
2-Butyl-2-tert-butyl-5,5-diethylpyrrolidine-1-oxyls: Synthesis and Properties

 , , ,
, , ,  and
and
Abstract
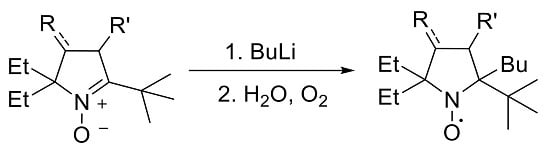
1. Introduction
2. Results
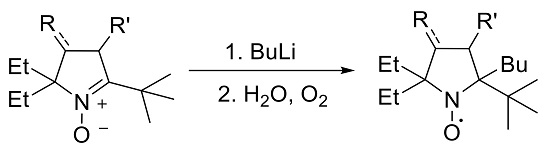
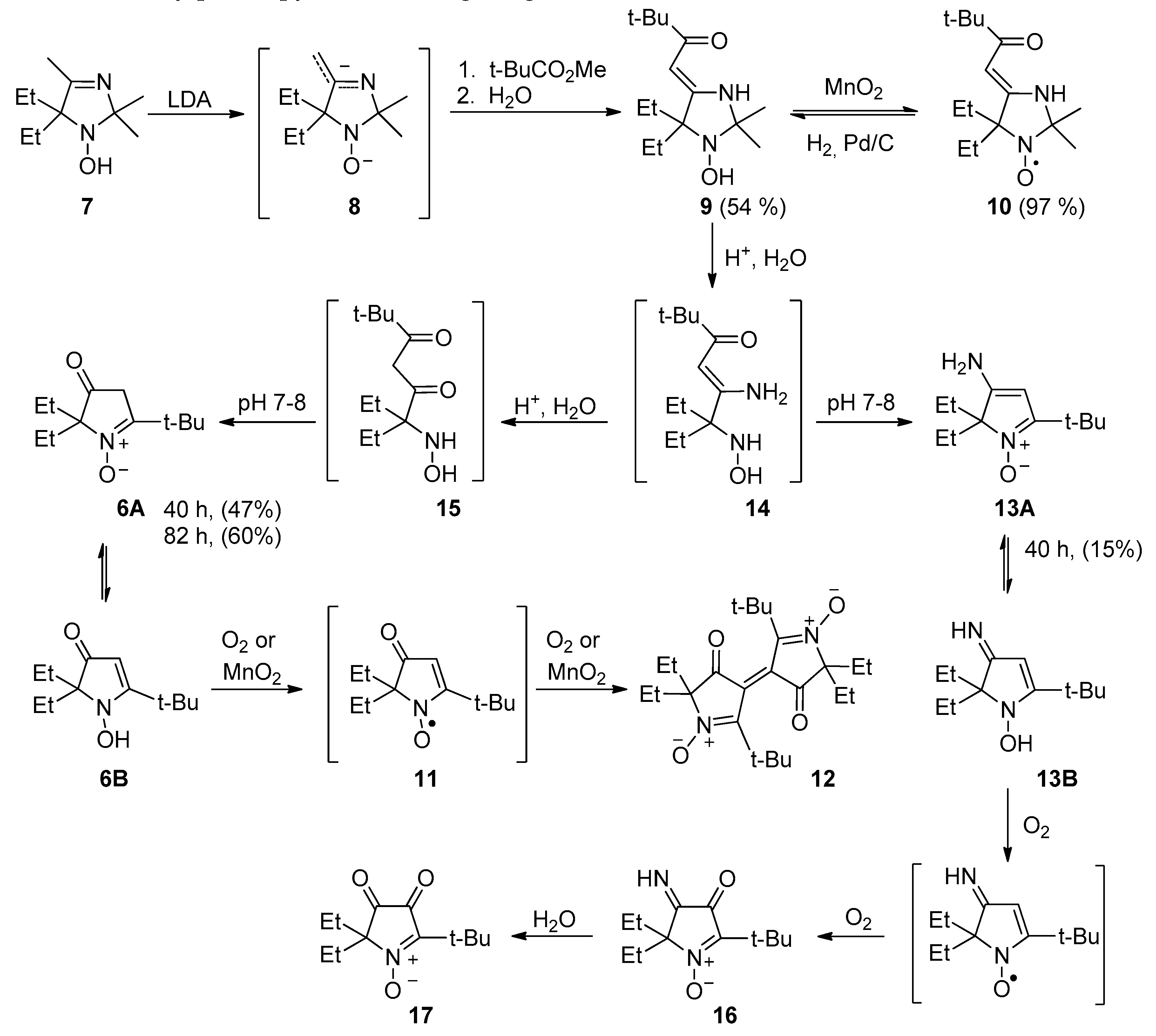
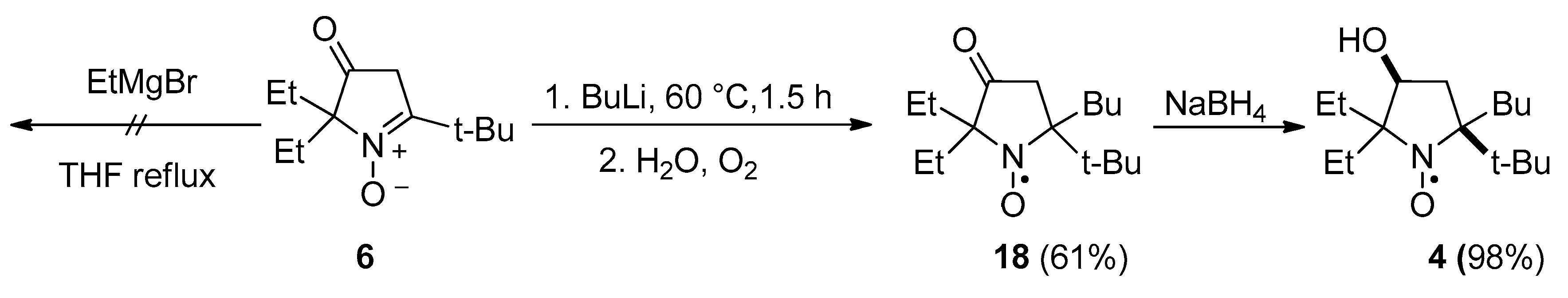
2.1. Synthesis of Nitroxide 4
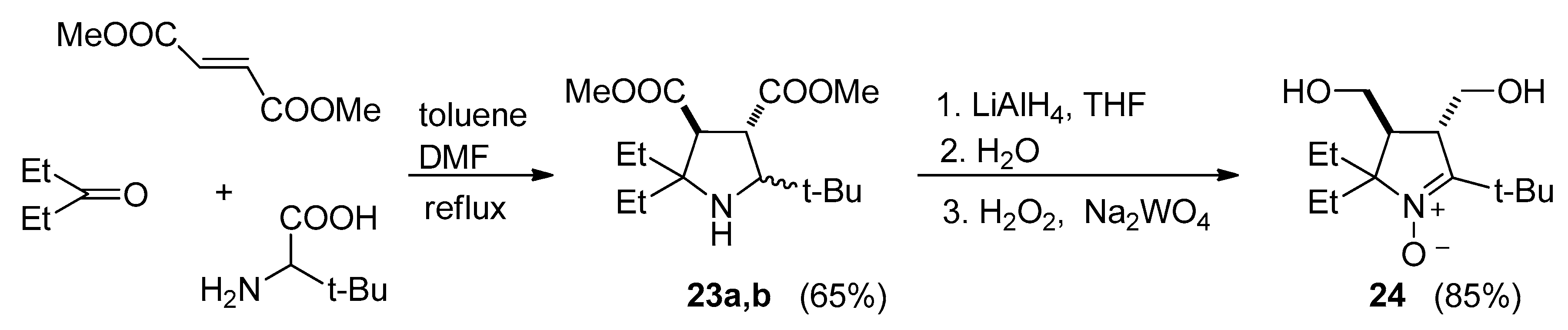
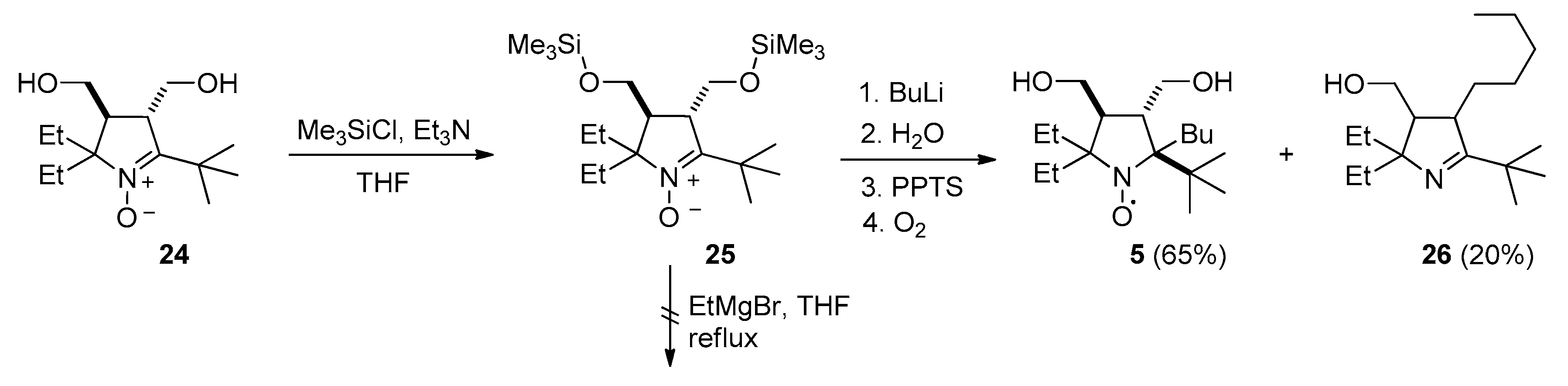
2.2. Synthesis of Nitroxide 5
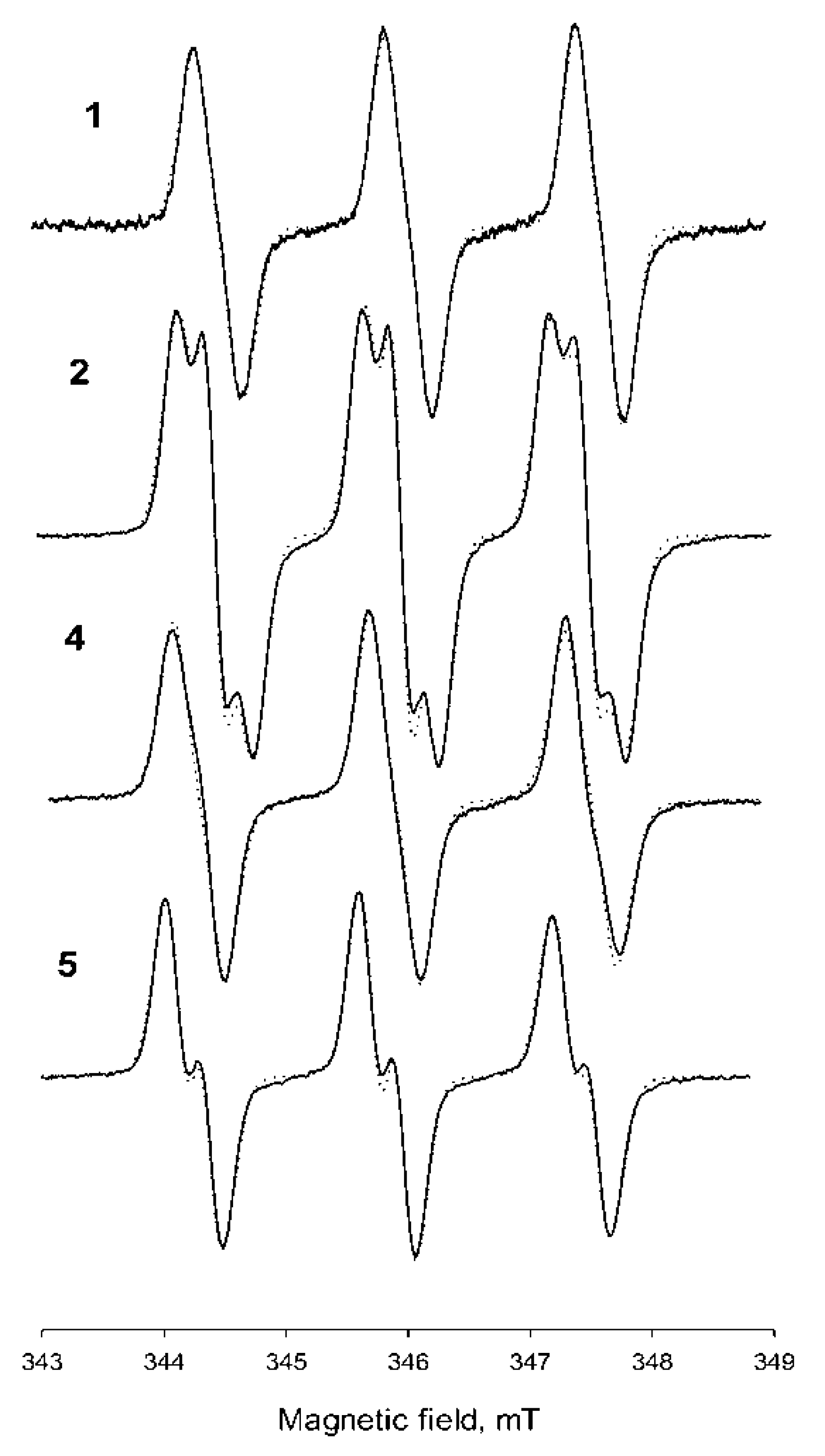
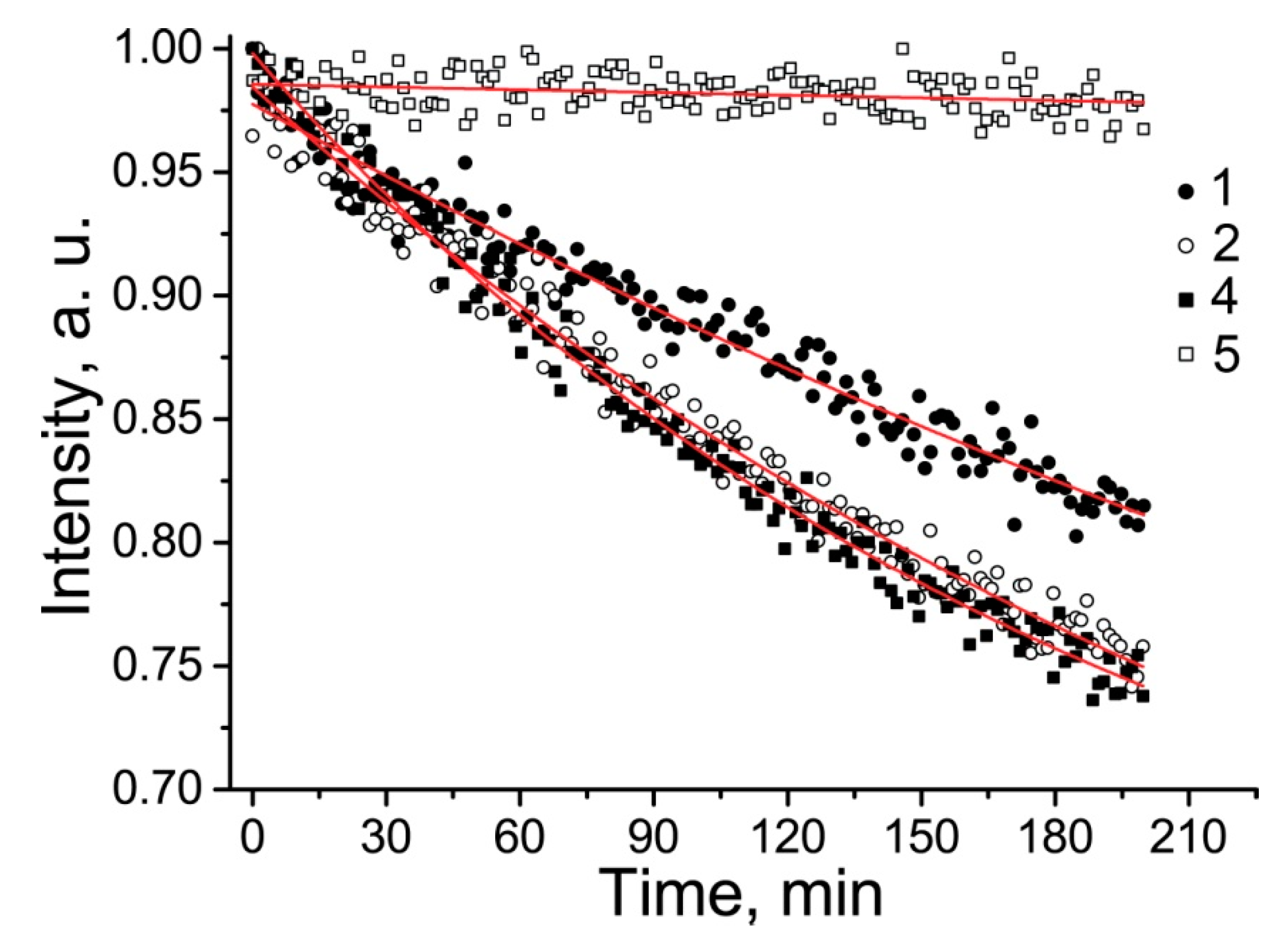
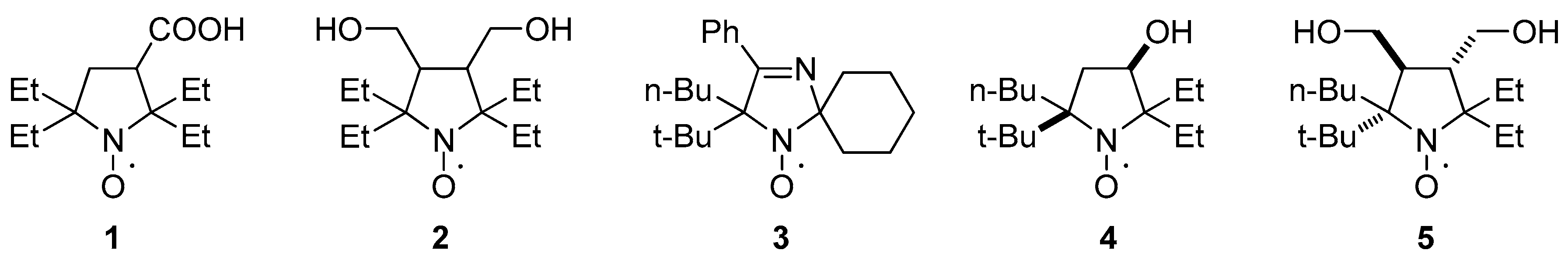
2.3. Properties of the New Nitroxides
3. Materials and Methods
3.1. General Information
3.2. Synthesis
3.2.1. 5,5-Diethyl-1-hydroxy-2,2,4-trimethyl-2,5-dihydro-1H-imidazole (7)
3.2.2. (Z)-4-(3,3-Dimethyl-2-oxobutylidene)-5,5-diethyl-2,2-dimethylimidazolidin-1-oxyl (10)
3.2.3. (Z)-1-(5,5-Diethyl-2,2-dimethyl-1-hydroxyimidazolidin-4-ylidene)-3,3-dimethylbutan-2-one (9)
3.2.4. Enaminoketone 9 Acid Hydrolysis and Recyclization
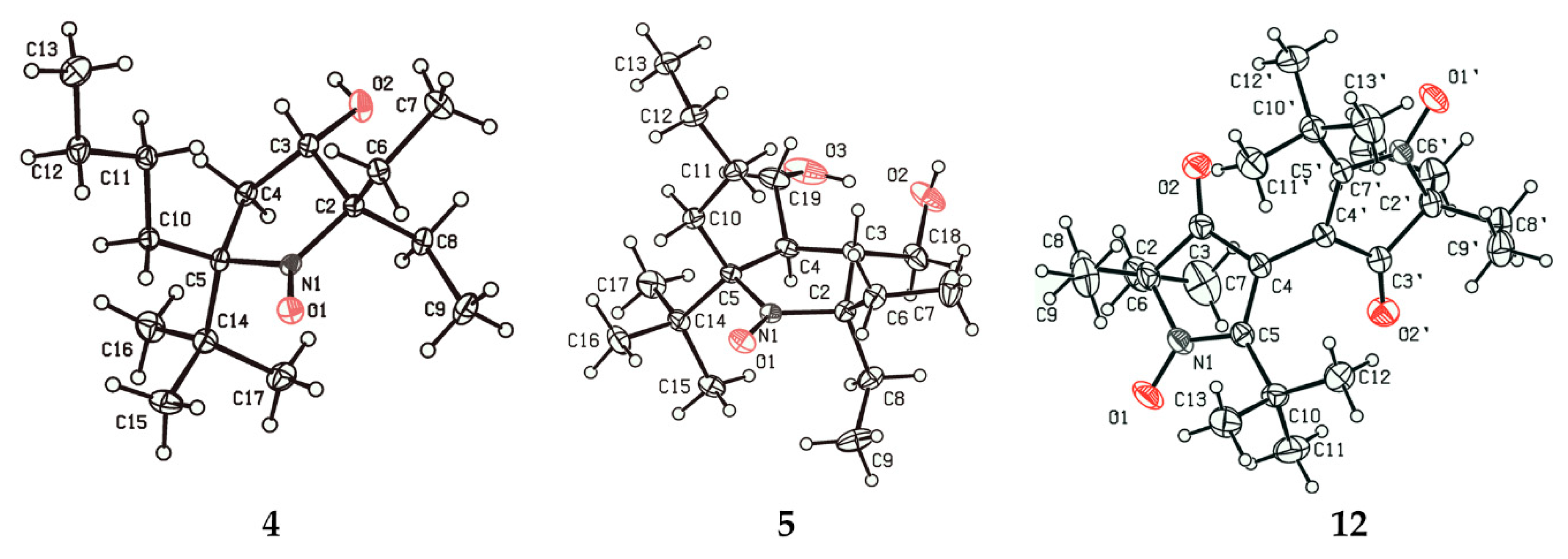
3.2.5. 3,3’-Bis(2-tert-butyl-5,5-diethyl-4-oxopyrrolinylidene) 1,1’-dioxide (12)
3.2.6. 5-(tert-Butyl)-5-butyl-2,2-diethyl-3-oxopyrrolidin-1-oxyl (18)
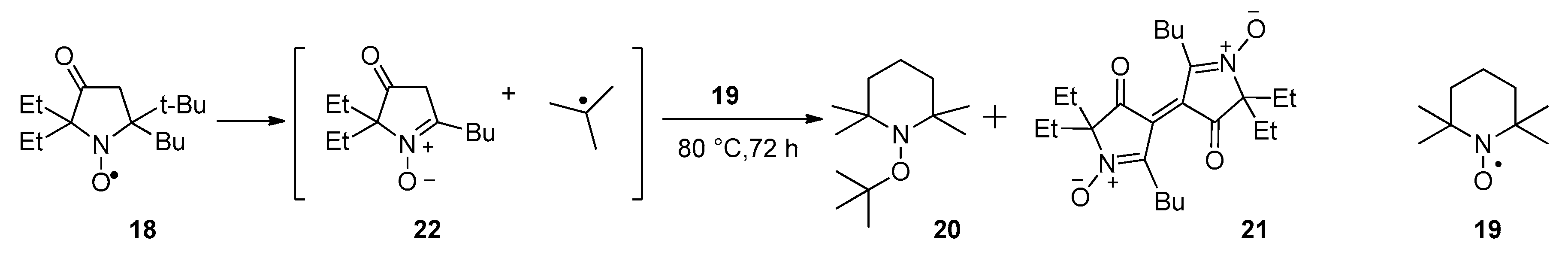
3.2.7. Thermal Decomposition of Nitroxide 18 in the Presence of TEMPO
3.2.8. 5-(tert-Butyl)-5-butyl-2,2-diethyl-3-hydroxypyrrolidin-1-oxyl (4)
3.2.9. 5-(tert-Butyl)-3,4-bis(methoxycarbonyl)-2,2-diethylpyrrolidine (23a,b)
3.2.10. 5-(tert-Butyl)-2,2-diethyl-3,4-bis(hydroxymethyl)-3,4-dihydro-2H-pyrrole 1-oxide (24)
3.2.11. 2-(tert-Butyl)-2-butyl-5,5-diethyl-3,4-bis(hydroxymethyl)pyrrolidin-1-oxyl (5)
3.2.12. The General Method for Nitroxides 4 and 5, Reduction with Zn in CF3COOH for NMR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hubbell, W.L.; López, C.J.; Altenbach, C.; Yang, Z. Technological advances in site-directed spin labelling. Curr. Opin. Struct. Biol. 2013, 23, 725–733. [Google Scholar] [CrossRef]
- Roser, P.; Schmidt, M.J.; Drescher, M.; Summerer, D. Site-directed spin labeling of proteins for distance measurements in vitro and in cells. Org. Biomol. Chem. 2016, 14, 5468–5476. [Google Scholar] [CrossRef] [PubMed]
- Endeward, B.; Marko, A.; Denysenkov, V.P.; Sigurdsson, S.T.; Prisner, T.F. Advanced EPR methods for studying conformational dynamics in nucleic acids. In Methods in Enzymology; Qin, P.Z., Warncke, K., Eds.; Publisher: Academic Press, 2015; Volume 564, pp. 403–425. [Google Scholar] [CrossRef]
- Babić, N.; Peyrot, F. Molecular Probes for Evaluation of Oxidative Stress by In Vivo EPR Spectroscopy and Imaging: State-of-the-Art and Limitations. Magnetochemistry 2019, 5, 13. [Google Scholar] [CrossRef]
- Soule, B.P.; Hyodo, F.; Matsumoto, K.I.; Simone, N.L.; Cook, J.A.; Krishna, M.C.; Mitchell, J.B. The Chemistry and Biology of Nitroxide Compounds. Free Radical Biol. Med. 2007, 42, 1632–1650. [Google Scholar] [CrossRef] [PubMed]
- Khramtsov, V.V. In vivo Spectroscopy and Imaging of Nitroxide Probes. In Nitroxides–Theory, Experiment and Applications; Kokorin, A.I., Ed.; InTech Publisher: Rijeca, Croatia, 2012; pp. 317–346. [Google Scholar]
- Karthikeyan, G.; Bonucci, A.; Casano, G.; Gerbaud, G.; Abel, S.; Thomé, V.; Kodjabachian, L.; Magalon, A.; Guigliarelli, B.; Belle, V.; et al. A bioresistant nitroxide spin label for in-cell EPR spectroscopy: In vitro and in oocytes protein structural dynamics studies. Angew. Chem. Int. Ed. 2018, 130, 1380–1384. [Google Scholar] [CrossRef]
- Bleicken, S.; Assafa, T.E.; Zhang, H.; Elsner, C.; Ritsch, I.; Pink, M.; Rajca, S.; Jeschke, G.; Rajca, A.; Bordignon, E. gem-Diethyl pyrroline nitroxide spin labels: Synthesis, EPR characterization, rotamer libraries and biocompatibility. ChemistryOpen 2019, 8, 1057–1065. [Google Scholar] [CrossRef]
- Komarov, D.A.; Ichikawa, Y.; Yamamoto, K.; Stewart, N.J.; Matsumoto, S.; Yasui, H.; Kirilyuk, I.A.; Khramtsov, V.V.; Inanami, O.; Hirata, H. In vivo extracellular pH mapping of tumors using electron paramagnetic resonance. Anal. Chem. 2018, 90, 13938–13945. [Google Scholar] [CrossRef]
- Audran, G.; Bosco, L.; Brémond, P.; Franconi, J.-M.; Koonjoo, N.; Marque, S.R.A.; Massot, P.; Mellet, P.; Parzy, E.; Thiaudière, E. Enzymatically shifting nitroxides for EPR spectroscopy and overhauser-enhanced magnetic resonance imaging. Angew. Chem. Int. Ed. 2015, 54, 13379–13384. [Google Scholar] [CrossRef]
- Couet, W.R.; Brasch, R.C.; Sosnovsky, G.; Tozer, T.N. Factors affecting nitroxide reduction in ascorbate solution and tissue homogenates. Magn. Res. Imaging 1985, 3, 83–88. [Google Scholar] [CrossRef]
- Kirilyuk, I.A.; Bobko, A.A.; Semenov, S.V.; Komarov, D.A.; Irtegova, I.G.; Grigor’ev, I.A.; Bagryanskaya, E.G. The effect of sterical shielding on the redox properties of imidazoline and imidazolidine nitroxides. J. Org. Chem. 2015, 80, 9118–9125. [Google Scholar] [CrossRef]
- Keana, J.F.W.; Van Nice, F.L. Influence of structure on the reduction of nitroxide MRI contrast-enhancing agents by ascorbate. Physiol. Chem. Phys. Med. NMR 1984, 16, 477–480. [Google Scholar] [PubMed]
- Morris, S.; Sosnovsky, G.; Hui, B.; Huber, C.O.; Rao, N.U.M.; Swartz, H.M. Chemical and electrochemical reduction rates of cyclic nitroxides (nitroxyls). J. Pharm. Sci. 1991, 80, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Marx, L.; Chiarelli, R.; Guiberteau, T.; Rassat, A. A comparative study of the reduction by ascorbate of 1, 1, 3, 3-tetraethylisoindolin-2-yloxyl and of 1, 1, 3, 3-tetramethylisoindolin-2-yloxyl. J. Chem. Soc., Perkin Trans. 1 2000, 1181–1182. [Google Scholar] [CrossRef]
- Kirilyuk, I.A.; Bobko, A.A.; Grigor’ev, I.A.; Khramtsov, V.V. Synthesis of the tetraethyl substituted pH-sensitive nitroxides of imidazole series with enhanced stability towards reduction. Org. Biomol. Chem. 2004, 2, 1025–1030. [Google Scholar] [CrossRef]
- Yamasaki, T.; Mito, F.; Ito, Y.; Pandian, S.; Kinoshita, Y.; Nakano, K.; Murugesan, R.; Sakai, K.; Utsumi, H.; Yamada, K. Structure− reactivity relationship of piperidine nitroxide: Electrochemical, ESR and computational studies. J. Org. Chem. 2011, 76, 435–440. [Google Scholar] [CrossRef]
- Paletta, J.T.; Pink, M.; Foley, B.; Rajca, S.; Rajca, A. Synthesis and reduction kinetics of sterically Sshielded pyrrolidine nitroxides. Org. Lett. 2012, 14, 5322–5325. [Google Scholar] [CrossRef]
- Jagtap, A.P.; Krstic, I.; Kunjir, N.C.; Hänsel, R.; Prisner, T.F.; Sigurdsson, S.T. Sterically shielded spin labels for in-cell EPR spectroscopy: Analysis of stability in reducing environment. Free Rad. Res. 2015, 49, 78–85. [Google Scholar] [CrossRef]
- Dobrynin, S.A.; Glazachev, Y.I.; Gatilov, Y.V.; Chernyak, E.I.; Salnikov, G.E.; Kirilyuk, I.A. Synthesis of 3,4-bis (hydroxymethyl)-2, 2, 5, 5-tetraethylpyrrolidin-1-oxyl via 1, 3-dipolar cycloaddition of azomethine ylide to activated alkene. J. Org. Chem. 2018, 83, 5392–5397. [Google Scholar] [CrossRef]
- Edeleva, M.V.; Kirilyuk, I.A.; Zubenko, D.P.; Zhurko, I.F.; Marque, S.R.; Gigmes, D.; Guillaneuf, Y.; Bagryanskaya, E.G. Kinetic study of H-atom transfer in imidazoline-, imidazolidine-, and pyrrolidine-based alkoxyamines: Consequences for nitroxide-mediated polymerization. J. Polym. Sci. Part A: Pol. Chem. 2009, 47, 6579–6595. [Google Scholar] [CrossRef]
- Nagura, K.; Takemoto, Y.; Yoshino, F.; Bogdanov, A.; Chumakova, N.; Vorobiev, A.K.; Imai, H.; Matsuda, T.; Shimono, S.; Kato, T.; et al. Magnetic mixed micelles composed of a non-ionic surfactant and nitroxide radicals containing a D-glucosamine unit: Preparation, stability, and biomedical application. Pharmaceutics 2019, 11, 42. [Google Scholar] [CrossRef]
- Nagura, K.; Takemoto, Y.; Moronaga, S.; Uchida, Y.; Shimono, S.; Shiino, A.; Tanigaki, K.; Amano, T.; Yoshino, F.; Noda, Y.; et al. Preparation of robust metal-free magnetic nanoemulsions encapsulating low-molecular-weight nitroxide radicals and hydrophobic drugs directed toward MRI-visible targeted delivery. Chemistry 2017, 23, 15713–15720. [Google Scholar] [CrossRef] [PubMed]
- Bye, N.; Hutt, O.E.; Hinton, T.M.; Acharya, D.P.; Waddington, L.J.; Moffat, B.A.; Wright, D.K.; Wang, H.X.; Mulet, X.; Muir, B.W. Nitroxide-loaded hexosomes provide MRI contrast in vivo. Langmuir 2014, 30, 8898–8906. [Google Scholar] [CrossRef] [PubMed]
- Reznikov, V.A.; Volodarskii, L.B. Recyclization of enamino ketones that are imidazolidine derivatives to 1-pyrrolin-4-one 1-oxides. Chem. Heterocycl. Compd. 1990, 26, 997–1000. [Google Scholar] [CrossRef]
- Reznikov, V.A.; Skuridin, N.G.; Khromovskikh, E.L.; Khramtsov, V.V. A new series of lipophilic pH-sensitive spin probes. Russ. Chem. Bull. 2003, 52, 2052–2056. [Google Scholar] [CrossRef]
- Zubenko, D.; Tsentalovich, Y.; Lebedeva, N.; Kirilyuk, I.; Roshchupkina, G.; Zhurko, I.; Reznikov, V.; Marque, S.R.A.; Bagryanskaya, E. Laser flash photolysis and CIDNP studies of steric effects on coupling rate constants of imidazolidine nitroxide with carbon-centered radicals, methyl isobutyrate-2-yl and tert-butyl propionate-2-yl. J. Org. Chem. 2006, 71, 6044–6052. [Google Scholar] [CrossRef]
- Reznikov, V.A.; Martin, V.V.; Volodarskii, L.B. Oxidative dimerization of heterocyclic nitrones, derivatives of pyrroline and imidazoline. Bull. Acad. Sci. USSR Div. Chem. Sci. 1990, 39, 1261–1267. [Google Scholar] [CrossRef]
- Rybalova, T.V.; Gatilov, Y.V.; Reznikov, V.A.; Pervukhina, N.V.; Burdukov, A.B. Investigation of conjugated dinitrones - derivatives of pyrroline oxide. J. Struct. Chem. 1997, 38, 648–656. [Google Scholar] [CrossRef]
- Shundrin, L.A.; Irtegova, I.G.; Rogachev, A.D.; Reznikov, V.A. Molecular ions of 3,3′-bi(2-R-5,5-dimethyl-4-oxopyrrolinylidene) 1,1′-dioxides. Russ. Chem. Bull. 2005, 54, 1178–1184. [Google Scholar] [CrossRef]
- Reznikov, V.A.; Volodarsky, L.B.; Rybalova, T.V.; Gatilov, Y.V. Stable vinylnitroxyl radicals, pyrroline derivatives. Russ. Chem. Bull. 2000, 49, 106–115. [Google Scholar] [CrossRef]
- Reznikov, V.A.; Roshchupkina, G.I.; Ribalova, T.V.; Gatilov, Y.V. Synthesis and properties of monoimines of α-diketones, derivatives of 3-imidazoline nitroxides. Rus. Chem. Bull. 2001, 50, 874–881. [Google Scholar] [CrossRef]
- Reznikov, V.A.; Vishnivetskaya, L.A.; Volodarskii, L.B. Reaction of 2-substituted 5,5-dimethyl-4-oxo-1-pyrroline-1-oxides with electrophilic reagents. Rus. Chem. Bull. 1990, 39, 335–340. [Google Scholar] [CrossRef]
- Reznikov, V.A.; Volodarskii, L.B. Reaction of 2-substituted 5,5-dimethyl-4-oxo-1-pyrroline-1-oxides with nucleophilic reagents and synthesis of nitroxyl radicals—derivatives of pyrrolidine. Bull. Acad. Sci. USSR, Div. Chem. Sci. 1990, 39, 329–334. [Google Scholar] [CrossRef]
- Bobko, A.A.; Kirilyuk, I.A.; Gritsan, N.P.; Polovyanenko, D.N.; Grigor’ev, I.A.; Khramtsov, V.V.; Bagryanskaya, E.G. EPR and quantum chemical studies of the pH-sensitive imidazoline and imidazolidine nitroxides with bulky substituents. Appl. Magn. Reson. 2010, 39, 437–451. [Google Scholar] [CrossRef]
- Bobko, A.A.; Kirilyuk, I.A.; Grigor’ev, I.A.; Zweier, J.L.; Khramtsov, V.V. Reversible reduction of nitroxides to hydroxylamines: Roles for ascorbate and glutathione. Free Radic. Biol. Med. 2007, 42, 404–412. [Google Scholar] [CrossRef]
- Reichardt, C.H. Solvent Effects in Organic Chemistry; Wiley-VCH: Marburg, Germany, 1990; pp. 147–181. ISBN 0-89573-584-5. [Google Scholar]
- Nkolo, P.; Audran, G.; Bikanga, R.; Bremond, P.; Marque, S.R.A.; Roubaud, V. C-ON bond homolysis when too high polarity is detrimental. Org. Biomol. Chem. 2017, 15, 6167–6176. [Google Scholar] [CrossRef]
- Dobrynin, S.A.; Khoroshunova, Y.V.; Kirilyuk, I.A. Synthesis of 2,2,5,5-tetraethyl-3-carboxypyrrolidine 1-oxyl. 2019, RU. Patent No. 2 702 331. In russian. Available online: https://www1.fips.ru/ofpstorage/Doc/IZPM/RUNWC1/000/000/002/702/331/%D0%98%D0%97-02702331-00001/document.pdf (accessed on 14 February 2020).
- Sheldrick, G.M. SADABS, v. 2008/1. Bruker AXS Inc.: Madison, WI, USA, 2008. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. Sect. C: Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef]
- Engel, P.S.; Wu, A. Photolysis of a J-Azo perester, a bifunctional initiator. The fragmentation rate of a β-peroxy ester radical determined by the cyclopropylcarbinyl clock method. J. Org. Chem. 1994, 59, 3969–3974. [Google Scholar] [CrossRef]
- Billera, C.F.; Dunn, T.B.; Barry, D.A.; Engel, P.S. Thermolysis of acylazo O-trimethylsilyl cyanohydrins: Azoalkanes yielding captodatively substituted radicals. J. Org. Chem. 1998, 63, 9763–9768. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1, 2, 4, 5 are available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
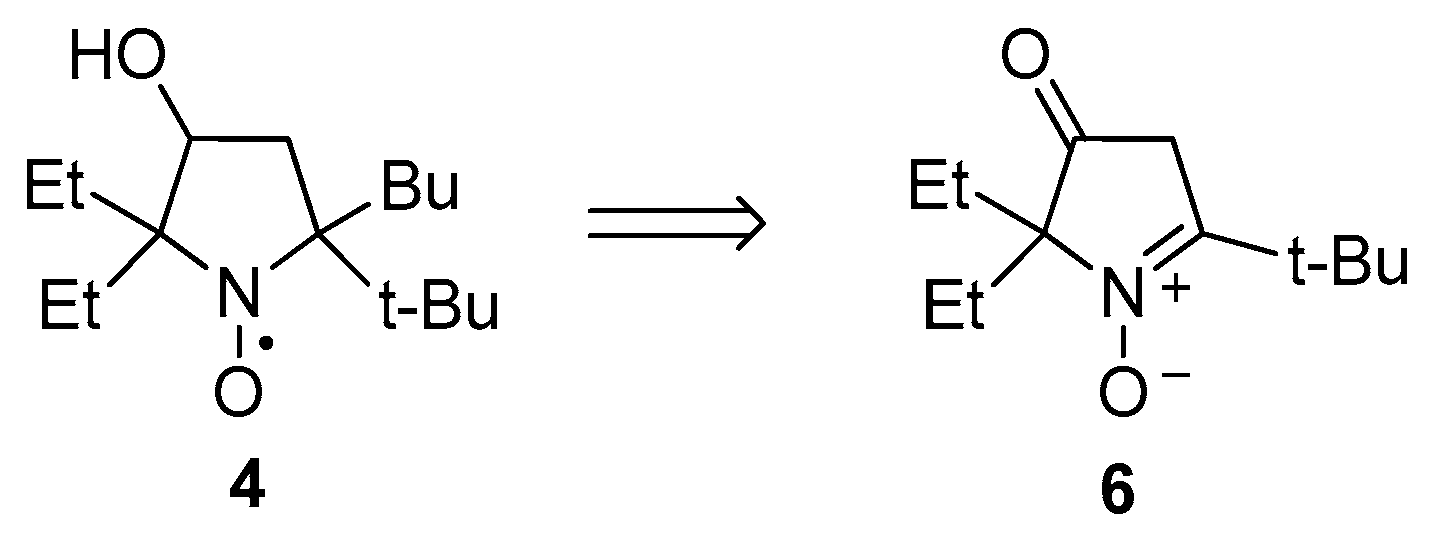{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nitroxide | Solvent | Parameters of EPR Spectra | Kp (Octanol–H2O) | kRed × 104, M−1c−1 | ||
|---|---|---|---|---|---|---|
| aN, mT | aH, mT | Hp-p, mT | ||||
| 1 | H2O | 1.56 | 0.2 | 0.125 | - | 12.5 ± 0.3 |
| 20 ± 10 [19] | ||||||
| H2O-MeOH 1:1 | 1.52 | 0.202 | 0.6 ± 0.2 | |||
| 2 | H2O | 1.527 | 0.225 | 0.12 | 3.8 ± 0.5 | |
| 3.3 ± 0.8 [20] | ||||||
| H2O-MeOH 1:1 | 1.493 | 0.198 | 1.2 ± 0.1 | |||
| 4 | H2O | 1.605 | 0.214 | 0.124 | 1000 ± 250 | - |
| H2O-MeOH 1:1 | 1.529 | 0.199 | 1.7 ± 0.1 | |||
| 5 | H2O | 1.586 | 0.251 | 0.117 | 850 ± 120 | - |
| H2O-MeOH 1:1 | 1.498 | 0.248 | 0.012 ± 0.03 | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhurko, I.F.; Dobrynin, S.; Gorodetskii, A.A.; Glazachev, Y.I.; Rybalova, T.V.; Chernyak, E.I.; Asanbaeva, N.; Bagryanskaya, E.G.; Kirilyuk, I.A. 2-Butyl-2-tert-butyl-5,5-diethylpyrrolidine-1-oxyls: Synthesis and Properties. Molecules 2020, 25, 845. https://doi.org/10.3390/molecules25040845
Zhurko IF, Dobrynin S, Gorodetskii AA, Glazachev YI, Rybalova TV, Chernyak EI, Asanbaeva N, Bagryanskaya EG, Kirilyuk IA. 2-Butyl-2-tert-butyl-5,5-diethylpyrrolidine-1-oxyls: Synthesis and Properties. Molecules. 2020; 25(4):845. https://doi.org/10.3390/molecules25040845
Chicago/Turabian StyleZhurko, Irina F., Sergey Dobrynin, Artem A. Gorodetskii, Yuri I. Glazachev, Tatyana V. Rybalova, Elena I. Chernyak, Nargiz Asanbaeva, Elena G. Bagryanskaya, and Igor A. Kirilyuk. 2020. "2-Butyl-2-tert-butyl-5,5-diethylpyrrolidine-1-oxyls: Synthesis and Properties" Molecules 25, no. 4: 845. https://doi.org/10.3390/molecules25040845
APA StyleZhurko, I. F., Dobrynin, S., Gorodetskii, A. A., Glazachev, Y. I., Rybalova, T. V., Chernyak, E. I., Asanbaeva, N., Bagryanskaya, E. G., & Kirilyuk, I. A. (2020). 2-Butyl-2-tert-butyl-5,5-diethylpyrrolidine-1-oxyls: Synthesis and Properties. Molecules, 25(4), 845. https://doi.org/10.3390/molecules25040845