Protein X-ray Crystallography and Drug Discovery



Abstract
1. Introduction
2. Theoretical Aspects
3. Workflow
- High-throughput screening (HTS) allows one to experimentally screen large chemical libraries, if an appropriate assay is available, resulting in a collection of potential ligands. Preliminary confirmation of direct binding to the protein target using biophysical methods will help to select the most appropriate compounds for structural studies.
- It is quite common that in the course of the characterization of a protein target, some knowledge is gained on potential ligands, such as substrate or cofactor analogues, which are often potent inhibitors.
- Success in target structure determination opens access to virtual screening methods, which aim at identifying potential ligands of a target using computational methods. This step serves often as a preliminary filter to identify a few hundred potential binders among huge virtual libraries, in order to reduce the experimental work needed to confirm the interaction. Experimentally confirmed ligands are then used in structural studies.
- Recently, fragment-based drug discovery (FBDD), also known as fragment-based drug design, and fragment-based ligand/lead discovery (FBLD) has emerged as a powerful tool to identify ligands, albeit with poor affinity given the reduced molecular weight of fragments (about 150–250 Da, containing 10 to 20 non-hydrogen atoms). With such compounds, detection of binding requires sensitive techniques, and X-ray crystallography has proved to be very effective at identifying weak binders, with affinity as low as a few mM. In most favorable cases, fragment libraries of a few hundreds of compounds can be screened using X-ray crystallography, delivering dozens of structures of complexes [11,25]. Although FBDD was initially used as an alternative approach to HTS, i.e., for targets without hits, it appears that these two methodologies are more and more often used in parallel. Despite numerous fragment-based screening technologies existing, X-ray crystallography and protein-based NMR are getting more and more popular since they provide direct and experimental structural information on fragment mode of binding. More recently, the concept of fragment-based screening has been extended to crystallographic screening of ultra-low-molecular weights compounds, called MiniFrags and typically containing 5 to 7 non-hydrogen atoms. Given the low affinity of these compounds, this new crystal-soaking methodology requires working at very high concentrations, typically 1 M [26]. As of October 2018, more than 40 FBLD-derived drugs have been in clinical development, three of them having been approved for clinical use (Table 1).
4. Practical Aspects
4.1. Crystallizing the Target
4.2. Preparation of Crystals of Complexes
4.2.1. Soaking
4.2.2. Co-crystallization
4.3. Crystal Mounting and Data Collection
4.4. Data Processing, Structure Determination, and Refinement
5. Potential Pitfalls
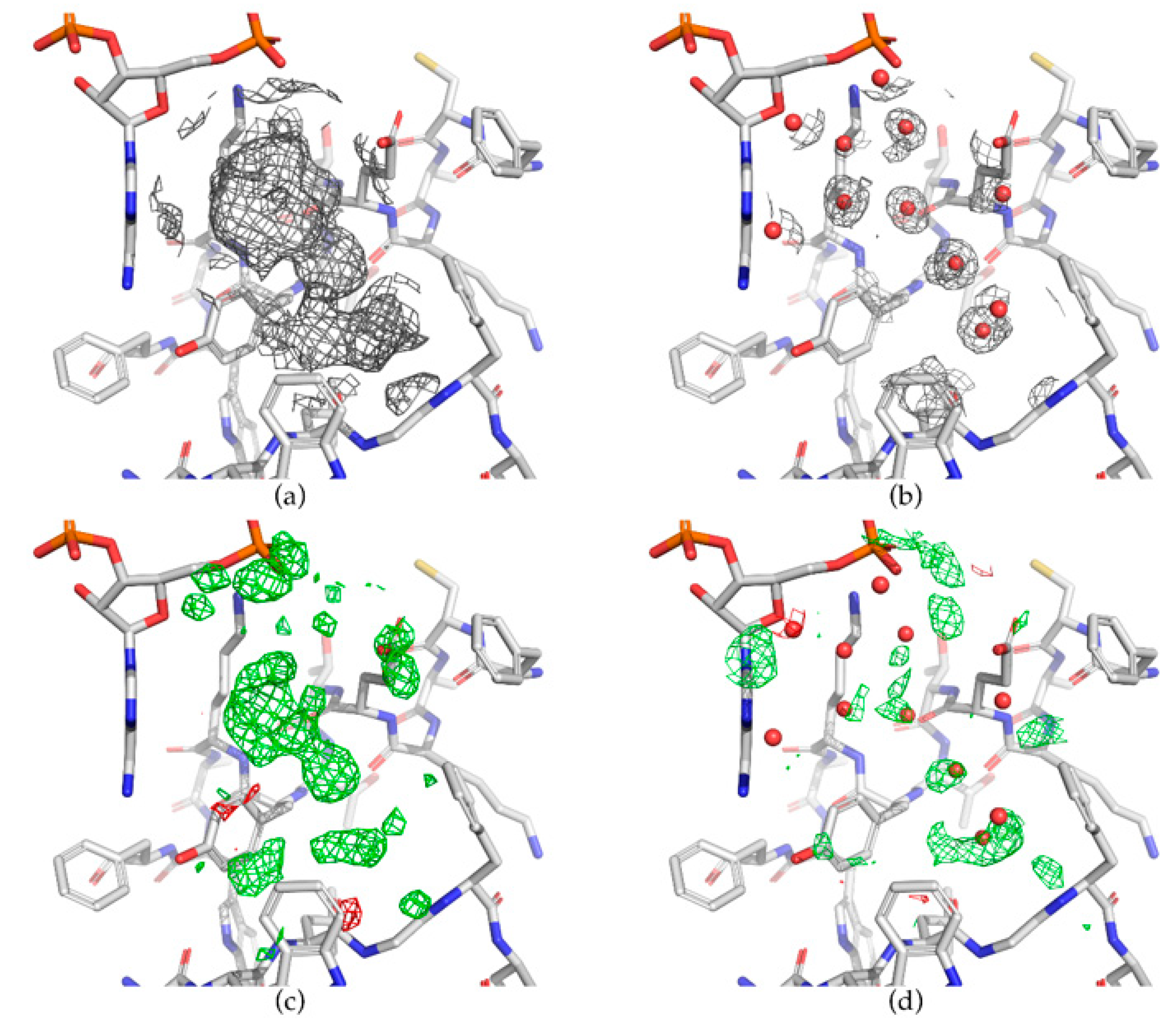
6. Examples
7. Discussion-Comparison with other Methods: serial Crystallography, cryo-EM, micro-ED, NMR, Neutron Crystallography
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bernal, J.D.; Crowfoot, D. X-ray photographs of crystalline pepsin. Nature 1934, 133, 794–795. [Google Scholar] [CrossRef]
- Clark, G.L.; Corrigan, K.E. The crystal structure of insulin. Phys. Rev. 1932, 40, 639. [Google Scholar] [CrossRef]
- Kendrew, J.C.; Dickerson, R.E.; Strandberg, B.E.; Hart, R.G.; Davies, D.R.; Phillips, D.C.; Shore, V.C. Structure of myoglobin: A three-dimensional fourier synthesis at 2 Å. Resolution. Nature 1960, 185, 422–427. [Google Scholar] [CrossRef]
- Hodgkin, D.C. The X-ray analysis of the structure of penicillin. Adv. Sci. 1949, 6, 85–89. [Google Scholar] [PubMed]
- Aitipamula, S.; Wong, A.B.H.; Kanaujia, P. Evaluating suspension formulations of theophylline cocrystals with artificial sweeteners. J. Pharm. Sci. 2018, 107, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Thakral, N.K.; Zanon, R.L.; Kelly, R.C.; Thakral, S. Applications of powder X-ray diffraction in small molecule pharmaceuticals: Achievements and aspirations. J. Pharm. Sci. 2018, 107, 2969–2982. [Google Scholar] [CrossRef]
- Beddell, C.R.; Goodford, P.J.; Norrington, F.E.; Wilkinson, S.; Wootton, R. Compounds designed to fit a site of known structure in human haemoglobin. Br. J. Pharmacol. 1976, 57, 201–209. [Google Scholar] [CrossRef]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.; Meyer, E.F., Jr.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The protein data bank: A computer-based archival file for macromolecular structures. J. Mol. Biol. 1977, 112, 535–542. [Google Scholar] [CrossRef]
- Hol, W.G.J. Protein crystallography and computer graphics—Toward rational drug design. Angew. Chem. Int. Ed. Eng. 1986, 25, 767–778. [Google Scholar] [CrossRef]
- Dauter, Z.; Wlodawer, A. Progress in protein crystallography. Protein Pept. Lett. 2016, 23, 201–210. [Google Scholar] [CrossRef]
- McIntyre, P.J.; Collins, P.M.; Vrzal, L.; Birchall, K.; Arnold, L.H.; Mpamhanga, C.; Coombs, P.J.; Burgess, S.G.; Richards, M.W.; Winter, A.; et al. Characterization of three druggable hot-spots in the Aurora-A/TPX2 interaction using biochemical, biophysical, and fragment-based approaches. ACS Chem. Biol. 2017, 12, 2906–2914. [Google Scholar] [CrossRef] [PubMed]
- Lesuisse, D.; Lange, G.; Deprez, P.; Benard, D.; Schoot, B.; Delettre, G.; Marquette, J.P.; Broto, P.; Jean-Baptiste, V.; Bichet, P.; et al. SAR and X-ray. A new approach combining fragment-based screening and rational drug design: Application to the discovery of nanomolar inhibitors of Src SH2. J. Med. Chem. 2002, 45, 2379–2387. [Google Scholar] [CrossRef] [PubMed]
- Hartshorn, M.J.; Murray, C.W.; Cleasby, A.; Frederickson, M.; Tickle, I.J.; Jhoti, H. Fragment-based lead discovery using X-ray crystallography. J. Med. Chem. 2005, 48, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.E.; Collins, P.; James, R.H.; Mendes, V.; Charoensutthivarakul, S.; Radoux, C.; Abell, C.; Coyne, A.G.; Floto, R.A.; von Delft, F.; et al. Structure-guided fragment-based drug discovery at the synchrotron: Screening binding sites and correlations with hotspot mapping. Philos. Trans. A Math. Phys. Eng. Sci. 2019, 377, 20180422. [Google Scholar] [CrossRef]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide protein data bank. Nat. Struct. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- Sun, Z.; Liu, Q.; Qu, G.; Feng, Y.; Reetz, M.T. Utility of B-factors in protein science: Interpreting rigidity, flexibility, and internal motion and engineering thermostability. Chem. Rev. 2019, 119, 1626–1665. [Google Scholar] [CrossRef]
- Baslé, A.; Lewis, R.J. Principles and practice in macromolecular X-ray crystallography. In Biomolecular and Bioanalytical Techniques; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2019; pp. 385–419. [Google Scholar]
- Rose, J.P.; Wang, B.-C.; Weiss, M.S. Native SAD is maturing. IUCrJ 2015, 2, 431–440. [Google Scholar] [CrossRef]
- Wlodawer, A. Stereochemistry and validation of macromolecular structures. Methods Mol. Biol. 2017, 1607, 595–610. [Google Scholar]
- Brünger, A.T. Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 1992, 355, 472–475. [Google Scholar] [CrossRef]
- Fitzpatrick, P.A.; Steinmetz, A.C.; Ringe, D.; Klibanov, A.M. Enzyme crystal structure in a neat organic solvent. Proc. Natl. Acad. Sci. USA 1993, 90, 8653–8657. [Google Scholar] [CrossRef]
- Cavasotto, C.N.; Phatak, S.S. Homology modeling in drug discovery: Current trends and applications. Drug Discov. Today 2009, 14, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.; Bergner, A.; Schwede, T. Modelling three-dimensional protein structures for applications in drug design. Drug Discov. Today 2014, 19, 890–897. [Google Scholar] [CrossRef]
- Du, H.; Brender, J.R.; Zhang, J.; Zhang, Y. Protein structure prediction provides comparable performance to crystallographic structures in docking-based virtual screening. Methods 2015, 71, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, P.; Hartman, A.M.; Hirsch, A.K.H.; Empting, M. Concepts and core principles of fragment-based drug design. Molecules 2019, 24, 4309. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.; Cleasby, A.; Davies, T.G.; Hall, R.J.; Ludlow, R.F.; Murray, C.W.; Tisi, D.; Jhoti, H. Crystallographic screening using ultra-low-molecular-weight ligands to guide drug design. Drug Discov. Today 2019, 24, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Muller, I. Guidelines for the successful generation of protein-ligand complex crystals. Acta Crystallogr. D Struct. Biol. 2017, 73, 79–92. [Google Scholar] [CrossRef]
- Bergfors, T. Seeds to crystals. J. Struct. Biol. 2003, 142, 66–76. [Google Scholar] [CrossRef]
- D’Arcy, A.; Bergfors, T.; Cowan-Jacob, S.W.; Marsh, M. Microseed matrix screening for optimization in protein crystallization: What have we learned? Acta Crystallogr. F Struct. Biol. Commun. 2014, 70, 1117–1126. [Google Scholar] [CrossRef]
- Maveyraud, L.; Mourey, L.; Kotra, L.P.; Pedelacq, J.-D.; Guillet, V.; Mobashery, S.; Samama, J.-P. Structural basis for clinical longevity of carbapenem antibiotics in the face of challenge by the common class A β-Lactamases from the antibiotic-resistant bacteria. J. Am. Chem. Soc. 1998, 120, 9748–9752. [Google Scholar] [CrossRef]
- Lusty, C. A gentle vapor-diffusion technique for cross-linking of protein crystals for cryocrystallography. J. Appl. Crystallogr. 1999, 32, 106–112. [Google Scholar] [CrossRef]
- Yan, E.K.; Lu, Q.Q.; Zhang, C.Y.; Liu, Y.L.; He, J.; Chen, D.; Wang, B.; Zhou, R.B.; Wu, P.; Yin, D.C. Preparation of cross-linked hen-egg white lysozyme crystals free of cracks. Sci. Rep. 2016, 6, 34770. [Google Scholar] [CrossRef] [PubMed]
- Andersen, O.A.; Schonfeld, D.L.; Toogood-Johnson, I.; Felicetti, B.; Albrecht, C.; Fryatt, T.; Whittaker, M.; Hallett, D.; Barker, J. Cross-linking of protein crystals as an aid in the generation of binary protein-ligand crystal complexes, exemplified by the human PDE10a-papaverine structure. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 872–874. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.M.; Ng, J.T.; Talon, R.; Nekrosiute, K.; Krojer, T.; Douangamath, A.; Brandao-Neto, J.; Wright, N.; Pearce, N.M.; von Delft, F. Gentle, fast and effective crystal soaking by acoustic dispensing. Acta Crystallogr. D Struct. Biol. 2017, 73, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Erlanson, D. Practical Fragments: Fragments in the Clinic: 2018 Edition. Available online: http://practicalfragments.blogspot.com/2018/10/fragments-in-clinic-2018-edition.html (accessed on 22 November 2019).
- le Maire, A.; Gelin, M.; Pochet, S.; Hoh, F.; Pirocchi, M.; Guichou, J.F.; Ferrer, J.L.; Labesse, G. In-plate protein crystallization, in situ ligand soaking and X-ray diffraction. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 747–755. [Google Scholar] [CrossRef]
- Gelin, M.; Delfosse, V.; Allemand, F.; Hoh, F.; Sallaz-Damaz, Y.; Pirocchi, M.; Bourguet, W.; Ferrer, J.L.; Labesse, G.; Guichou, J.F. Combining ‘dry’ co-crystallization and in situ diffraction to facilitate ligand screening by X-ray crystallography. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 1777–1787. [Google Scholar] [CrossRef]
- Garman, E. Cool data: Quantity AND quality. Acta Crystallogr. D Biol. Crystallogr. 1999, 55, 1641–1653. [Google Scholar] [CrossRef]
- Deller, M.C.; Rupp, B. Approaches to automated protein crystal harvesting. Acta Crystallogr. F Struct. Biol. Commun. 2014, 70, 133–155. [Google Scholar] [CrossRef]
- Zander, U.; Hoffmann, G.; Cornaciu, I.; Marquette, J.P.; Papp, G.; Landret, C.; Seroul, G.; Sinoir, J.; Rower, M.; Felisaz, F.; et al. Automated harvesting and processing of protein crystals through laser photoablation. Acta Crystallogr. D Struct. Biol. 2016, 72, 454–466. [Google Scholar] [CrossRef]
- Bowler, M.W.; Nurizzo, D.; Barrett, R.; Beteva, A.; Bodin, M.; Caserotto, H.; Delageniere, S.; Dobias, F.; Flot, D.; Giraud, T.; et al. MASSIF-1: A beamline dedicated to the fully automatic characterization and data collection from crystals of biological macromolecules. J. Synchrotron Radiat. 2015, 22, 1540–1547. [Google Scholar] [CrossRef]
- Hutin, S.; Van Laer, B.; Mueller-Dieckmann, C.; Leonard, G.; Nurizzo, D.; Bowler, M.W. Fully autonomous characterization and data collection from crystals of biological macromolecules. J. Vis. Exp. 2019. [Google Scholar] [CrossRef]
- Broecker, J.; Morizumi, T.; Ou, W.L.; Klingel, V.; Kuo, A.; Kissick, D.J.; Ishchenko, A.; Lee, M.Y.; Xu, S.; Makarov, O.; et al. High-throughput in situ X-ray screening of and data collection from protein crystals at room temperature and under cryogenic conditions. Nat. Protoc. 2018, 13, 260–292. [Google Scholar] [CrossRef] [PubMed]
- Vonrhein, C.; Flensburg, C.; Keller, P.; Sharff, A.; Smart, O.; Paciorek, W.; Womack, T.; Bricogne, G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Krug, M.; Weiss, M.S.; Heinemann, U.; Mueller, U. XDSAPP: A graphical user interface for the convenient processing of diffraction data using XDS. J. Appl. Crystallogr. 2012, 45, 568–572. [Google Scholar] [CrossRef]
- Sparta, K.M.; Krug, M.; Heinemann, U.; Mueller, U.; Weiss, M.S. XDSAPP2.0. J. Appl. Crystallogr. 2016, 49, 1085–1092. [Google Scholar] [CrossRef]
- Winter, G. Xia2: An expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 2010, 43, 186–190. [Google Scholar] [CrossRef]
- Monaco, S.; Gordon, E.; Bowler, M.W.; Delageniere, S.; Guijarro, M.; Spruce, D.; Svensson, O.; McSweeney, S.M.; McCarthy, A.A.; Leonard, G.; et al. Automatic processing of macromolecular crystallography X-ray diffraction data at the ESRF. J. Appl. Crystallogr. 2013, 46, 804–810. [Google Scholar] [CrossRef]
- Kabsch, W. Xds. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Battye, T.G.; Kontogiannis, L.; Johnson, O.; Powell, H.R.; Leslie, A.G. iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 271–281. [Google Scholar] [CrossRef]
- Winter, G.; Waterman, D.G.; Parkhurst, J.M.; Brewster, A.S.; Gildea, R.J.; Gerstel, M.; Fuentes-Montero, L.; Vollmar, M.; Michels-Clark, T.; Young, I.D.; et al. DIALS: Implementation and evaluation of a new integration package. Acta Crystallogr. D Struct. Biol. 2018, 74, 85–97. [Google Scholar] [CrossRef]
- Huschmann, F.U.; Linnik, J.; Sparta, K.; Uhlein, M.; Wang, X.; Metz, A.; Schiebel, J.; Heine, A.; Klebe, G.; Weiss, M.S.; et al. Structures of endothiapepsin-fragment complexes from crystallographic fragment screening using a novel, diverse and affordable 96-compound fragment library. Acta Crystallogr. F Struct. Biol. Commun. 2016, 72, 346–355. [Google Scholar] [CrossRef]
- Pearce, N.M.; Bradley, A.R.; Krojer, T.; Marsden, B.D.; Deane, C.M.; von Delft, F. Partial-occupancy binders identified by the pan-dataset density analysis method offer new chemical opportunities and reveal cryptic binding sites. Struct. Dyn. 2017, 4, 032104. [Google Scholar] [CrossRef]
- Xue, Y.; Guo, H.; Hillertz, P. Fragment screening of RORgammat using cocktail crystallography: Identification of simultaneous binding of multiple fragments. ChemMedChem 2016, 11, 1881–1885. [Google Scholar] [CrossRef]
- Echols, N.; Moriarty, N.W.; Klei, H.E.; Afonine, P.V.; Bunkoczi, G.; Headd, J.J.; McCoy, A.J.; Oeffner, R.D.; Read, R.J.; Terwilliger, T.C.; et al. Automating crystallographic structure solution and refinement of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 144–154. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef]
- Blanc, E.; Roversi, P.; Vonrhein, C.; Flensburg, C.; Lea, S.M.; Bricogne, G. Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2210–2221. [Google Scholar] [CrossRef]
- Schiebel, J.; Krimmer, S.G.; Röwer, K.; Knörlein, A.; Wang, X.; Park, A.Y.; Stieler, M.; Ehrmann, F.R.; Fu, K.; Radeva, N.; et al. High-throughput crystallography: Reliable and efficient identification of fragment hits. Structure 2016, 24, 1398–1409. [Google Scholar] [CrossRef]
- Hanson, M.A.; Oost, T.K.; Sukonpan, C.; Rich, D.H.; Stevens, R.C. Structural basis for BABIM inhibition of botulinum neurotoxin type B protease. J. Am. Chem. Soc. 2000, 122, 11268–11269. [Google Scholar] [CrossRef]
- Davis, A.M.; St-Gallay, S.A.; Kleywegt, G.J. Limitations and lessons in the use of X-ray structural information in drug design. Drug Discov. Today 2008, 13, 831–841. [Google Scholar] [CrossRef]
- Davis, A.M.; Teague, S.J.; Kleywegt, G.J. Application and limitations of X-ray crystallographic data in structure-based ligand and drug design. Angew. Chem. Int. Ed. Engl. 2003, 42, 2718–2736. [Google Scholar] [CrossRef]
- Pearce, N.M.; Krojer, T.; Bradley, A.R.; Collins, P.; Nowak, R.P.; Talon, R.; Marsden, B.D.; Kelm, S.; Shi, J.; Deane, C.M.; et al. A multi-crystal method for extracting obscured crystallographic states from conventionally uninterpretable electron density. Nat. Commun. 2017, 8, 15123. [Google Scholar] [CrossRef]
- Pearce, N.M.; Krojer, T.; von Delft, F. Proper modelling of ligand binding requires an ensemble of bound and unbound states. Acta Crystallogr. D Struct. Biol. 2017, 73, 256–266. [Google Scholar] [CrossRef]
- Schuttelkopf, A.W.; van Aalten, D.M.F. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. Sect. D 2004, 60, 1355–1363. [Google Scholar] [CrossRef]
- Krojer, T.; Talon, R.; Pearce, N.; Collins, P.; Douangamath, A.; Brandao-Neto, J.; Dias, A.; Marsden, B.; von Delft, F. The XChemExplorer graphical workflow tool for routine or large-scale protein-ligand structure determination. Acta Crystallogr. D Struct. Biol. 2017, 73, 267–278. [Google Scholar] [CrossRef]
- Mooij, W.T.; Hartshorn, M.J.; Tickle, I.J.; Sharff, A.J.; Verdonk, M.L.; Jhoti, H. Automated protein-ligand crystallography for structure-based drug design. ChemMedChem 2006, 1, 827–838. [Google Scholar] [CrossRef]
- Wang, H.; Feng, L.; Webb, G.I.; Kurgan, L.; Song, J.; Lin, D. Critical evaluation of bioinformatics tools for the prediction of protein crystallization propensity. Brief Bioinform 2018, 19, 838–852. [Google Scholar] [CrossRef]
- Ehrmann, F.R.; Stojko, J.; Metz, A.; Debaene, F.; Barandun, L.J.; Heine, A.; Diederich, F.; Cianferani, S.; Reuter, K.; Klebe, G. Soaking suggests “alternative facts”: Only co-crystallization discloses major ligand-induced interface rearrangements of a homodimeric tRNA-binding protein indicating a novel mode-of-inhibition. PLoS ONE 2017, 12, e0175723. [Google Scholar] [CrossRef]
- Renaud, J.P.; Chung, C.W.; Danielson, U.H.; Egner, U.; Hennig, M.; Hubbard, R.E.; Nar, H. Biophysics in drug discovery: Impact, challenges and opportunities. Nat. Rev. Drug Discov. 2016, 15, 679–698. [Google Scholar] [CrossRef]
- Cushman, J.C. Characterization and expression of a NADP-malic enzyme cDNA induced by salt stress from the facultative crassulacean acid metabolism plant, Mesembryanthemum crystallinum. Eur. J. Biochem. 1992, 208, 259–266. [Google Scholar] [CrossRef]
- Sugrue, M.F. Pharmacological and ocular hypotensive properties of topical carbonic anhydrase inhibitors. Prog. Retin. Eye Res. 2000, 19, 87–112. [Google Scholar] [CrossRef]
- Cowan-Jacob, S.W.; Fendrich, G.; Floersheimer, A.; Furet, P.; Liebetanz, J.; Rummel, G.; Rheinberger, P.; Centeleghe, M.; Fabbro, D.; Manley, P.W. Structural Biology Contributions to the Discovery of Drugs to Treat Chronic Myelogenous Leukemia; Springer: Dordrecht, The Netherlands, 2009; pp. 37–61. [Google Scholar]
- Zhang, J.; Adrián, F.J.; Jahnke, W.; Cowan-Jacob, S.W.; Li, A.G.; Iacob, R.E.; Sim, T.; Powers, J.; Dierks, C.; Sun, F.; et al. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature 2010, 463, 501. [Google Scholar] [CrossRef]
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Gillette, S.; Kong, J.; Haass, N.K.; et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3041–3046. [Google Scholar] [CrossRef]
- Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: The first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 2012, 11, 873. [Google Scholar] [CrossRef]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef]
- Leverson, J.D.; Sampath, D.; Souers, A.J.; Rosenberg, S.H.; Fairbrother, W.J.; Amiot, M.; Konopleva, M.; Letai, A. Found in translation: How preclinical research is guiding the clinical development of the BCL2-selective inhibitor venetoclax. Cancer Discov. 2017, 7, 1376–1393. [Google Scholar] [CrossRef]
- FDA Approves First Targeted Therapy for Metastatic Bladder Cancer. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-targeted-therapy-metastatic-bladder-cancer (accessed on 27 November 2019).
- Perera, T.P.S.; Jovcheva, E.; Mevellec, L.; Vialard, J.; De Lange, D.; Verhulst, T.; Paulussen, C.; Van De Ven, K.; King, P.; Freyne, E.; et al. Discovery and pharmacological characterization of JNJ-42756493 (Erdafitinib), a functionally selective small-molecule FGFR family inhibitor. Mol. Cancer 2017, 16, 1010–1020. [Google Scholar] [CrossRef]
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [CrossRef]
- Liu, H.; Lee, W. The XFEL protein crystallography: Developments and perspectives. Int. J. Mol. Sci. 2019, 20, 3421. [Google Scholar] [CrossRef]
- Neutze, R.; Wouts, R.; van der Spoel, D.; Weckert, E.; Hajdu, J. Potential for biomolecular imaging with femtosecond X-ray pulses. Nature 2000, 406, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Chapman, H.N.; Fromme, P.; Barty, A.; White, T.A.; Kirian, R.A.; Aquila, A.; Hunter, M.S.; Schulz, J.; DePonte, D.P.; Weierstall, U.; et al. Femtosecond X-ray protein nanocrystallography. Nature 2011, 470, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Boutet, S.; Lomb, L.; Williams, G.J.; Barends, T.R.; Aquila, A.; Doak, R.B.; Weierstall, U.; DePonte, D.P.; Steinbrener, J.; Shoeman, R.L.; et al. High-resolution protein structure determination by serial femtosecond crystallography. Science 2012, 337, 362–364. [Google Scholar] [CrossRef]
- Schlichting, I.; Miao, J. Emerging opportunities in structural biology with X-ray free-electron lasers. Curr. Opin. Struct. Biol. 2012, 22, 613–626. [Google Scholar] [CrossRef]
- Johansson, L.C.; Arnlund, D.; Katona, G.; White, T.A.; Barty, A.; DePonte, D.P.; Shoeman, R.L.; Wickstrand, C.; Sharma, A.; Williams, G.J.; et al. Structure of a photosynthetic reaction centre determined by serial femtosecond crystallography. Nat. Commun. 2013, 4, 2911. [Google Scholar] [CrossRef]
- Liu, W.; Wacker, D.; Gati, C.; Han, G.W.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; Katritch, V.; Barty, A.; et al. Serial femtosecond crystallography of G protein-coupled receptors. Science 2013, 342, 1521–1524. [Google Scholar] [CrossRef]
- Weierstall, U.; James, D.; Wang, C.; White, T.A.; Wang, D.; Liu, W.; Spence, J.C.; Bruce Doak, R.; Nelson, G.; Fromme, P.; et al. Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat. Commun. 2014, 5, 3309. [Google Scholar] [CrossRef]
- Suga, M.; Akita, F.; Hirata, K.; Ueno, G.; Murakami, H.; Nakajima, Y.; Shimizu, T.; Yamashita, K.; Yamamoto, M.; Ago, H.; et al. Native structure of photosystem II at 1.95 A resolution viewed by femtosecond X-ray pulses. Nature 2015, 517, 99–103. [Google Scholar] [CrossRef]
- Sawaya, M.R.; Cascio, D.; Gingery, M.; Rodriguez, J.; Goldschmidt, L.; Colletier, J.P.; Messerschmidt, M.M.; Boutet, S.; Koglin, J.E.; Williams, G.J.; et al. Protein crystal structure obtained at 2.9 A resolution from injecting bacterial cells into an X-ray free-electron laser beam. Proc. Natl. Acad. Sci. USA 2014, 111, 12769–12774. [Google Scholar] [CrossRef]
- Diederichs, K.; Wang, M. Serial Synchrotron X-ray crystallography (SSX). Methods Mol. Biol. 2017, 1607, 239–272. [Google Scholar]
- Cheng, R.K.Y.; Abela, R.; Hennig, M. X-ray free electron laser: Opportunities for drug discovery. Essays Biochem. 2017, 61, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.M.; Higgins, A.J.; Muench, S.P. Emerging role of electron microscopy in drug discovery. Trends Biochem. Sci. 2019, 44, 897–898. [Google Scholar] [CrossRef] [PubMed]
- Renaud, J.P.; Chari, A.; Ciferri, C.; Liu, W.T.; Remigy, H.W.; Stark, H.; Wiesmann, C. Cryo-EM in drug discovery: Achievements, limitations and prospects. Nat. Rev. Drug Discov. 2018, 17, 471–492. [Google Scholar] [CrossRef]
- Wong, W.; Bai, X.C.; Sleebs, B.E.; Triglia, T.; Brown, A.; Thompson, J.K.; Jackson, K.E.; Hanssen, E.; Marapana, D.S.; Fernandez, I.S.; et al. Mefloquine targets the Plasmodium falciparum 80S ribosome to inhibit protein synthesis. Nat. Microbiol. 2017, 2, 17031. [Google Scholar] [CrossRef]
- Garreau de Loubresse, N.; Prokhorova, I.; Holtkamp, W.; Rodnina, M.V.; Yusupova, G.; Yusupov, M. Structural basis for the inhibition of the eukaryotic ribosome. Nature 2014, 513, 517–522. [Google Scholar] [CrossRef]
- Nannenga, B.L.; Gonen, T. The cryo-EM method microcrystal electron diffraction (MicroED). Nat. Methods 2019, 16, 369–379. [Google Scholar] [CrossRef]
- Shi, D.; Nannenga, B.L.; Iadanza, M.G.; Gonen, T. Three-dimensional electron crystallography of protein microcrystals. eLife 2013, 2, e01345. [Google Scholar] [CrossRef]
- Nannenga, B.L.; Shi, D.; Leslie, A.G.W.; Gonen, T. High-resolution structure determination by continuous-rotation data collection in MicroED. Nat. Methods 2014, 11, 927–930. [Google Scholar] [CrossRef]
- Gruene, T.; Wennmacher, J.T.C.; Zaubitzer, C.; Holstein, J.J.; Heidler, J.; Fecteau-Lefebvre, A.; De Carlo, S.; Muller, E.; Goldie, K.N.; Regeni, I.; et al. Rapid structure determination of microcrystalline molecular compounds using electron diffraction. Angew. Chem. Int. Ed. Eng. 2018, 57, 16313–16317. [Google Scholar] [CrossRef]
- Purdy, M.D.; Shi, D.; Chrustowicz, J.; Hattne, J.; Gonen, T.; Yeager, M. MicroED structures of HIV-1 Gag CTD-SP1 reveal binding interactions with the maturation inhibitor bevirimat. Proc. Natl. Acad. Sci. USA 2018, 115, 13258–13263. [Google Scholar] [CrossRef]
- Spiliopoulou, M.; Valmas, A.; Triandafillidis, D.-P.; Kosinas, C.; Fitch, A.; Karavassili, F.; Margiolaki, I. Applications of X-ray powder diffraction in protein crystallography and drug screening. Crystals 2020, 10, 54. [Google Scholar] [CrossRef]
- Williamson, M.P.; Havel, T.F.; Wuthrich, K. Solution conformation of proteinase inhibitor IIA from bull seminal plasma by 1H nuclear magnetic resonance and distance geometry. J. Mol. Biol. 1985, 182, 295–315. [Google Scholar] [CrossRef]
- Fiaux, J.; Bertelsen, E.B.; Horwich, A.L.; Wuthrich, K. NMR analysis of a 900K GroEL GroES complex. Nature 2002, 418, 207–211. [Google Scholar] [CrossRef]
- Gossert, A.D.; Jahnke, W. NMR in drug discovery: A practical guide to identification and validation of ligands interacting with biological macromolecules. Prog. Nucl. Magn. Reson. Spectrosc. 2016, 97, 82–125. [Google Scholar] [CrossRef]
- Shuker, S.B.; Hajduk, P.J.; Meadows, R.P.; Fesik, S.W. Discovering high-affinity ligands for proteins: SAR by NMR. Science 1996, 274, 1531–1534. [Google Scholar] [CrossRef]
- O’Dell, W.B.; Bodenheimer, A.M.; Meilleur, F. Neutron protein crystallography: A complementary tool for locating hydrogens in proteins. Arch. Biochem. Biophys. 2016, 602, 48–60. [Google Scholar] [CrossRef]
- Kovalevsky, A.; Aggarwal, M.; Velazquez, H.; Cuneo, M.J.; Blakeley, M.P.; Weiss, K.L.; Smith, J.C.; Fisher, S.Z.; McKenna, R. “To be or not to be” protonated: Atomic details of human carbonic anhydrase-clinical drug complexes by neutron crystallography and simulation. Structure 2018, 26, 383–390. [Google Scholar] [CrossRef]
- Manzoni, F.; Wallerstein, J.; Schrader, T.E.; Ostermann, A.; Coates, L.; Akke, M.; Blakeley, M.P.; Oksanen, E.; Logan, D.T. Elucidation of hydrogen bonding patterns in ligand-free, lactose- and glycerol-bound galectin-3C by neutron crystallography to guide drug design. J. Med. Chem. 2018, 61, 4412–4420. [Google Scholar] [CrossRef]
- Forster, A.; Schulze-Briese, C. A shared vision for macromolecular crystallography over the next five years. Struct. Dyn. 2019, 6, 064302. [Google Scholar] [CrossRef]
- Bricogne, G. High-throughput macromolecular crystallography in drug discovery. In Structural Biology in Drug Discovery; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2020; pp. 211–251. [Google Scholar]



{kind=link}
{kind=link}
| Drug | Company | Target name, Function |
|---|---|---|
| Approved | ||
| Vemurafenib/ZELBORAF | Plexxikon/Roche | BRAF, protein kinase (mutation V600E) |
| Venetoclax/VENCLEXTA | AbbVie/Genentech | BCL-2, apoptosis suppressor protein |
| Erdafitinib/BALVERSA | Janssen Pharmaceutical Companies | FGFRs, fibroblast growth factor receptors |
| Phase III | ||
| Asciminib/ABL001 | Novartis | Bcr-Abl, oncoprotein |
| Lanabecestat 1 | AstraZeneca/Eli Lilly and Company | BACE-1, β-site amyloid precursor protein-cleaving enzyme 1 |
| Pexidartinib/TURALIOTM | Plexxikon/Daiichi Sankyo | CSF1R, colony stimulating factor 1 receptor KIT, proto-oncogene receptor tyrosine kinase |
| Verubecestat 1 | Merck | BACE-1, β-site amyloid precursor protein-cleaving enzyme 1 |
| Phase II | ||
| AT7519 2 | Astex | CDKs, cyclin-dependent kinases |
| AT9283 | Astex | Aurora and JAK2, kinases |
| Luminespib/AUY-922 1 | Vernalis/Novartis | HSP90, heat shock protein |
| Capivasertib/AZD5363 | AstraZeneca/Astex | AKT, serine/threonine protein kinase |
| CPI-0610 | Constellation | BET, bromodomain and extra-terminal protein |
| DG-051 2 | deCODE | LTA4H, leukotriene A4 hydrolase |
| eFT508 | eFFECTOR | MNK1/2, kinases |
| Indeglitazar 2 | Plexxikon | PPARs, peroxisome proliferator-activated receptors |
| LY2886721 2 | Eli Lilly and Company | BACE-1, β-site amyloid precursor protein-cleaving enzyme 1 |
| LY517717 2 | Eli Lilly and Company | FXa, Factor Xa |
| Navitoclax/ABT-263 | Abbott | BCL-2/BCL-XL, apoptosis suppressor proteins |
| Onalespib/AT13387 | Astex | HSP90, heat shock protein |
| PF-06650833 | Pfizer | IRAK4, interleukin-1 receptor-associated kinase |
| PF-06835919 | Pfizer | KHK, ketohexokinase |
| Phase I3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maveyraud, L.; Mourey, L. Protein X-ray Crystallography and Drug Discovery. Molecules 2020, 25, 1030. https://doi.org/10.3390/molecules25051030
Maveyraud L, Mourey L. Protein X-ray Crystallography and Drug Discovery. Molecules. 2020; 25(5):1030. https://doi.org/10.3390/molecules25051030
Chicago/Turabian StyleMaveyraud, Laurent, and Lionel Mourey. 2020. "Protein X-ray Crystallography and Drug Discovery" Molecules 25, no. 5: 1030. https://doi.org/10.3390/molecules25051030
APA StyleMaveyraud, L., & Mourey, L. (2020). Protein X-ray Crystallography and Drug Discovery. Molecules, 25(5), 1030. https://doi.org/10.3390/molecules25051030