholar] [CrossRef]
- Nagul, E.A.; McKelvie, I.D.; Worsfold, P.; Kolev, S.D. The molybdenum blue reaction for the determination of orthophosphate revisited: Opening the black box. Anal. Chim. Acta 2015, 890, 60–82. [Google Scholar] [CrossRef]
- Young, L. The determination of phytic acid1. Biochem. J. 1936, 30, 252–257. [Google Scholar] [CrossRef]
- Davies, N.T.; Reid, H. An evaluation of the phytate, zinc, copper, iron and manganese contents of, and Zn availability from, soya-based textured-vegetable-protein meat-substitutes or meat-extenders. Br. J. Nutr. 1979, 41, 579–589. [Google Scholar] [CrossRef]
- Haug, W.; Lantzsch, H.-J. Sensitive method for the rapid determination of phytate in cereals and cereal products. J. Sci. Food Agric. 1983, 34, 1423–1426. [Google Scholar] [CrossRef]
- Holt, R. Studies on dried peas I—The determination of phytate phosphorus. J. Sci. Food Agric. 1955, 6, 136–142. [Google Scholar] [CrossRef]
- Reeves, R.J.; Carroll, R.T.; Gennaro, G.P. Titration of phytic acid. Talanta 1979, 26, 1033–1034. [Google Scholar] [CrossRef]
- Thompson, D.B.; Erdman, J.W. Phytic Acid Determination in Soybeans. J. Food Sci. 1982, 47, 513–517. [Google Scholar] [CrossRef]
- Xu, P.; Price, J.; Aggett, P.J. Recent advances in methodology for analysis of phytate and inositol phosphates in foods. Prog. Food Nutr. Sci. 1992, 16, 245–262. [Google Scholar]
- Crea, F.; De Stefano, C.; Milea, D.; Sammartano, S. Formation and stability of phytate complexes in solution. Coord. Chem. Rev. 2008, 252, 1108–1120. [Google Scholar] [CrossRef]
- Burgos-Luján, I.; Tong, A.Z. Determination of Phytic Acid in Juices and Milks by Developing a Quick Complexometric-Titration Method. Food Anal. Methods 2015, 8, 1836–1841. [Google Scholar] [CrossRef]
- Marolt, G.; Pihlar, B. Potentiometric determination of phytic acid and investigations of phytate interactions with some metal ions. Acta Chim. Slov. 2015, 62, 319–327. [Google Scholar] [CrossRef][Green Version]
- Santiviago Petzoldt, C.; Peralta Lezcano, J.; López Moreda, I. Removal of orthophosphate and dissolved organic phosphorus from synthetic wastewater in a combined struvite precipitation-adsorption system. J. Environ. Chem. Eng. 2020, 8, 103923. [Google Scholar] [CrossRef]
- Lolas, G.M.; Markakis, P. Phytic acid and other phosphorus compounds of beans (Phaseolus vulgaris L.). J. Agric. Food Chem. 1975, 23, 13–15. [Google Scholar] [CrossRef]
- Duhan, A.; Chauhan, B.M.; Punia, D.; Kapoor, A.C. Phytic acid content of chickpea (Cicer arietinum) and black gram (Vigna mungo): Varietal differences and effect of domestic processing and cooking methods. J. Sci. Food Agric. 1989, 49, 449–455. [Google Scholar] [CrossRef]
- Desjobert, A.; Petek, F. Chromatographie sur papier des esters phosphoriques de l’inositol; application a l’étude de la dégradation hydrolytique de l’inositolhexaphosphate. Bull. Soc. Chim. Biol. 1956, 38, 871–883. [Google Scholar]
- Posternak, S.; Posternak, T. Sur la configuration de l’inosite inactive. Helv. Chim. Acta 1929, 12, 1165–1181. [Google Scholar] [CrossRef]
- Smith, D.H.; Clark, F.E. Chromatographic Separations of Inositol Phosphorus Compounds. Soil Sci. Soc. Am. J. 1952, 16, 170–172. [Google Scholar] [CrossRef]
- Cosgrove, D.J. The Isolation of Myoinositol Pentaphosphates from Hydrolysates of Phytic Acid. Biochem. J. 1963, 89, 172–175. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Isaacks, R.E.; Harkness, D.R.; Froeman, G.A.; Sussman, S.A. Studies on avian erythrocyte metabolism. I. Procedure for separation and quantitation of the major phosphorylated metabolic intermediates by anion exchange chromatography. Comp. Biochem. Physiol. 1975, 53, 95–99. [Google Scholar] [CrossRef]
- Bartlett, G.R. Phosphorus assay in column chromatography. J. Biol. Chem. 1959, 234, 466–468. [Google Scholar] [CrossRef]
- Tangendjaja, B.; Buckle, K.A.; Wootton, M. Analysis of phytic acid by high-performance liquid chromatography. J. Chromatogr. A 1980, 197, 274–277. [Google Scholar] [CrossRef]
- Camire, A.L.; Clydesdale, F.M. Analysis of Phytic Acid in Foods by HPLC. J. Food Sci. 1982, 47, 575–578. [Google Scholar] [CrossRef]
- Knuckles, B.E.; Kuzmicky, D.D.; Betschart, A.A. HPLC Analysis of Phytic Acid in Selected Foods and Biological Samples. J. Food Sci. 1982, 47, 1257–1258. [Google Scholar] [CrossRef]
- Graf, E.; Dintzis, F.R. Determination of phytic acid in foods by high-performance liquid chromatography. J. Agric. Food Chem. 1982, 30, 1094–1097. [Google Scholar] [CrossRef]
- Foda, N.H. High performance liquid chromatographic determination of nalidixic acid in tablets. J. Liq. Chromatogr. 1995, 18, 4135–4147. [Google Scholar] [CrossRef]
- Sandberg, A.-S.; Ahderinne, R. HPLC Method for Determination of inositol Tri-, Tetra-, Penta-, and Hexaphosphates in Foods and Intestinal Contents. J. Food Sci. 1986, 51, 547–550. [Google Scholar] [CrossRef]
- Burbano, C.; Muzquiz, M.; Osagie, A.; Ayet, G.; Cuadrado, C. Determination of phytate and lower inositol phosphates in Spanish legumes by HPLC methodology. Food Chem. 1995, 52, 321–325. [Google Scholar] [CrossRef]
- Lehrfeld, J. High-performance liquid chromatography analysis of phytic acid on a pH-stable, macroporous polymer column. Cereal Chem. 1989, 66, 510–515. [Google Scholar]
- Berridge, M.J.; Irvine, R.F. Inositol trisphosphate, a novel second messenger in cellular signal transduction. Nature 1984, 312, 315–321. [Google Scholar] [CrossRef]
- Heslop, J.P.; Irvine, R.F.; Tashjian, A.H.; Berridge, M.J. Inositol tetrakis- and pentakisphosphates in GH4 cells. J. Exp. Biol. 1985, 119, 395–401. [Google Scholar]
- Morgan, R.O.; Chang, J.P.; Catt, K.J. Novel aspects of gonadotropin-releasing hormone action on inositol polyphosphate metabolism in cultured pituitary gonadotrophs. J. Biol. Chem. 1987, 262, 1166–1171. [Google Scholar] [CrossRef]
- Sulpice, J.-C.; Gascard, P.; Journet, E.; Rendu, F.; Renard, D.; Poggioli, J.; Giraud, F. The separation of [32P] inositol phosphates by ion-pair chromatography: Optimization of the method and biological applications. Anal. Biochem. 1989, 179, 90–97. [Google Scholar] [CrossRef]
- Irth, H.; Lamoree, M.; de Jong, G.J.; Brinkman, U.A.T.; Frei, R.W.; Kornfeldt, R.A.; Persson, L. Determination of d-myo-1,2,6-inositol trisphosphate by ion-pair reversed-phase liquid chromatography with post-column ligand exchange and fluorescence detection. J. Chromatogr. A 1990, 499, 617–625. [Google Scholar] [CrossRef]
- Letcher, A.J.; Schell, M.J.; Irvine, R.F. Do mammals make all their own inositol hexakisphosphate? Biochem. J. 2008, 416, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, C.; Saiardi, A. Extraction and analysis of soluble inositol polyphosphates from yeast. Nat. Protoc. 2006, 1, 2416–2422. [Google Scholar] [CrossRef] [PubMed]
- Laha, D.; Johnen, P.; Azevedo, C.; Dynowski, M.; Weiß, M.; Capolicchio, S.; Mao, H.; Iven, T.; Steenbergen, M.; Freyer, M.; et al. VIH2 Regulates the Synthesis of Inositol Pyrophosphate InsP 8 and Jasmonate-Dependent Defenses in Arabidopsis. Plant. Cell 2015, 27, 1082–1097. [Google Scholar] [CrossRef] [PubMed]
- Phillippy, B.O.; Johnston, M.R. Determination of Phytic Acid in Foods by Ion Chromatography with Post-Column Derivatization. J. Food Sci. 2006, 50, 541–542. [Google Scholar] [CrossRef]
- Smith, R.; Martell, A. Critical Stability Constants Volume 4: Inorganic Complexes; Springer: Berlin/Heidelberg, Germany, 1976; Volume 4, ISBN 9781475755084. [Google Scholar]
- Fruhbeck, G.; Alonso, R.; Marzo, F.; Santidrian, S. A Modified Method for the Indirect Quantitative Analysis of Phytate in Foodstuffs. Anal. Biochem. 1995, 225, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Vaintraub, I.A.; Lapteva, N.A. Colorimetric determination of phytate in unpurified extracts of seeds and the products of their processing. Anal. Biochem. 1988, 175, 227–230. [Google Scholar] [CrossRef]
- Costa-Bauza, A.; Grases, F.; Gomila, I.; Rodriguez, A.; Prieto, R.M.; Tur, F. A simple and rapid colorimetric method for determination of phytate in urine. Urol. Res. 2012, 40, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, A.-S.; Larsen, T.; Sandström, B. High Dietary Calcium Level Decreases Colonic Phytate Degradation in Pigs Fed a Rapeseed Diet. J. Nutr. 1993, 123, 559–566. [Google Scholar] [CrossRef]
- Thies, W. Determination of the Phytic Acid and Sinapic Acid Esters in Seeds of Rapeseed and Selection of Genotypes with Reduced Concentrations of these Compounds. Fett Wiss. Technol. Sci. Technol. 1991, 93, 49–52. [Google Scholar] [CrossRef]
- Cilliers, J.J.L.; Van Niekerk, P.J. LC determination of phytic acid in food by postcolumn colorimetric detection. J. Agric. Food Chem. 1986, 34, 680–683. [Google Scholar] [CrossRef]
- Rounds, M.A.; Nielsen, S.S. Anion-exchange high-performance liquid chromatography with post-column detection for the analysis of phytic acid and other inositol phosphates. J. Chromatogr. A 1993, 653, 148–152. [Google Scholar] [CrossRef]
- Bos, K.D.; Verbeek, C.; Van Eeden, C.H.P.; Slump, P.; Wolters, M.G.E. Improved determination of phytate by ion-exchange chromatography. J. Agric. Food Chem. 1991, 39, 1770–1772. [Google Scholar] [CrossRef]
- Li, N.; Hefferren, J.J.; Li, K. Quantitative Chemical Analysis; World Scientific: Singapore, 2013; ISBN 978-981-4452-28-1. [Google Scholar]
- Meek, J.L. Inositol bis-, tris-, and tetrakis(phosphate)s: Analysis in tissues by HPLC. Proc. Natl. Acad. Sci. USA 1986, 83, 4162–4166. [Google Scholar] [CrossRef] [PubMed]
- Mayr, G.W. A novel metal-dye detection system permits picomolar-range h.p.l.c. analysis of inositol polyphosphates from non-radioactively labelled cell or tissue specimens. Biochem. J. 1988, 254, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Guse, A.H.; Emmrich, F. T-cell receptor-mediated metabolism of inositol polyphosphates in Jurkat T-lymphocytes. Identification of a D-myo-inositol 1,2,3,4,6-pentakisphosphate-2-phosphomonoesterase activity, a D-myo-inositol 1,3,4,5,6-pentakisphosphate-1/3-phosphatase activity an. J. Biol. Chem. 1991, 266, 24498–24502. [Google Scholar] [CrossRef] [PubMed]
- Mayr, G.W.; Thieleczek, R. Masses of inositol phosphates in resting and tetanically stimulated vertebrate skeletal muscles. Biochem. J. 1991, 280, 631–640. [Google Scholar] [CrossRef]
- Freund, W.-D.; Mayr, G.W.; Tietz, C.; Schultz, J.E. Metabolism of inositol phosphates in the protozoan Paramecium. Characterization of a novel inositol-hexakisphosphate-dephosphorylating enzyme. Eur. J. Biochem. 1992, 207, 359–367. [Google Scholar] [CrossRef]
- Schlemmer, U.; Jany, K.-D.; Berk, A.; Schulz, E.; Rechkemmer, G. Degradation of phytate in the gut of pigs—Pathway of gastrointestinal inositol phosphate hydrolysis and enzymes involved. Arch. Tierernaehrung 2001, 55, 255–280. [Google Scholar] [CrossRef]
- Guse, A.H.; Goldwich, A.; Weber, K.; Mayr, G.W. Non-radioactive, isomer-specific inositol phosphate mass determinations: High-performance liquid chromatography-micro-metal-dye detection strongly improves speed and sensitivity of analyses from cells and micro-enzyme assays. J. Chromatogr. B Biomed. Sci. Appl. 1995, 672, 189–198. [Google Scholar] [CrossRef]
- Skoglund, E.; Carlsson, N.G.; Sandberg, A.S. Determination of Isomers of Inositol Mono- to Hexaphosphates in Selected Foods and Intestinal Contents Using High-Performance Ion Chromatography. J. Agric. Food Chem. 1997, 431–436. [Google Scholar] [CrossRef]
- Phillippy, B.Q.; Bland, J.M. Gradient ion chromatography of inositol phosphates. Anal. Biochem. 1988, 175, 162–166. [Google Scholar] [CrossRef]
- Phillippy, B.Q.; Bland, J.M.; Evens, T.J. Ion Chromatography of Phytate in Roots and Tubers. J. Agric. Food Chem. 2003, 51, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Skoglund, E.; Carlsson, N.G.; Sandberg, A.S. High-Performance Chromatographic Separation of Inositol Phosphate Isomers on Strong Anion Exchange Columns. J. Agric. Food Chem. 1998, 46, 1877–1882. [Google Scholar] [CrossRef]
- Smith, R.E.; MacQuarrie, R.A. Determination of inositol phosphates and other biologically important anions by ion chromatography. Anal. Biochem. 1988, 170, 308–315. [Google Scholar] [CrossRef]
- Smith, R.E.; MacQuarrie, R.A.; Jope, R.S. Determination of Inositol Phosphates and Other Anions in Rat Brain. J. Chromatogr. Sci. 1989, 27, 491–495. [Google Scholar] [CrossRef]
- Talamond, P.; Gallon, G.; Treche, S. Rapid and sensitive liquid chromatographic method using a conductivity detector for the determination of phytic acid in food. J. Chromatogr. A 1998, 805, 143–147. [Google Scholar] [CrossRef]
- Skoglund, E.; Carlsson, N.G.; Sandberg, A.S. Analysis of Inositol Mono- and Diphosphate Isomers Using High-Performance Ion Chromatography and Pulsed Amperometric Detection. J. Agric. Food Chem. 1997, 45, 4668–4673. [Google Scholar] [CrossRef]
- Guse, A.H.; Emmrich, F. Determination of inositol polyphosphates from human T-lymphocyte cell lines by anion-exchange high-performance liquid chromatography and post-column derivatization. J. Chromatogr. A 1992, 593, 157–163. [Google Scholar] [CrossRef]
- Hull, S.R.; Montgomery, R. myo-Inositol Phosphates in Corn Steep Water. J. Agric. Food Chem. 1995, 43, 1516–1523. [Google Scholar] [CrossRef]
- Chen, Q.-C.; Li, B.W. Separation of phytic acid and other related inositol phosphates by high-performance ion chromatography and its applications. J. Chromatogr. A 2003, 1018, 41–52. [Google Scholar] [CrossRef]
- Michalski, R. Ion Chromatography Applications in Wastewater Analysis. Separations 2018, 5, 16. [Google Scholar] [CrossRef]
- Chen, Q. Determination of Phytic Acid and Inositol Pentakisphosphates in Foods by High-Performance Ion Chromatography. J. Agric. Food Chem. 2004, 52, 4604–4613. [Google Scholar] [CrossRef]
- Harland, B.F.; Smikle-Williams, S.; Oberleas, D. High performance liquid chromatography analysis of phytate (IP6) in selected foods. J. Food Compos. Anal. 2004, 17, 227–233. [Google Scholar] [CrossRef]
- Pourghasem, G.B.; Mahboub, S.A.; Razavieh, S.V. Phytic acid and its molar ratio to zinc in consumed breads in tabriz. J. Urmia Univ. Med. Sci. 2005, 16, 136–142. [Google Scholar]
- Sekiguchi, Y.; Matsunaga, A.; Yamamoto, A.; Inoue, Y. Analysis of condensed phosphates in food products by ion chromatography with an on-line hydroxide eluent generator. J. Chromatogr. A 2000, 881, 639–644. [Google Scholar] [CrossRef]
- Shintani, H.; Dasgupta, P.K. Gradient anion chromatography with hydroxide and carbonate eluents using simultaneous conductivity and pH detection. Anal. Chem. 1987, 59, 802–808. [Google Scholar] [CrossRef]
- Matsunaga, A.; Yamamoto, A.; Kurokawa, H.; Sekiguchi, Y. Determination of condensed phosphates in food stuffs by ion chromatography. J. Food Hyg. Soc. Japan 1998, 39, 1–6. [Google Scholar] [CrossRef][Green Version]
- Strong, D.L.; Dasgupta, P.K.; Friedman, K.; Stillian, J.R. Electrodialytic Eluent Production and Gradient Generation in Ion Chromatography. Anal. Chem. 1991, 63, 480–486. [Google Scholar] [CrossRef]
- Blaabjerg, K.; Hansen-Møller, J.; Poulsen, H.D. High-performance ion chromatography method for separation and quantification of inositol phosphates in diets and digesta. J. Chromatogr. B 2010, 878, 347–354. [Google Scholar] [CrossRef]
- Blaabjerg, K.; Poulsen, H.D. Microbial phytase and liquid feeding increase phytate degradation in the gastrointestinal tract of growing pigs. Livest. Sci. 2010, 134, 88–90. [Google Scholar] [CrossRef]
- Liu, X.; Villalta, P.W.; Sturla, S.J. Simultaneous determination of inositol and inositol phosphates in complex biological matrices: Quantitative ion-exchange chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2009, 23, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Kindt, E.; Shum, Y.; Badura, L.; Snyder, P.J.; Brant, A.; Fountain, S.; Szekely-Klepser, G. Development and validation of an LC/MS/MS procedure for the quantification of endogenous myo-inositol concentrations in rat brain tissue homogenates. Anal. Chem. 2004, 76, 4901–4908. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Jaisi, D.P. Distribution of inositol phosphates in animal feed grains and excreta: Distinctions among isomers and phosphate oxygen isotope compositions. Plant. Soil 2018, 430, 291–305. [Google Scholar] [CrossRef]
- von Sperber, C.; Tamburini, F.; Brunner, B.; Bernasconi, S.M.; Frossard, E. The oxygen isotope composition of phosphate released from phytic acid by the activity of wheat and Aspergillus niger phytase. Biogeosciences 2015, 12, 4175–4184. [Google Scholar] [CrossRef]
- Ito, M.; Fujii, N.; Wittwer, C.; Sasaki, A.; Tanaka, M.; Bittner, T.; Jessen, H.J.; Saiardi, A.; Takizawa, S.; Nagata, E. Hydrophilic interaction liquid chromatography–tandem mass spectrometry for the quantitative analysis of mammalian-derived inositol poly/pyrophosphates. J. Chromatogr. A 2018, 1573, 87–97. [Google Scholar] [CrossRef]
- Kennington, A.S.; Hill, C.R.; Craig, J.; Bogardus, C.; Raz, I.; Ortmeyer, H.K.; Hansen, B.C.; Romero, G.; Larner, J. Low Urinary chiro -Inositol Excretion in Non-Insulin-Dependent Diabetes Mellitus. N. Engl. J. Med. 1990, 323, 373–378. [Google Scholar] [CrossRef]
- Haga, H.; Nakajima, T. Determination of polyol profiles in human urine by capillary gas chromatography. Biomed. Chromatogr. 1989, 3, 68–71. [Google Scholar] [CrossRef]
- Jansen, G.; Muskiet, F.A.J.; Schierbeek, H.; Berger, R.; van der Slik, W. Capillary gas chromatographic profiling of urinary, plasma and erythrocyte sugars and polyols as their trimethylsilyl derivatives, preceded by a simple and rapid prepurification method. Clin. Chim. Acta 1986, 157, 277–293. [Google Scholar] [CrossRef]
- de Koning, A.J. Determination of myo-inositol and phytic acid by gas chromatography using scyllitol as internal standard. Analyst 1994, 119, 1319. [Google Scholar] [CrossRef]
- Roberts, R.N.; Johnston, J.A.; Fuhr, B.W. A method for the quantitative estimation of myoinositol by gas-liquid chromatography. Anal. Biochem. 1965, 10, 282–289. [Google Scholar] [CrossRef]
- March, J.; Simonet, B..; Grases, F. Determination of phytic acid by gas chromatography–mass spectroscopy: Application to biological samples. J. Chromatogr. B Biomed. Sci. Appl. 2001, 757, 247–255. [Google Scholar] [CrossRef]
- March, J.G.; Forteza, R.; Grases, F. Determination of inositol isomers and arabitol in human urine by gas chromatography-mass spectrometry. Chromatographia 1996, 42, 329–331. [Google Scholar] [CrossRef]
- Park, H.-R.; Ahn, H.-J.; Kim, S.-H.; Lee, C.-H.; Byun, M.-W.; Lee, G.-W. Determination of the phytic acid levels in infant foods using different analytical methods. Food Control. 2006, 17, 727–732. [Google Scholar] [CrossRef]
- Harland, B.F.; Oberleas, D.; Ellis, R.; Gelroth, J.; Gordon, D.; Phillips, K.; Ranhotra, G.; Shah, B.G.; Stoecker, B.; Trick, K.D.; et al. Anion-Exchange Method for Determination of Phytate in Foods: Collaborative Study. J. AOAC Int. 1986, 69, 667–670. [Google Scholar] [CrossRef]
- Lehrfeld, J.; Morris, E.R. Overestimation of phytic acid in foods by the AOAC anion-exchange method. J. Agric. Food Chem. 1992, 40, 2208–2210. [Google Scholar] [CrossRef]
- Schormüller, J.; Würdig, G. Über das Vorkommen von Phytin, insbesondere in Getreide und Getreideprodukten. Deut. Leb. Rundschau 1957, 53, 1. [Google Scholar]
- Angyal, S.J.; Russell, A.F. Cyclitols. XXVIII. Methyl esters of inositol phosphates. The structure of phytic acid. Aust. J. Chem. 1969, 22, 383. [Google Scholar] [CrossRef]
- Emilsson, A.; Sundler, R. Differential activation of phosphatidylinositol deacylation and a pathway via diphosphoinositide in macrophages responding to zymosan and ionophore A23187. J. Biol. Chem. 1984, 259, 3111–3116. [Google Scholar] [CrossRef]
- Kates, M. Techniques of Lipidology. Laboratory Techniques in Biochemistry and Molecular Biology; Laboratory Techniques in Biochemistry and Molecular Biology; North-Holland Publishing Company: Amsterdam, The Netherlands, 1972; Volume 3, p. 267. ISBN 9780444103505. [Google Scholar]
- Hatzack, F.; Rasmussen, S.K. High-performance thin-layer chromatography method for inositol phosphate analysis. J. Chromatogr. B Biomed. Sci. Appl. 1999, 736, 221–229. [Google Scholar] [CrossRef]
- Bandurski, R.S.; Axelrod, B. The chromatographic identification of some biologically important phosphate esters. J. Biol. Chem. 1951, 193, 405–410. [Google Scholar]
- Shi, J.; Wang, H.; Hazebroek, J.; Ertl, D.S.; Harp, T. The maize low-phytic acid 3 encodes a myo-inositol kinase that plays a role in phytic acid biosynthesis in developing seeds. Plant. J. 2005, 42, 708–719. [Google Scholar] [CrossRef] [PubMed]
- Arnold, P.W. Paper ionophoresis of inositol phosphates, with a note on the acid hydrolysates of phytic acid. Biochim. Biophys. Acta 1956, 19, 552–554. [Google Scholar] [CrossRef]
- Seiffert, U.B.; Agranoff, B.W. Isolation and separation of inositol phosphates from hydrolysates of rat tissues. Biochim. Biophys. Acta Lipids Lipid Metab. 1965, 98, 574–581. [Google Scholar] [CrossRef]
- Tate, M.E. Separation of myoinositol pentaphosphates by moving paper electrophoresis (MPE). Anal. Biochem. 1968, 23, 141–149. [Google Scholar] [CrossRef]
- Jackson, J.F.; Jones, G.; Linskens, H.F. Phytic acid in pollen. Phytochemistry 1982, 21, 1255–1258. [Google Scholar] [CrossRef]
- Kikunaga, S.; Takahashi, M.; Huzisige, H. Accurate and simple measurement of phytic acid contents in cereal grains. Plant. Cell Physiol. 1985, 26, 1323–1330. [Google Scholar] [CrossRef]
- Blatny, P.; Kvasnicka, F.; Kenndler, E. Determination of Phytic Acid in Cereal Grains, Legumes, and Feeds by Capillary Isotachophoresis. J. Agric. Food Chem. 1995, 43, 129–133. [Google Scholar] [CrossRef]
- Nardi, A.; Cristalli, M.; Desiderio, C.; Ossicini, L.; Shukla, S.K.; Fanali, S. Indirect UV photometric detection in capillary zone electrophoresis for the determination of phytate in soybeans. J. Microcolumn Sep. 1992, 4, 9–11. [Google Scholar] [CrossRef]
- Kvasnička, F.; Ševčík, R.; Voldřich, M.; Krátká, J. Determination of aristolochic acid by capillary zone electrophoresis. Open Chem. 2004, 2, 417–424. [Google Scholar] [CrossRef]
- Kvasnička, F.; Čopíková, J.; Ševčík, R.; Václavíková, E.; Synytsya, A.; Vaculová, K.; Voldřich, M. Determination of phytic acid and inositolphosphates in barley. Electrophoresis 2011, 32, 1090–1093. [Google Scholar] [CrossRef]
- Losito, O.; Szijgyarto, Z.; Resnick, A.C.; Saiardi, A. Inositol pyrophosphates and their unique metabolic complexity: Analysis by gel electrophoresis. PLoS ONE 2009, 4, e5580. [Google Scholar] [CrossRef]
- Alimohammadi, M.; Ali, N.; Khodakovskaya, M. Quantitative Detection of Inositol Hexakisphosphate (InsP6) in Crop Plants Using Polyacrylamide Gel Electrophoresis (PAGE). Am. J. Plant. Sci. 2013, 4, 1–6. [Google Scholar] [CrossRef]
- Giridhari, A.C.; Sujatha, R.; Deeshma, K.P. Detection and quantification of phytic acid in black pepper variety panniyur-1 using polyacrylamide gel electrophoresis. J. Trop. Agric. 2017, 55, 96–98. [Google Scholar]
- Wilson, M.S.C.; Bulley, S.J.; Pisani, F.; Irvine, R.F.; Saiardi, A. A novel method for the purification of inositol phosphates from biological samples reveals that no phytate is present in human plasma or urine. Open Biol. 2015, 5, 150014. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.; Saiardi, A. Inositol Phosphates Purification Using Titanium Dioxide Beads. Bio Protoc. 2018, 8. [Google Scholar] [CrossRef]
- Qiu, D.; Wilson, M.S.; Eisenbeis, V.B.; Harmel, R.K.; Riemer, E.; Haas, T.M.; Wittwer, C.; Jork, N.; Gu, C.; Shears, S.B.; et al. Analysis of inositol phosphate metabolism by capillary electrophoresis electrospray ionization mass spectrometry. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Pavlovic, I.; Thakor, D.T.; Vargas, J.R.; McKinlay, C.J.; Hauke, S.; Anstaett, P.; Camunã, R.C.; Bigler, L.; Gasser, G.; Schultz, C.; et al. Cellular delivery and photochemical release of a caged inositol-pyrophosphate induces PH-domain translocation in cellulo. Nat. Commun. 2016, 7, 10622. [Google Scholar] [CrossRef]
- Harmel, R.K.; Puschmann, R.; Nguyen Trung, M.; Saiardi, A.; Schmieder, P.; Fiedler, D. Harnessing 13 C-labeled myo -inositol to interrogate inositol phosphate messengers by NMR. Chem. Sci. 2019, 10, 5267–5274. [Google Scholar] [CrossRef]
- Lolas, G.M.; Palamidis, N.; Markakis, P. Phytic acid total phosphorus relationship relationship in barley, oats, soybeans and wheat. Cereal Chem. 1976, 53, 867–871. [Google Scholar]
- March, J.G.; Villacampa, A.I.; Grases, F. Enzymatic—spectrophotometric determination of phytic acid with phytase from Aspergillus ficuum. Anal. Chim. Acta 1995, 300, 269–272. [Google Scholar] [CrossRef]
- Raheja, R.K.; Kaur, C.; Singh, A.; Bhatia, I.S. New calorimetric method for the quantitative estimation of phospholipids without acid digestion. J. Lipid Res. 1973, 14, 695–697. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.I.; Perera, P.A.J.; Hafez, Y.S. New Chromophore for Phytic Acid Determination. Cereal Chem. 1986, 63, 475–478. [Google Scholar]
- Santiviago, C.; Peralta, J.; López, I. A simple spectrophotometric method to determine phytic acid in poultry wastewater without acid digestion. Int. J. Environ. Anal. Chem. 2020, 1–11. [Google Scholar] [CrossRef]
- Carneiro, J.M.T.; Zagatto, E.A.G.; Santos, J.L.M.; Lima, J.L.F.C. Spectrophotometric determination of phytic acid in plant extracts using a multi-pumping flow system. Anal. Chim. Acta 2002, 474, 161–166. [Google Scholar] [CrossRef]
- Agostinho, A.J.; de Souza Oliveira, W.; Anunciação, D.S.; Santos, J.C.C. Simple and Sensitive Spectrophotometric Method for Phytic Acid Determination in Grains. Food Anal. Methods 2016, 9, 2087–2096. [Google Scholar] [CrossRef]
- Lee, M.H. Official Methods of Analysis of AOAC International, 16th ed.; AOAC International: Rockville, MD, USA, 1995; Volume I, ISBN 0935584544. [Google Scholar]
- Frølich, W.; Drakenberg, T.; Asp, N.-G. Enzymic degradation of phytate (myo-inositol Hexaphosphate) in whole grain flour suspension and dough. A comparison between 31P NMR spectroscopy and a ferric ion method. J. Cereal Sci. 1986, 4, 325–334. [Google Scholar] [CrossRef]
- McKie, V.A.; McCleary, B. V A Novel and Rapid Colorimetric Method for Measuring Total Phosphorus and Phytic Acid in Foods and Animal Feeds. J. AOAC Int. 2016, 99, 738–743. [Google Scholar] [CrossRef]
- Perera, I.; Fukushima, A.; Arai, M.; Yamada, K.; Nagasaka, S.; Seneweera, S.; Hirotsu, N. Identification of low phytic acid and high Zn bioavailable rice (Oryza sativa L.) from 69 accessions of the world rice core collection. J. Cereal Sci. 2019, 85, 206–213. [Google Scholar] [CrossRef]
- Kamaya, M.; Furuki, T.; Nagashima, K.; Ishii, E.; Saito, H. Indirect spectrophotometric determination of phytic acid with zinc chloranilate. Phytochem. Anal. 1995, 6, 251–254. [Google Scholar] [CrossRef]
- March, J.G.; Simonet, B.M.; Grases, F. Fluorimetric determination of phytic acid based on the activation of the oxidation of 2,2′-dipyridyl ketone hydrazone catalysed by Cu(II). Analyst 1999, 124, 897–900. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, J.; Ma, K.; Cao, S.; Chen, X. Fluorimetric determination of phytic acid in urine based on replacement reaction. Anal. Chim. Acta 2007, 605, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Dong, N.; Chen, J. Synchronous fluorescence determination of phytic acid in FOODSTUFFS and urine based on replacement reaction. Phytochem. Anal. 2011, 22, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Kolozsvari, B.; Parisi, F.; Saiardi, A. Inositol phosphates induce DAPI fluorescence shift. Biochem. J. 2014, 460, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Kolozsvari, B.; Firth, S.; Saiardi, A. Raman Spectroscopy Detection of Phytic Acid in Plant Seeds Reveals the Absence of Inorganic Polyphosphate. Mol. Plant. 2015, 8, 826–828. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, Y.; Chen, J.; Luo, Z.; Ma, K.; Chen, X. Synchronous fluorescence analysis of phytate in food. Microchim. Acta 2009, 164, 35–40. [Google Scholar] [CrossRef]
- Qu, Z.; Na, W.; Nie, Y.; Su, X. A novel fluorimetric sensing strategy for highly sensitive detection of phytic acid and hydrogen peroxide. Anal. Chim. Acta 2018, 1039, 74–81. [Google Scholar] [CrossRef]
- Bebot-Brigaud, A.; Dange, C.; Fauconnier, N.; Gérard, C. 31P NMR, potentiometric and spectrophotometric studies of phytic acid ionization and complexation properties toward Co2+, Ni2+, Cu2+, Zn2+ and Cd2+. J. Inorg. Biochem. 1999, 75, 71–78. [Google Scholar] [CrossRef]
- Šala, M.; Makuc, D.; Kolar, J.; Plavec, J.; Pihlar, B. Potentiometric and 31P NMR studies on inositol phosphates and their interaction with iron(III) ions. Carbohydr. Res. 2011, 346, 488–494. [Google Scholar] [CrossRef]
- Tse, R.S.; Wong, S.C.; Yuen, C.P. Determination of deuterium/hydrogen ratios in natural waters by Fourier transform nuclear magnetic resonance spectrometry. Anal. Chem. 1980, 52, 2445. [Google Scholar] [CrossRef]
- Harland, B.F.; Oberleas, D. A modified method for phytate analysis using an ion-exchange procedure: Application to textured vegetable proteins [Soybeans]. Cereal Chem. 1977, 54, 827–832. [Google Scholar]
- Mazzola, E.P.; Phillippy, B.Q.; Harland, B.F.; Miller, T.H.; Potemra, J.M.; Katsimpiris, E.W. Phosphorus-31 nuclear magnetic resonance spectroscopic determination of phytate in foods. J. Agric. Food Chem. 1986, 34, 60–62. [Google Scholar] [CrossRef]
- Johnson, K.; Barrientos, L.G.; Le, L.; Murthy, P.P.N. Application of Two-Dimensional Total Correlation Spectroscopy for Structure Determination of Individual Inositol Phosphates in a Mixture. Anal. Biochem. 1995, 231, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.L. Inositol phosphates in soil: Amounts, forms and significance of the phosphorylated inositol stereoisomers. In Inositol Phosphates: Linking Agriculture and the Environment; CABI: Wallingford, UK, 2007; pp. 186–206. [Google Scholar]
- Doolette, A.L.; Smernik, R.J.; Dougherty, W.J. Overestimation of the importance of phytate in NaOH–EDTA soil extracts as assessed by 31P NMR analyses. Org. Geochem. 2011, 42, 955–964. [Google Scholar] [CrossRef]
- Wu, J.; Paudel, P.; Sun, M.; Joshi, S.R.; Stout, L.M.; Greiner, R.; Jaisi, D.P. Mechanisms and Pathways of Phytate Degradation: Evidence from Oxygen Isotope Ratios of Phosphate, HPLC, and Phosphorus-31 NMR Spectroscopy. Soil Sci. Soc. Am. J. 2015, 79, 1615–1628. [Google Scholar] [CrossRef]
- Watson, F.T.; Smernik, R.J.; Doolette, A.L. Thermal degradation of phytate produces all four possible inositol pentakisphosphates as determined by ion chromatography and 1 H and 31 P NMR spectroscopy. Phosphorus. Sulfur. Silicon Relat. Elem. 2019, 194, 1140–1148. [Google Scholar] [CrossRef]
- Plaami, S.; Kumpulainen, J. Determination of Phytic Acid in Cereals Using ICP-AES to Determine Phosphorus. J. AOAC Int. 1991, 74, 32–36. [Google Scholar] [CrossRef]
- Saastamoinen, M.; Plaami, S.; Kumpulainen, J. β-Glucan and Phytic Acid Content of Oats Cultivated in Finland. Acta Agric. Scand. Sect. B Soil Plant. Sci. 1992, 42, 6–11. [Google Scholar] [CrossRef]
- Grases, F.; Llobera, A. Determination of Phytic Acid in Urine by ICP Atomic Emission Spectrometry. Anal. Lett. 1996, 29, 1193–1199. [Google Scholar] [CrossRef]
- Grases, F.; Perelló, J.; Isern, B.; Prieto, R.M. Determination of myo-inositol hexakisphosphate (phytate) in urine by inductively coupled plasma atomic emission spectrometry. Anal. Chim. Acta 2004, 510, 41–43. [Google Scholar] [CrossRef]
- Muñoz, J.A.; Valiente, M. Determination of Phytic Acid in Urine by Inductively Coupled Plasma Mass Spectrometry. Anal. Chem. 2003, 75, 6374–6378. [Google Scholar] [CrossRef]
- Muñoz, J.A.; López-Mesas, M.; Valiente, M. Minimum handling method for the analysis of phosphorous inhibitors of urolithiasis (pyrophosphate and phytic acid) in urine by SPE-ICP techniques. Anal. Chim. Acta 2010, 658, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; He, L.; Valiente, M.; López-Mesas, M. Fast determination of bioactive phytic acid and pyrophosphate in walnuts using microwave accelerated extraction. Food Chem. 2017, 221, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Mak, W.C.; Ng, Y.M.; Chan, C.; Kwong, W.K.; Renneberg, R. Novel biosensors for quantitative phytic acid and phytase measurement. Biosens. Bioelectron. 2004, 19, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Caseli, L.; Moraes, M.L.; Zucolotto, V.; Ferreira, M.; Nobre, T.M.; Zaniquelli, M.E.D.; Rodrigues Filho, U.P.; Oliveira, O.N. Fabrication of Phytic Acid Sensor Based on Mixed Phytase−Lipid Langmuir−Blodgett Films. Langmuir 2006, 22, 8501–8508. [Google Scholar] [CrossRef] [PubMed]
- Troitsky, V.I.; Berzina, T.S.; Pastorino, L.; Bernasconi, E.; Nicolini, C. A new approach to the deposition of nanostructured biocatalytic films. Nanotechnology 2003, 14, 597–602. [Google Scholar] [CrossRef]
- Moraes, M.L.; Oliveira, O.N., Jr.; Filho, U.P.R.; Ferreira, M. Phytase immobilization on modified electrodes for amperometric biosensing. Sens. Actuators B Chem. 2008, 131, 210–215. [Google Scholar] [CrossRef]
- Moraes, M.L.; Maki, R.M.; Paulovich, F.V.; Rodrigues Filho, U.P.; de Oliveira, M.C.F.; Riul, A., Jr.; de Souza, N.C.; Ferreira, M.; Gomes, H.L.; Oliveira, O.N. Strategies to Optimize Biosensors Based on Impedance Spectroscopy to Detect Phytic Acid Using Layer-by-Layer Films. Anal. Chem. 2010, 82, 3239–3246. [Google Scholar] [CrossRef]
- Esmaeili, C.; Norouzi, P.; Zar, M.S.; Eskandari, M.; Faridbod, F.; Ganjali, M.R. A FFT Square Wave Voltammetry Sensing Method for Highly Sensitive Detection of Phytic Acid Using a Cerium Oxide Nanoparticles Decorated Graphene Oxide. J. Electrochem. Soc. 2019, 166, B1630–B1636. [Google Scholar] [CrossRef]
- Shi, H.; Zhang, A.; Du, H.; Zhang, M.; Zhang, Y.; Huang, H.; Xiao, Y.; Zhang, Y.; He, X.; Wang, K. A novel fluorescent nanosensor based on small-sized conjugated polyelectrolyte dots for ultrasensitive detection of phytic acid. Talanta 2019, 202, 214–220. [Google Scholar] [CrossRef]
- Lee, M.; Moon, J.H.; Jun, E.J.; Kim, G.; Kwon, Y.-U.; Lee, J.Y.; Yoon, J. A tetranaphthoimidazolium receptor as a fluorescent chemosensor for phytate. Chem. Commun. 2014, 50, 5851–5853. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, X.; Wen, Y.; Zheng, Y.; Duan, G.; Zhang, Z.; Yang, H. Phytic Acid-Based Layer-by-Layer Assembly for Fabrication of Mesoporous Gold Film and Its Biosensor Application. J. Electrochem. Soc. 2010, 157, K5. [Google Scholar] [CrossRef]
- Dai, H.; Wang, N.; Wang, D.; Ma, H.; Lin, M. An electrochemical sensor based on phytic acid functionalized polypyrrole/graphene oxide nanocomposites for simultaneous determination of Cd(II) and Pb(II). Chem. Eng. J. 2016, 299, 150–155. [Google Scholar] [CrossRef]
- Wang, K.; Liu, P.; Ye, Y.; Li, J.; Zhao, W.; Huang, X. Fabrication of a novel laccase biosensor based on silica nanoparticles modified with phytic acid for sensitive detection of dopamine. Sens. Actuators B Chem. 2014, 197, 292–299. [Google Scholar] [CrossRef]
- Yang, L.; Wang, H.; Lü, H.; Hui, N. Phytic acid doped poly(3,4-ethylenedioxythiophene) modified with copper nanoparticles for enzymeless amperometric sensing of glucose. Microchim. Acta 2020, 187, 49. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methodology | Detection | Analyte | Sample Material | Sample Preparation | LOD 1 (S/N = 3) | Year, Ref. |
|---|---|---|---|---|---|---|
| Precipitation titration (Fe3+) | Endpoint color indication (NH4SCN) | InsP6 | Cereals | HCl extraction | 0.3 mg/g | 1914 [33] |
| Precipitation (Fe3+), digestion (Kjeldahl) | Colorimetric as PO43− (molybdenum blue) | InsP6 | Dried food | HCl extraction | 0.1 mg/g 2 | 1935 [37] |
| Precipitation (Fe3+) | Colorimetric by residual Fe3+ (2,2′-bipyridine) | InsP6 | Soya, cereals | HCl extraction | 2 µg/g | 1983 [43] |
| Differential titrimetric standardization (OH−) | Potentiometric by derivatives (∂pH/∂nOH-) | InsP6 | Phytate salt | Cation-exchange chromatography | 1 µmol | 2015 [50] |
| Ion-pair HPLC (C18 RP column) | Refractive index | InsP3–InsP6 | Legumes | Anion-exchange chromatography | 100 mg/L | 1995 [66] |
| Ion-pair HPLC (10-SAX column) | Fluorescence by isotopic labeling (32P) | InsP1–InsP4, phosphates | Cultured cells | Charcoal pretreatment | 0.2 nmol | 1989 [71] |
| HPLC (10 SAX WCS) | Radioactivity by enzymatic 32P-labeling | InsP6 | HeLa cells, rat tissue, serum, etc. | Trichloroacetic acid extraction, centrifugation | 0.25 pmol (5 nM) | 2008 [73] |
| HILIC (HILICpak VG-50 column) | Tandem mass spectrometry (HILIC-MS/MS) | InsP6, InsP7 | Human blood, HEK293, mouse brain | Anion-exchange chromatography | 2 pmol | 2018 [119] |
| IC anion-exchange (SAX column) | Colorimetric (post-column reaction by Wade reagent) | InsP2–InsP6 | Plants | HCl extraction | 0.2 mg/g | 1993 [84] |
| IC anion-exchange (Mono Q HR 5/5) | Colorimetric as PO43− (post-column enzymatic hydrolysis) | InsP2–InsP4 stereoisomers | Rat tissue | HClO4 extraction, charcoal | 1 nmol | 1986 [87] |
| IC anion-exchange (Mono 1, HCl elution) | Colorimetric (post-column reaction by Y3+-PAR) | InsP1–InsP6 stereoisomers | Cultured cells, tissue | HClO4 extraction, charcoal | 0.1 µmol | 1995 [93] |
| IC anion-exchange (PAX-100, HCl & NaOH elution) | UV (post-column reaction by Fe(NO3)3); Suppressed conductometry | InsP1–InsP6, isomers of InsP4–InsP5 | Food, digesta | HCl extraction, ion-exchange purification | 0.5 µg | 1998 [97] |
| IC anion-exchange (AS7) | Evaporative light scattering | InsP6 | Roots, tubers | HCl extraction | 1.0 µg (30 µg/g) | 2003 [96] |
| IC anion-exchange (CarboPac PA-10, NaOH elution) | Pulsed amperometric detection (PAD) | Ins, InsP1–InsP2 (isomers) | Food, digesta | HCl extraction, ion-exchange purification | 0.04 pmol | 1997 [101] |
| IC anion-exchange (CarboPac PA-100, HCl/KCl elution) | UV (post-column reaction by Fe(NO3)3) | InsP2–InsP6 (27 isomers) | Nuts, beans | HCl extraction, (solid phase cartridge) | 2 µM | 2003 [104,106] |
| IC anion-exchange (AS-11, online-generated KOH elution) | Suppressed conductometry | InsP3, InsP6, (poly)phosph-ates (P1–P52) | Ham, fish, cheese | Trichloroacetic acid extraction, cation-exchange | 0.15 µM | 2000 [109] |
| IC anion-exchange (PA-1, online MSA elution) | UV (post-column reaction by Fe(NO3)3), ICP-MS (corr. factors) | InsP2–InsP6 (23 isomers) | Wheat, soybean, digesta | HCl extraction, cation-exchange purification | 0.1 mg/L | 2010 [113] |
| IC anion-exchange (Biobasic AX, (NH4)2CO3 elution) | Tandem mass spectrometry (ESI-MS/MS) | InsP1–InsP6, Ins | Nut, grain, cultured cells | Acetic acid & hexane extraction | 0.1 pmol | 2009 [115] |
| IC anion-exchange (CarboPac PA-100, HCl elution) | Isotope ratio-mass spectrometry (δ18O IRMS) | InsP2–InsP6, phosphate | soy, corn, animal excreta | HCl extraction, anion-exchange purification | 20 µg/g | 2018 [117] |
| GC (hydrolysis, derivatization by trimethylsilyl) | Mass spectrometry (MS) | InsP6 | Rat tissue, urine, plasma | Anion-exchange purification | 9 µg/L | 2001 [125] |
| GC (hydrolysis derivatization by HMDS, TMCS) | Flame ionization (FID) | InsP6 | Infant food (flour and paste) | Anion-exchange purification, hexane extraction | 4 ng | 2006 [127] |
| TLC - Thin layer chromatography (cellulose glass plates) | UV (reaction by molybdate reagent) | InsP1–InsP6, organic phosphates | Barley grains | EDTA, diethyl ether, charcoal | 0.1 nmol | 1999 [134] |
| cITP/CZE - Capillary isotachophoresis/Zone electrophoresis | Conductivity detection | InsP6, phosphate | Barley, meat additives | HCl extraction | 20 µg/L | 2004 [144,145] |
| PAGE - Polyacrylamide gel electrophoresis | Toluidine blue, DAPI | InsP4–InsP13, | Tomato, rice, tobacco | HCl extraction, centrifugation | 0.5 nmol | 2009 [146,147] |
| PAGE - Polyacrylamide gel electrophoresis | Toluidine blue | InsP6–InsP8, nucleotides | Mammalian cells, tissue, plasma | TiO2 beads purification | 0.25 nmol | 2015 [149] |
| Spectrophotometric (AOAC method) | Colorimetric as PO43− (acidic/enzymatic hydrolysis) | InsP6 | Various foodstuffs | Ion-exchange purification | 0.9 mg/g | 1986 [128] |
| Fluorimetric | Cu2+ catalyzed oxidation of 2,2-dipyridyl ketone | InsP6 | Oat, wheat, grape, almond | HCl extraction, anion-exchange purification | 30 µg/L | 1999 [166] |
| NMR | 2-dimensional techniques (31P, 1H) | InsP1–InsP6 stereoisomers | Soils | Alkaline extraction | 3 µg/g | 2007 [179] |
| ICP-AES | Atomic emission spectroscopy | InsP6 | Urine | Anion-exchange purification | 64 µg/L | 2004 [186] |
| ICP-MS | Mass spectrometry | InsP6 | Walnuts | Microwave extraction (H2SO4/HCl) | 5 µg/L | 2017 [189] |
| Enzymatic sensor | Amperometry | InsP6 | InsP6 standard | Not required | 1 mg/L | 2004 [190] |
| Enzymatic sensor (CeO2NPs, graphene oxide, glassy carbon) | FFT-SWV (Fast Fourier transform-Square wave voltammetry) | InsP6 | Corn flour | Not reported | 0.07 ng/g | 2019 [195] |
| Fluorescence nanoprobe | Fluorescence quenching (Fe3+, polyelectrolyte dots) | InsP6 | Live cells | Digestion (trypsin) | 7 µg/L | 2019 [196] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marolt, G.; Kolar, M. Analytical Methods for Determination of Phytic Acid and Other Inositol Phosphates: A Review. Molecules 2021, 26, 174. https://doi.org/10.3390/molecules26010174
Marolt G, Kolar M. Analytical Methods for Determination of Phytic Acid and Other Inositol Phosphates: A Review. Molecules. 2021; 26(1):174. https://doi.org/10.3390/molecules26010174
Chicago/Turabian StyleMarolt, Gregor, and Mitja Kolar. 2021. "Analytical Methods for Determination of Phytic Acid and Other Inositol Phosphates: A Review" Molecules 26, no. 1: 174. https://doi.org/10.3390/molecules26010174
APA StyleMarolt, G., & Kolar, M. (2021). Analytical Methods for Determination of Phytic Acid and Other Inositol Phosphates: A Review. Molecules, 26(1), 174. https://doi.org/10.3390/molecules26010174