Proteasome Inhibitors: Harnessing Proteostasis to Combat Disease

Abstract
1. Introduction
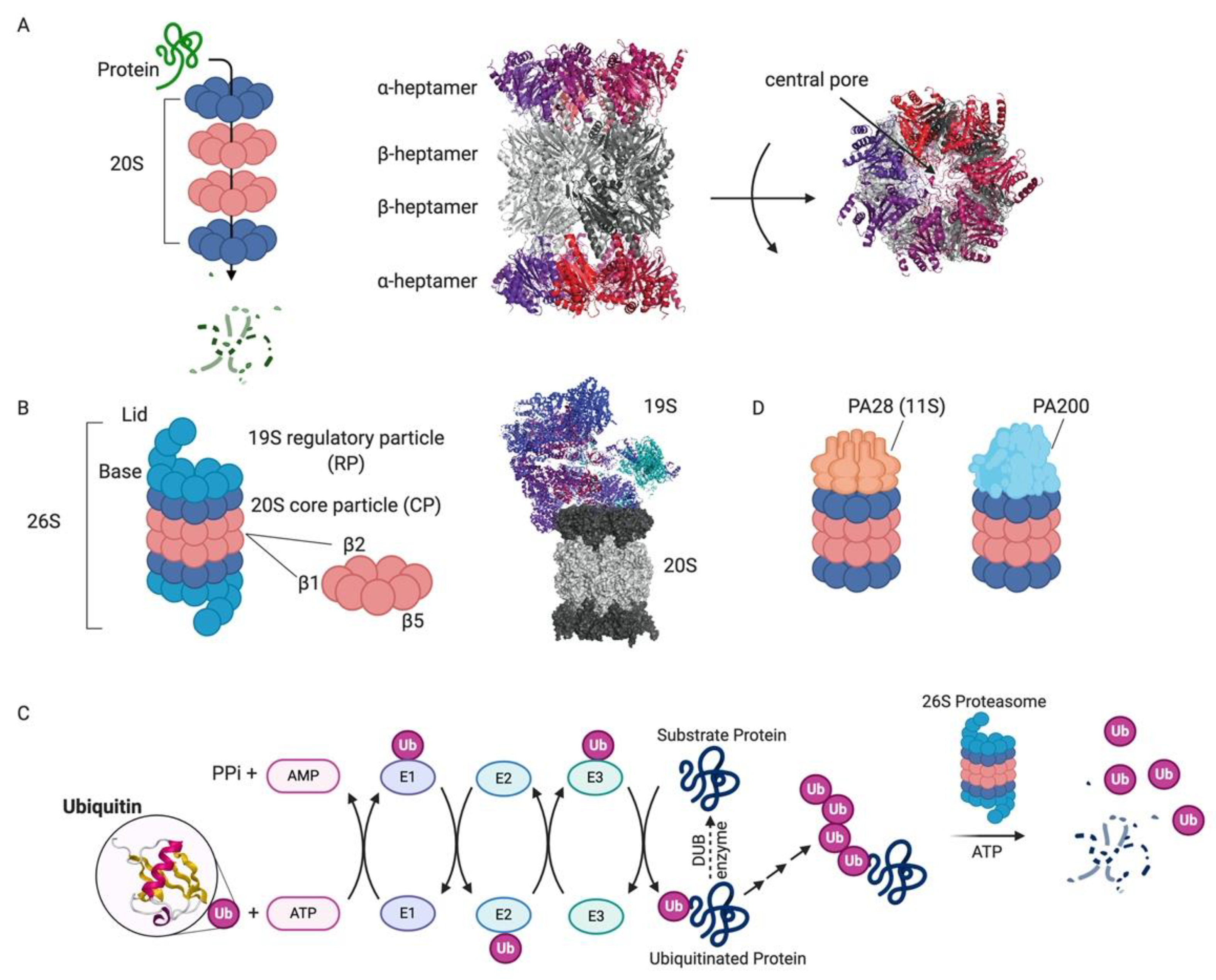
1.1. Proteasome: Structure and Function
1.1.1. 26S Proteasome
1.1.2. Alternative Proteasome Complexes
1.1.3. Immunoproteasome
2. Proteasome Inhibitors
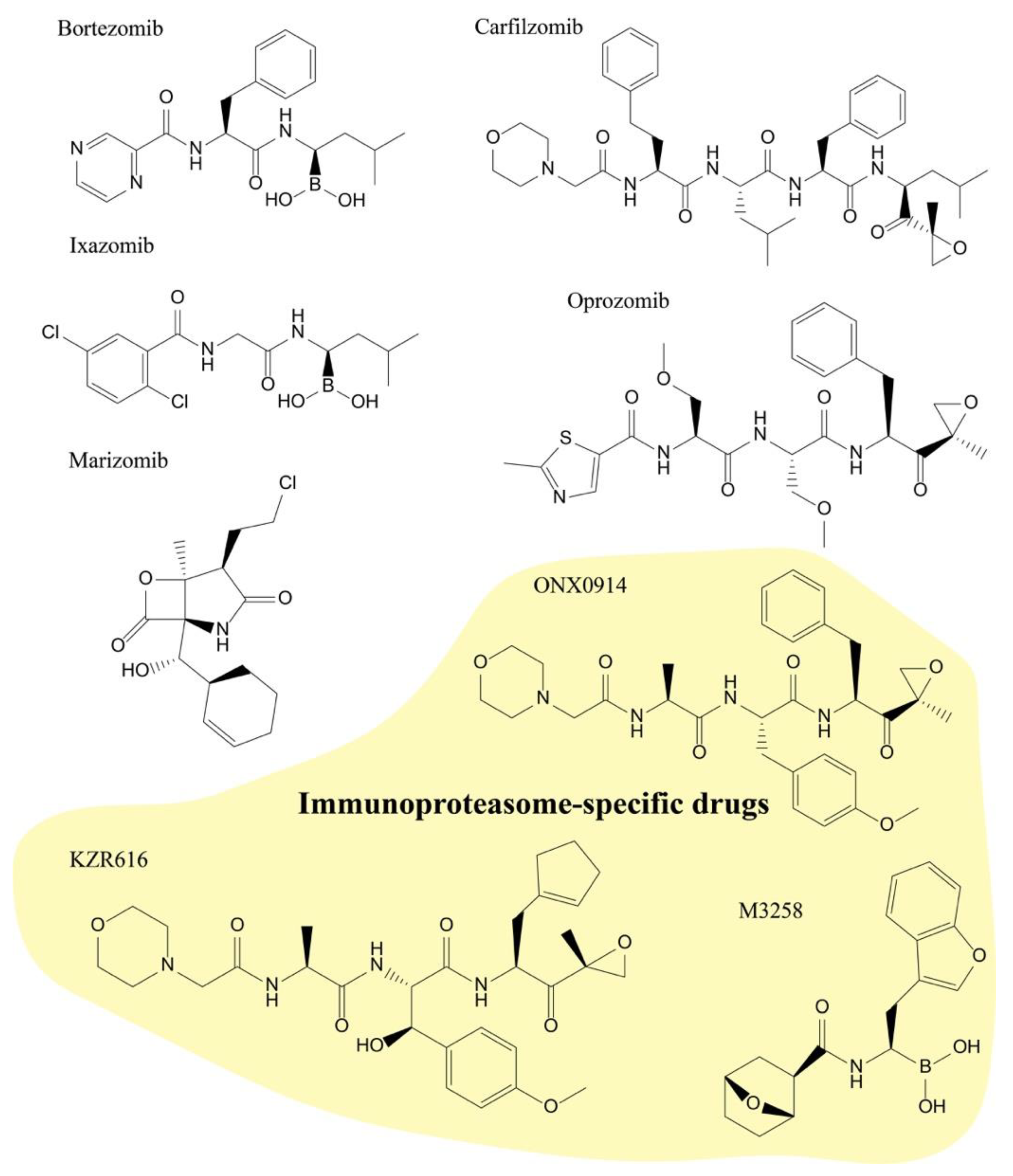
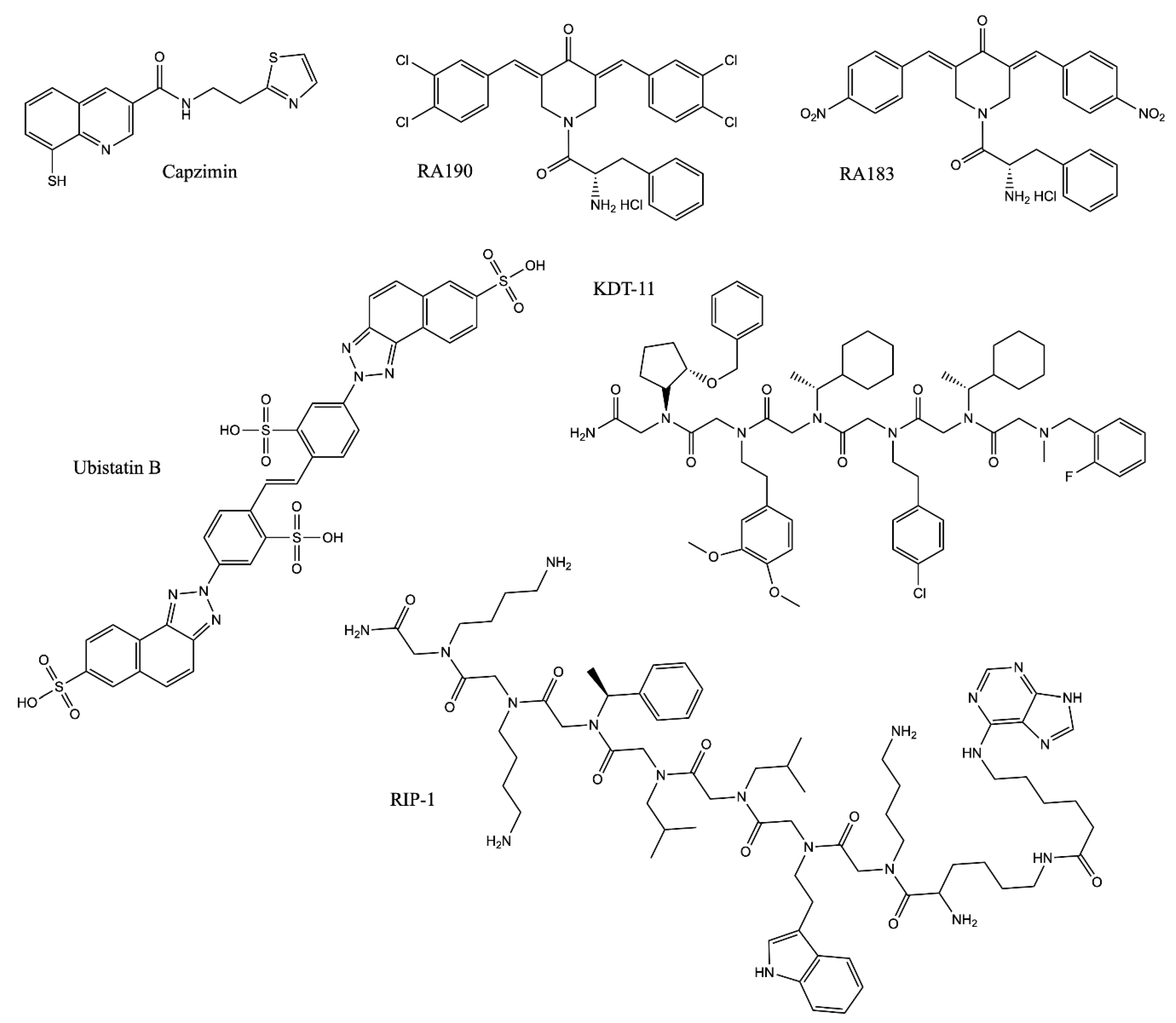
2.1. Structural Characteristics of Proteasome Inhibitors
2.2. Proteasome Inhibitors in Clinic
2.2.1. Bortezomib (Velcade)
2.2.2. Carfilzomib (Kyprolis)
2.2.3. Ixazomib (Ninlaro)
2.3. Proteasome Inhibitors in Clinical Trials
2.3.1. Marizomib
2.3.2. Oprozomib
2.3.3. KZR-616

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Type | IC50 (nM) Against Immuno- and Constitutive-Proteasome [141] | |||||
|---|---|---|---|---|---|---|---|
| LMP7 (β5i) | β5 | LMP2 (β1i) | β1 | MECL-1 (β2i) | β2 | ||
| ONX0914 | Epoxyketone | 39 | 422 | 287 | >12700 | 902 | 927 |
| KZR-616 | Epoxyketone | 39 | 688 | 131 | >10600 | 623 | 604 |
2.4. Proteasome Inhibitors in Lab
2.4.1. Block the Removal of Ubiquitin Chains from Substrates: Rpn11 Inhibitors
2.4.2. Block the Interaction between Substrate and the Proteasome
| Name | Target | IC50/Kd | Mechanism |
|---|---|---|---|
| Capzimin | Rpn11 | 0.3 µM (In vitro Rpn11 assay); 0.6 µM (UbG76V-GFP degradation assay in cells) | Chelating the zinc ion in Rpn11, inactivating its DUB activity [40] |
| Ubistatin B | polyUb | 10 µM (CFTR ubiquitination assay in cells) | Binding to the polyUb chain and blocking the substrate/Ub receptor interaction [151,152] |
| RA190 | Rpn13 | Not Applicable | Covalently binding to cysteine residue 88 (Cys88) of Rpn13 when it is not bound to the proteasome and Uch37 at the proteasome [158,159,160] |
| RA183 | |||
| KDT-11 | Rpn13 | 1.7 µM (Kd) | Peptoid ligand binding to Rpn13 [161] |
| RIP-1 | Rpt4 | 3.0 µM (proteasome-mediated stripping of the Gal4-VP16 protein from DNA in vitro) | Inhibiting the protein unfolding activity of the 19S RP [167,168] |
2.4.3. Block Substrate Translocation into the 20S CP: Proteasome ATPase inhibitors
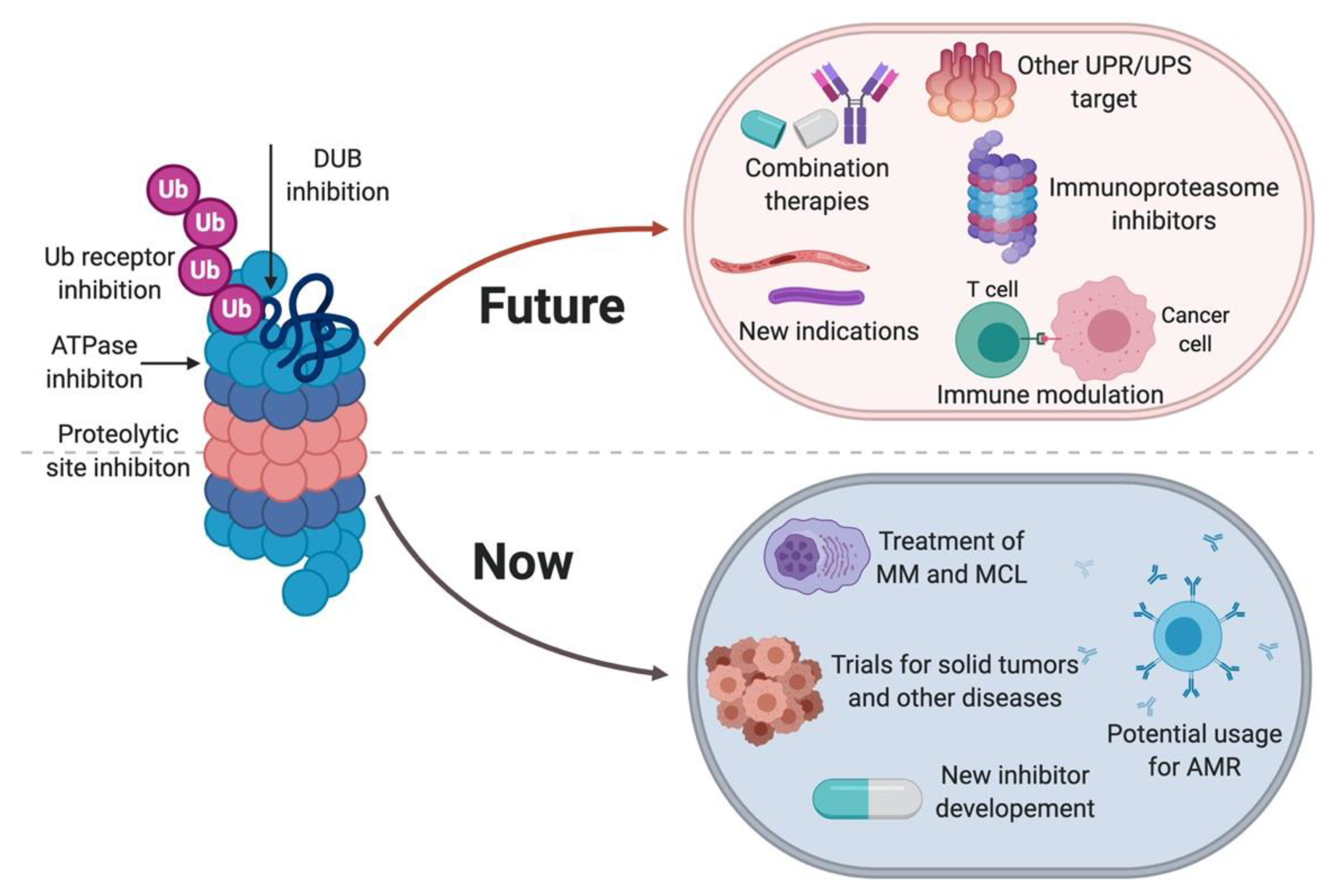
3. Perspective on the Use of Proteasome Inhibitors in Clinic
3.1. Combatting Proteasome Inhibitor Resistance
3.2. Other UPS Inhibitors
3.2.1. VCP/p97 Inhibition
3.2.2. Inhibiting other UPS Components
3.3. Other Indications for Proteasome Inhibitors
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Harper, J.W.; Bennett, E.J. Proteome complexity and the forces that drive proteome imbalance. Nature 2016, 537, 328–338. [Google Scholar] [CrossRef]
- Hershey, J.W.; Sonenberg, N.; Mathews, M.B. Principles of translational control: An overview. Cold Spring Harb. Perspect. Biol. 2012. [Google Scholar] [CrossRef]
- Hetz, C.; Glimcher, L.H. Protein homeostasis networks in physiology and disease. Curr. Opin. Cell Biol. 2011, 23, 123–125. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, A.F.; Goldberg, A.L. Proteasome inhibitors: From research tools to drug candidates. Chem. Biol. 2001, 8, 739–758. [Google Scholar] [CrossRef]
- Finley, D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.C.; Weissman, A.M. The Unfolded Protein Response, Degradation from Endoplasmic Reticulum and Cancer. Genes Cancer 2010, 1, 764–778. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Baldwin, A.S. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-kappaB. J. Clin. Investig. 2001, 107, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Piva, R.; Belardo, G.; Santoro, M.G. NF-kappaB: A stress-regulated switch for cell survival. Antioxid. Redox Signal. 2006, 8, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Opoku-Nsiah, K.A.; Gestwicki, J.E. Aim for the core: Suitability of the ubiquitin-independent 20S proteasome as a drug target in neurodegeneration. Transl. Res. 2018, 198, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.C.; Schuman, E.M. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat. Rev. Neurosci. 2008, 9, 826–838. [Google Scholar] [CrossRef]
- Ramachandran, K.V.; Margolis, S.S. A mammalian nervous-system-specific plasma membrane proteasome complex that modulates neuronal function. Nat. Struct. Mol. Biol. 2017, 24, 419–430. [Google Scholar] [CrossRef]
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef]
- Thibaudeau, T.A.; Smith, D.M. A Practical Review of Proteasome Pharmacology. Pharm. Rev. 2019, 71, 170–197. [Google Scholar] [CrossRef]
- Kisselev, A.F.; van der Linden, W.A.; Overkleeft, H.S. Proteasome inhibitors: An expanding army attacking a unique target. Chem. Biol. 2012, 19, 99–115. [Google Scholar] [CrossRef]
- Schmidt, M.; Finley, D. Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta 2014, 1843, 13–25. [Google Scholar] [CrossRef]
- Cromm, P.M.; Crews, C.M. The Proteasome in Modern Drug Discovery: Second Life of a Highly Valuable Drug Target. ACS Cent. Sci. 2017, 3, 830–838. [Google Scholar] [CrossRef]
- Lobanova, E.S.; Finkelstein, S.; Li, J.; Travis, A.M.; Hao, Y.; Klingeborn, M.; Skiba, N.P.; Deshaies, R.J.; Arshavsky, V.Y. Increased proteasomal activity supports photoreceptor survival in inherited retinal degeneration. Nat. Commun. 2018. [Google Scholar] [CrossRef] [PubMed]
- Etlinger, J.D.; Goldberg, A.L. A soluble ATP-dependent proteolytic system responsible for the degradation of abnormal proteins in reticulocytes. Proc. Natl. Acad. Sci. USA 1977, 74, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Yoshimura, T.; Kumatori, A.; Ichihara, A.; Ikai, A.; Nishigai, M.; Kameyama, K.; Takagi, T. Proteasomes (multi-protease complexes) as 20 S ring-shaped particles in a variety of eukaryotic cells. J. Biol. Chem. 1988, 263, 16209–16217. [Google Scholar] [PubMed]
- Arrigo, A.P.; Tanaka, K.; Goldberg, A.L.; Welch, W.J. Identity of the 19S “prosome” particle with the large multifunctional protease complex of mammalian cells (the proteasome). Nature 1988, 331, 192–194. [Google Scholar] [CrossRef] [PubMed]
- Sauer, R.T.; Baker, T.A. AAA+ proteases: ATP-fueled machines of protein destruction. Annu. Rev. Biochem. 2011, 80, 587–612. [Google Scholar] [CrossRef] [PubMed]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef]
- Bogyo, M.; Gaczynska, M.; Ploegh, H.L. Proteasome inhibitors and antigen presentation. Biopolymers 1997, 43, 269–280. [Google Scholar] [CrossRef]
- Groll, M.; Bajorek, M.; Kohler, A.; Moroder, L.; Rubin, D.M.; Huber, R.; Glickman, M.H.; Finley, D. A gated channel into the proteasome core particle. Nat. Struct. Biol. 2000, 7, 1062–1067. [Google Scholar] [CrossRef]
- Whitby, F.G.; Masters, E.I.; Kramer, L.; Knowlton, J.R.; Yao, Y.; Wang, C.C.; Hill, C.P. Structural basis for the activation of 20S proteasomes by 11S regulators. Nature 2000, 408, 115–120. [Google Scholar] [CrossRef]
- Finley, D.; Chen, X.; Walters, K.J. Gates, Channels, and Switches: Elements of the Proteasome Machine. Trends Biochem. Sci. 2016, 41, 77–93. [Google Scholar] [CrossRef]
- Kish-Trier, E.; Hill, C.P. Structural biology of the proteasome. Annu. Rev. Biophys. 2013, 42, 29–49. [Google Scholar] [CrossRef] [PubMed]
- Thrower, J.S.; Hoffman, L.; Rechsteiner, M.; Pickart, C.M. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000, 19, 94–102. [Google Scholar] [CrossRef]
- Verma, R.; Deshaies, R.J. A proteasome howdunit: The case of the missing signal. Cell 2000, 101, 341–344. [Google Scholar] [CrossRef]
- Glickman, M.H.; Rubin, D.M.; Fried, V.A.; Finley, D. The regulatory particle of the Saccharomyces cerevisiae proteasome. Mol. Cell Biol. 1998, 18, 3149–3362. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Aravind, L.; Oania, R.; McDonald, W.H.; Yates, J.R.; Koonin, E.V.; Deshaies, R.J. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 2002, 298, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Worden, E.J.; Dong, K.C.; Martin, A. An AAA Motor-Driven Mechanical Switch in Rpn11 Controls Deubiquitination at the 26S Proteasome. Mol. Cell 2017, 67, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Nalepa, G.; Wade Harper, J. Therapeutic anti-cancer targets upstream of the proteasome. Cancer Treat. Rev. 2003, 29 (Suppl. 1), 49–57. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.; Da Silva Sil Dos Santos, B.; Wang, F.; Ma, Y.; Perez, C.; Yang, Y.; Peng, J.; Cohen, S.M.; Chou, T.F.; et al. Epidithiodiketopiperazines Inhibit Protein Degradation by Targeting Proteasome Deubiquitinase Rpn11. Cell Chem. Biol. 2018, 25, 1350–1358. [Google Scholar] [CrossRef]
- Lauinger, L.; Li, J.; Shostak, A.; Cemel, I.A.; Ha, N.; Zhang, Y.; Merkl, P.E.; Obermeyer, S.; Stankovic-Valentin, N.; Schafmeier, T.; et al. Thiolutin is a zinc chelator that inhibits the Rpn11 and other JAMM metalloproteases. Nat. Chem. Biol. 2017, 13, 709–714. [Google Scholar] [CrossRef]
- Li, J.; Yakushi, T.; Parlati, F.; Mackinnon, A.L.; Perez, C.; Ma, Y.; Carter, K.P.; Colayco, S.; Magnuson, G.; Brown, B.; et al. Capzimin is a potent and specific inhibitor of proteasome isopeptidase Rpn11. Nat. Chem. Biol. 2017, 13, 486–493. [Google Scholar] [CrossRef]
- Noda, C.; Tanahashi, N.; Shimbara, N.; Hendil, K.B.; Tanaka, K. Tissue distribution of constitutive proteasomes, immunoproteasomes, and PA28 in rats. Biochem. Biophys. Res. Commun. 2000, 277, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.P.; Slaughter, C.A.; DeMartino, G.N. Identification, purification, and characterization of a protein activator (PA28) of the 20 S proteasome (macropain). J. Biol. Chem. 1992, 267, 10515–10523. [Google Scholar] [PubMed]
- Dubiel, W.; Pratt, G.; Ferrell, K.; Rechsteiner, M. Purification of an 11 S regulator of the multicatalytic protease. J. Biol. Chem. 1992, 267, 22369–22377. [Google Scholar] [PubMed]
- Knowlton, J.R.; Johnston, S.C.; Whitby, F.G.; Realini, C.; Zhang, Z.; Rechsteiner, M.; Hill, C.P. Structure of the proteasome activator REGalpha (PA28alpha). Nature 1997, 390, 639–643. [Google Scholar] [CrossRef]
- Realini, C.; Jensen, C.C.; Zhang, Z.; Johnston, S.C.; Knowlton, J.R.; Hill, C.P.; Rechsteiner, M. Characterization of recombinant REGalpha, REGbeta, and REGgamma proteasome activators. J. Biol. Chem. 1997, 272, 25483–25492. [Google Scholar] [CrossRef] [PubMed]
- Sijts, A.; Sun, Y.; Janek, K.; Kral, S.; Paschen, A.; Schadendorf, D.; Kloetzel, P.M. The role of the proteasome activator PA28 in MHC class I antigen processing. Mol. Immunol. 2002, 39, 165–169. [Google Scholar] [CrossRef]
- Groettrup, M.; Soza, A.; Eggers, M.; Kuehn, L.; Dick, T.P.; Schild, H.; Rammensee, H.G.; Koszinowski, U.H.; Kloetzel, P.M. A role for the proteasome regulator PA28alpha in antigen presentation. Nature 1996, 381, 166–168. [Google Scholar] [CrossRef]
- Li, X.; Amazit, L.; Long, W.; Lonard, D.M.; Monaco, J.J.; O’Malley, B.W. Ubiquitin- and ATP-independent proteolytic turnover of p21 by the REGgamma-proteasome pathway. Mol. Cell 2007, 26, 831–842. [Google Scholar] [CrossRef]
- Chen, X.; Barton, L.F.; Chi, Y.; Clurman, B.E.; Roberts, J.M. Ubiquitin-independent degradation of cell-cycle inhibitors by the REGgamma proteasome. Mol. Cell 2007, 26, 843–852. [Google Scholar] [CrossRef]
- Sadre-Bazzaz, K.; Whitby, F.G.; Robinson, H.; Formosa, T.; Hill, C.P. Structure of a Blm10 complex reveals common mechanisms for proteasome binding and gate opening. Mol. Cell 2010, 37, 728–735. [Google Scholar] [CrossRef]
- Fehlker, M.; Wendler, P.; Lehmann, A.; Enenkel, C. Blm3 is part of nascent proteasomes and is involved in a late stage of nuclear proteasome assembly. EMBO Rep. 2003, 4, 959–963. [Google Scholar] [CrossRef]
- Doherty, K.; Pramanik, A.; Pride, L.; Lukose, J.; Moore, C.W. Expression of the expanded YFL007w ORF and assignment of the gene name BLM10. Yeast 2004, 21, 1021–1023. [Google Scholar] [CrossRef] [PubMed]
- Ustrell, V.; Hoffman, L.; Pratt, G.; Rechsteiner, M. PA200, a nuclear proteasome activator involved in DNA repair. EMBO J. 2002, 21, 3516–3525. [Google Scholar] [CrossRef] [PubMed]
- Dange, T.; Smith, D.; Noy, T.; Rommel, P.C.; Jurzitza, L.; Cordero, R.J.; Legendre, A.; Finley, D.; Goldberg, A.L.; Schmidt, M. Blm10 protein promotes proteasomal substrate turnover by an active gating mechanism. J. Biol. Chem. 2011, 286, 42830–42839. [Google Scholar] [CrossRef]
- Fabre, B.; Lambour, T.; Garrigues, L.; Ducoux-Petit, M.; Amalric, F.; Monsarrat, B.; Burlet-Schiltz, O.; Bousquet-Dubouch, M.P. Label-free quantitative proteomics reveals the dynamics of proteasome complexes composition and stoichiometry in a wide range of human cell lines. J. Proteome Res. 2014, 13, 3027–3037. [Google Scholar] [CrossRef] [PubMed]
- Welk, V.; Coux, O.; Kleene, V.; Abeza, C.; Trumbach, D.; Eickelberg, O.; Meiners, S. Inhibition of Proteasome Activity Induces Formation of Alternative Proteasome Complexes. J. Biol. Chem. 2016, 291, 13147–13159. [Google Scholar] [CrossRef] [PubMed]
- Toste Rêgo, A.; da Fonseca, P.C.A. Characterization of Fully Recombinant Human 20S and 20S-PA200 Proteasome Complexes. Mol. Cell 2019, 76, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Kopp, F.; Dahlmann, B.; Kuehn, L. Reconstitution of hybrid proteasomes from purified PA700-20 S complexes and PA28alphabeta activator: Ultrastructure and peptidase activities. J. Mol. Biol. 2001, 313, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Dahlmann, B. Mammalian proteasome subtypes: Their diversity in structure and function. Arch. Biochem. Biophys. 2016, 591, 132–140. [Google Scholar] [CrossRef]
- Cascio, P.; Call, M.; Petre, B.M.; Walz, T.; Goldberg, A.L. Properties of the hybrid form of the 26S proteasome containing both 19S and PA28 complexes. EMBO J. 2002, 21, 2636–2645. [Google Scholar] [CrossRef]
- Baugh, J.M.; Viktorova, E.G.; Pilipenko, E.V. Proteasomes can degrade a significant proportion of cellular proteins independent of ubiquitination. J. Mol. Biol. 2009, 386, 814–827. [Google Scholar] [CrossRef] [PubMed]
- Erales, J.; Coffino, P. Ubiquitin-independent proteasomal degradation. Biochim. Biophys. Acta 2014, 1843, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Pickart, C.M.; Coffino, P. Determinants of proteasome recognition of ornithine decarboxylase, a ubiquitin-independent substrate. EMBO J. 2003, 22, 1488–1496. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Matsufuji, S.; Kameji, T.; Hayashi, S.; Igarashi, K.; Tamura, T.; Tanaka, K.; Ichihara, A. Ornithine decarboxylase is degraded by the 26S proteasome without ubiquitination. Nature 1992, 360, 597–599. [Google Scholar] [CrossRef] [PubMed]
- Ferrington, D.A.; Gregerson, D.S. Immunoproteasomes: Structure, function, and antigen presentation. Prog. Mol. Biol. Transl. Sci. 2012, 109, 75–112. [Google Scholar] [PubMed]
- Kimura, H.; Caturegli, P.; Takahashi, M.; Suzuki, K. New Insights into the Function of the Immunoproteasome in Immune and Nonimmune Cells. J. Immunol. Res. 2015. [Google Scholar] [CrossRef]
- Rock, K.L.; York, I.A.; Saric, T.; Goldberg, A.L. Protein degradation and the generation of MHC class I-presented peptides. Adv. Immunol. 2002, 80, 1–70. [Google Scholar]
- Tanaka, K. Role of proteasomes modified by interferon-gamma in antigen processing. J. Leukoc. Biol. 1994, 56, 571–575. [Google Scholar] [CrossRef]
- Van Kaer, L.; Ashton-Rickardt, P.G.; Eichelberger, M.; Gaczynska, M.; Nagashima, K.; Rock, K.L.; Goldberg, A.L.; Doherty, P.C.; Tonegawa, S. Altered peptidase and viral-specific T cell response in LMP2 mutant mice. Immunity 1994, 1, 533–541. [Google Scholar] [CrossRef]
- Gaczynska, M.; Rock, K.L.; Goldberg, A.L. Gamma-interferon and expression of MHC genes regulate peptide hydrolysis by proteasomes. Nature 1993, 365, 264–267. [Google Scholar] [CrossRef]
- Eskandari, S.K.; Seelen, M.A.J.; Lin, G.; Azzi, J.R. The immunoproteasome: An old player with a novel and emerging role in alloimmunity. Am. J. Transpl. 2017, 17, 3033–3039. [Google Scholar] [CrossRef] [PubMed]
- Murata, S.; Takahama, Y.; Kasahara, M.; Tanaka, K. The immunoproteasome and thymoproteasome: Functions, evolution and human disease. Nat. Immunol. 2018, 19, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Heink, S.; Ludwig, D.; Kloetzel, P.-M.; Krüger, E. IFN-gamma-induced immune adaptation of the proteasome system is an accelerated and transient response. Proc. Natl. Acad. Sci. USA 2005, 102, 9241–9246. [Google Scholar] [CrossRef]
- Griffin, T.A.; Nandi, D.; Cruz, M.; Fehling, H.J.; Kaer, L.V.; Monaco, J.J.; Colbert, R.A. Immunoproteasome assembly: Cooperative incorporation of interferon gamma (IFN-gamma)-inducible subunits. J. Exp. Med. 1998, 187, 97–104. [Google Scholar] [CrossRef]
- Groll, M.; Huber, R. Inhibitors of the eukaryotic 20S proteasome core particle: A structural approach. Biochim. Biophys. Acta 2004, 1695, 33–44. [Google Scholar] [CrossRef]
- Kuhn, D.J.; Orlowski, R.Z.; Bjorklund, C.C. Second generation proteasome inhibitors: Carfilzomib and immunoproteasome-specific inhibitors (IPSIs). Curr. Cancer Drug Targets 2011, 11, 285–295. [Google Scholar] [CrossRef]
- Basler, M.; Mundt, S.; Bitzer, A.; Schmidt, C.; Groettrup, M. The immunoproteasome: A novel drug target for autoimmune diseases. Clin. Exp. Rheumatol. 2015, 33, 74–79. [Google Scholar]
- Santos, R.L.A.; Bai, L.; Singh, P.K.; Murakami, N.; Fan, H.; Zhan, W.; Zhu, Y.; Jiang, X.; Zhang, K.; Assker, J.P.; et al. Structure of human immunoproteasome with a reversible and noncompetitive inhibitor that selectively inhibits activated lymphocytes. Nat. Commun. 2017. [Google Scholar] [CrossRef]
- Huber, E.M.; Basler, M.; Schwab, R.; Heinemeyer, W.; Kirk, C.J.; Groettrup, M.; Groll, M. Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 2012, 148, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Murata, S.; Sasaki, K.; Kishimoto, T.; Niwa, S.; Hayashi, H.; Takahama, Y.; Tanaka, K. Regulation of CD8+ T cell development by thymus-specific proteasomes. Science 2007, 316, 1349–1353. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Takada, K.; Ohte, Y.; Kondo, H.; Sorimachi, H.; Tanaka, K.; Takahama, Y.; Murata, S. Thymoproteasomes produce unique peptide motifs for positive selection of CD8(+) T cells. Nat. Commun. 2015. [Google Scholar] [CrossRef] [PubMed]
- Kniepert, A.; Groettrup, M. The unique functions of tissue-specific proteasomes. Trends Biochem. Sci. 2014, 39, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Khor, B.; Bredemeyer, A.L.; Huang, C.-Y.; Turnbull, I.R.; Evans, R.; Maggi, L.B.; White, J.M.; Walker, L.M.; Carnes, K.; Hess, R.A.; et al. Proteasome activator PA200 is required for normal spermatogenesis. Mol. Cell Biol. 2006, 26, 2999–3007. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.-X.; Pang, Y.; Liu, C.H.; Haratake, K.; Du, B.-Y.; Ji, D.-Y.; Wang, G.-F.; Zhu, Q.-Q.; Song, W.; Yu, Y.; et al. Acetylation-mediated proteasomal degradation of core histones during DNA repair and spermatogenesis. Cell 2013, 153, 1012–1024. [Google Scholar] [CrossRef] [PubMed]
- BioRender.com, version 2017; M5V 2B7, Created Graphical Illustrations, BioRender 2017–2020; BioRender Customer Service: Toronto, ON, Canada, 2017.
- Lee, D.H.; Goldberg, A.L. Proteasome inhibitors: Valuable new tools for cell biologists. Trends Cell Biol. 1998, 8, 397–403. [Google Scholar] [CrossRef]
- Wiest, D.L.; Burkhardt, J.K.; Hester, S.; Hortsch, M.; Meyer, D.I.; Argon, Y. Membrane biogenesis during B cell differentiation: Most endoplasmic reticulum proteins are expressed coordinately. J. Cell Biol. 1990, 110, 1501–1511. [Google Scholar] [CrossRef]
- Meister, S.; Schubert, U.; Neubert, K.; Herrmann, K.; Burger, R.; Gramatzki, M.; Hahn, S.; Schreiber, S.; Wilhelm, S.; Herrmann, M.; et al. Extensive immunoglobulin production sensitizes myeloma cells for proteasome inhibition. Cancer Res. 2007, 67, 1783–1792. [Google Scholar] [CrossRef]
- Deshaies, R.J. Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol. 2014. [Google Scholar] [CrossRef]
- Smith, M.H.; Ploegh, H.L.; Weissman, J.S. Road to ruin: Targeting proteins for degradation in the endoplasmic reticulum. Science 2011, 334, 1086–1090. [Google Scholar] [CrossRef]
- Sosič, I.; Gobec, M.; Brus, B.; Knez, D.; Živec, M.; Konc, J.; Lešnik, S.; Ogrizek, M.; Obreza, A.; Žigon, D.; et al. Nonpeptidic Selective Inhibitors of the Chymotrypsin-Like (β5 i) Subunit of the Immunoproteasome. Angew. Chem. Int. Ed Engl. 2016, 55, 5745–5748. [Google Scholar] [CrossRef]
- Cui, H.; Baur, R.; Le Chapelain, C.; Dubiella, C.; Heinemeyer, W.; Huber, E.M.; Groll, M. Structural Elucidation of a Nonpeptidic Inhibitor Specific for the Human Immunoproteasome. Chembiochem Eur. J. Chem. Biol. 2017, 18, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Sonneveld, P.; Schuster, M.W.; Irwin, D.; Stadtmauer, E.A.; Facon, T.; Harousseau, J.-L.; Ben-Yehuda, D.; Lonial, S.; Goldschmidt, H.; et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2005, 352, 2487–2498. [Google Scholar] [CrossRef]
- Kane, R.C.; Farrell, A.T.; Sridhara, R.; Pazdur, R. United States Food and Drug Administration approval summary: Bortezomib for the treatment of progressive multiple myeloma after one prior therapy. Clin. Cancer Res. 2006, 12, 2955–2960. [Google Scholar] [CrossRef]
- Sonneveld, P.; Schmidt-Wolf, I.G.H.; van der Holt, B.; El Jarari, L.; Bertsch, U.; Salwender, H.; Zweegman, S.; Vellenga, E.; Broyl, A.; Blau, I.W.; et al. Bortezomib induction and maintenance treatment in patients with newly diagnosed multiple myeloma: Results of the randomized phase III HOVON-65/GMMG-HD4 trial. J. Clin. Oncol. 2012, 30, 2946–2955. [Google Scholar] [CrossRef]
- Demo, S.D.; Kirk, C.J.; Aujay, M.A.; Buchholz, T.J.; Dajee, M.; Ho, M.N.; Jiang, J.; Laidig, G.J.; Lewis, E.R.; Parlati, F.; et al. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res. 2007, 67, 6383–6391. [Google Scholar] [CrossRef]
- Kane, R.C.; Bross, P.F.; Farrell, A.T.; Pazdur, R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncology 2003, 8, 508–513. [Google Scholar] [CrossRef]
- Aghajanian, C.; Soignet, S.; Dizon, D.S.; Pien, C.S.; Adams, J.; Elliott, P.J.; Sabbatini, P.; Miller, V.; Hensley, M.L.; Pezzulli, S.; et al. A phase I trial of the novel proteasome inhibitor PS341 in advanced solid tumor malignancies. Clin. Cancer Res. 2002, 8, 2505–2511. [Google Scholar]
- Annunziata, C.M.; Davis, R.E.; Demchenko, Y.; Bellamy, W.; Gabrea, A.; Zhan, F.; Lenz, G.; Hanamura, I.; Wright, G.; Xiao, W.; et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007, 12, 115–130. [Google Scholar] [CrossRef]
- Keats, J.J.; Fonseca, R.; Chesi, M.; Schop, R.; Baker, A.; Chng, W.-J.; Van Wier, S.; Tiedemann, R.; Shi, C.-X.; Sebag, M.; et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell 2007, 12, 131–144. [Google Scholar] [CrossRef]
- Hideshima, T.; Richardson, P.; Chauhan, D.; Palombella, V.J.; Elliott, P.J.; Adams, J.; Anderson, K.C. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001, 61, 3071–3076. [Google Scholar]
- Hideshima, T.; Chauhan, D.; Richardson, P.; Mitsiades, C.; Mitsiades, N.; Hayashi, T.; Munshi, N.; Dang, L.; Castro, A.; Palombella, V.; et al. NF-kappa B as a therapeutic target in multiple myeloma. J. Biol. Chem. 2002, 277, 16639–16647. [Google Scholar] [CrossRef] [PubMed]
- Kupperman, E.; Lee, E.C.; Cao, Y.; Bannerman, B.; Fitzgerald, M.; Berger, A.; Yu, J.; Yang, Y.; Hales, P.; Bruzzese, F.; et al. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010, 70, 1970–1980. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-J.; Aujay, M.A.; Bennett, M.K.; Dajee, M.; Demo, S.D.; Fang, Y.; Ho, M.N.; Jiang, J.; Kirk, C.J.; Laidig, G.J.; et al. Design and synthesis of an orally bioavailable and selective peptide epoxyketone proteasome inhibitor (PR-047). J. Med. Chem. 2009, 52, 3028–3038. [Google Scholar] [CrossRef]
- Oerlemans, R.; Franke, N.E.; Assaraf, Y.G.; Cloos, J.; van Zantwijk, I.; Berkers, C.R.; Scheffer, G.L.; Debipersad, K.; Vojtekova, K.; Lemos, C.; et al. Molecular basis of bortezomib resistance: Proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008, 112, 2489–2499. [Google Scholar] [CrossRef]
- Lü, S.; Yang, J.; Song, X.; Gong, S.; Zhou, H.; Guo, L.; Song, N.; Bao, X.; Chen, P.; Wang, J. Point mutation of the proteasome beta5 subunit gene is an important mechanism of bortezomib resistance in bortezomib-selected variants of Jurkat T cell lymphoblastic lymphoma/leukemia line. J. Pharm. Exp. Ther. 2008, 326, 423–431. [Google Scholar] [CrossRef]
- Rückrich, T.; Kraus, M.; Gogel, J.; Beck, A.; Ovaa, H.; Verdoes, M.; Overkleeft, H.S.; Kalbacher, H.; Driessen, C. Characterization of the ubiquitin-proteasome system in bortezomib-adapted cells. Leukemia 2009, 23, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, D.J.; Berkova, Z.; Jones, R.J.; Woessner, R.; Bjorklund, C.C.; Ma, W.; Davis, R.E.; Lin, P.; Wang, H.; Madden, T.L.; et al. Targeting the insulin-like growth factor-1 receptor to overcome bortezomib resistance in preclinical models of multiple myeloma. Blood 2012, 120, 3260–3270. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, N.; Mitsiades, C.S.; Poulaki, V.; Chauhan, D.; Fanourakis, G.; Gu, X.; Bailey, C.; Joseph, M.; Libermann, T.A.; Treon, S.P.; et al. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proc. Natl. Acad. Sci. USA 2002, 99, 14374–14379. [Google Scholar] [CrossRef] [PubMed]
- Niewerth, D.; Jansen, G.; Assaraf, Y.G.; Zweegman, S.; Kaspers, G.J.L.; Cloos, J. Molecular basis of resistance to proteasome inhibitors in hematological malignancies. Drug Resist. Updat. Rev. Comment Antimicrob. Anticancer Chemother. 2015, 18, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Weller, E.; Lonial, S.; Jakubowiak, A.J.; Jagannath, S.; Raje, N.S.; Avigan, D.E.; Xie, W.; Ghobrial, I.M.; Schlossman, R.L.; et al. Lenalidomide, bortezomib, and dexamethasone combination therapy in patients with newly diagnosed multiple myeloma. Blood 2010, 116, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wu, Y.; Zhou, X.; Xu, J.; Zhu, W.; Shu, Y.; Liu, P. Efficacy of therapy with bortezomib in solid tumors: A review based on 32 clinical trials. Future Oncol. Lond. Engl. 2014, 10, 1795–1807. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.B.; Crews, C.M. From epoxomicin to carfilzomib: Chemistry, biology, and medical outcomes. Nat. Prod. Rep. 2013, 30, 600–604. [Google Scholar] [CrossRef]
- Harshbarger, W.; Miller, C.; Diedrich, C.; Sacchettini, J. Crystal structure of the human 20S proteasome in complex with carfilzomib. Struct. Lond. Engl. 1993 2015, 23, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Moreau, P.; Palumbo, A.; Joshua, D.; Pour, L.; Hájek, R.; Facon, T.; Ludwig, H.; Oriol, A.; Goldschmidt, H.; et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): A randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016, 17, 27–38. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Goldschmidt, H.; Niesvizky, R.; Joshua, D.; Chng, W.-J.; Oriol, A.; Orlowski, R.Z.; Ludwig, H.; Facon, T.; Hajek, R.; et al. Carfilzomib or bortezomib in relapsed or refractory multiple myeloma (ENDEAVOR): An interim overall survival analysis of an open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 1327–1337. [Google Scholar] [CrossRef]
- Landgren, O.; Sonneveld, P.; Jakubowiak, A.; Mohty, M.; Iskander, K.S.; Mezzi, K.; Siegel, D.S. Carfilzomib with immunomodulatory drugs for the treatment of newly diagnosed multiple myeloma. Leukemia 2019, 33, 2127–2143. [Google Scholar] [CrossRef]
- Waxman, A.J.; Clasen, S.; Hwang, W.-T.; Garfall, A.; Vogl, D.T.; Carver, J.; O’Quinn, R.; Cohen, A.D.; Stadtmauer, E.A.; Ky, B.; et al. Carfilzomib-Associated Cardiovascular Adverse Events: A Systematic Review and Meta-analysis. JAMA Oncol. 2018. [Google Scholar] [CrossRef]
- Jakubowiak, A.J.; DeCara, J.M.; Mezzi, K. Cardiovascular events during carfilzomib therapy for relapsed myeloma: Practical management aspects from two case studies. Hematol. Amst. Neth. 2017, 22, 585–591. [Google Scholar] [CrossRef]
- Lendvai, N.; Tsakos, I.; Devlin, S.M.; Schaffer, W.L.; Hassoun, H.; Lesokhin, A.M.; Landau, H.; Korde, N.; Mailankody, S.; Smith, E.; et al. Predictive biomarkers and practical considerations in the management of carfilzomib-associated cardiotoxicity. Leuk. Lymphoma 2018, 59, 1981–1985. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Burris, H.A.; Gordon, M.; Lee, P.; Sausville, E.A.; Rosen, P.J.; Patnaik, A.; Cutler, R.E.; Wang, Z.; Lee, S.; et al. A phase I/II study of carfilzomib 2-10-min infusion in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013, 72, 861–868. [Google Scholar] [CrossRef]
- Shirley, M. Ixazomib: First Global Approval. Drugs 2016, 76, 405–411. [Google Scholar] [CrossRef]
- Dick, L.R.; Fleming, P.E. Building on bortezomib: Second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov. Today 2010, 15, 243–249. [Google Scholar] [CrossRef]
- Spencer, A.; Harrison, S.; Zonder, J.; Badros, A.; Laubach, J.; Bergin, K.; Khot, A.; Zimmerman, T.; Chauhan, D.; Levin, N.; et al. A phase 1 clinical trial evaluating marizomib, pomalidomide and low-dose dexamethasone in relapsed and refractory multiple myeloma (NPI-0052-107): Final study results. Br. J. Haematol. 2018, 180, 41–51. [Google Scholar] [CrossRef]
- Ruiz, S.; Krupnik, Y.; Keating, M.; Chandra, J.; Palladino, M.; McConkey, D. The proteasome inhibitor NPI-0052 is a more effective inducer of apoptosis than bortezomib in lymphocytes from patients with chronic lymphocytic leukemia. Mol. Cancer Ther. 2006, 5, 1836–1843. [Google Scholar] [CrossRef]
- Groll, M.; Huber, R.; Potts, B.C.M. Crystal structures of Salinosporamide A (NPI-0052) and B (NPI-0047) in complex with the 20S proteasome reveal important consequences of beta-lactone ring opening and a mechanism for irreversible binding. J. Am. Chem. Soc. 2006, 128, 5136–5141. [Google Scholar] [CrossRef]
- Potts, B.C.; Albitar, M.X.; Anderson, K.C.; Baritaki, S.; Berkers, C.; Bonavida, B.; Chandra, J.; Chauhan, D.; Cusack, J.C.; Fenical, W.; et al. Marizomib, a proteasome inhibitor for all seasons: Preclinical profile and a framework for clinical trials. Curr. Cancer Drug Targets 2011, 11, 254–284. [Google Scholar] [CrossRef]
- Macherla, V.R.; Mitchell, S.S.; Manam, R.R.; Reed, K.A.; Chao, T.-H.; Nicholson, B.; Deyanat-Yazdi, G.; Mai, B.; Jensen, P.R.; Fenical, W.F.; et al. Structure-activity relationship studies of salinosporamide A (NPI-0052), a novel marine derived proteasome inhibitor. J. Med. Chem. 2005, 48, 3684–3687. [Google Scholar] [CrossRef]
- Harrison, S.J.; Mainwaring, P.; Price, T.; Millward, M.J.; Padrik, P.; Underhill, C.R.; Cannell, P.K.; Reich, S.D.; Trikha, M.; Spencer, A. Phase I Clinical Trial of Marizomib (NPI-0052) in Patients with Advanced Malignancies Including Multiple Myeloma: Study NPI-0052-102 Final Results. Clin. Cancer Res. 2016, 22, 4559–4566. [Google Scholar] [CrossRef]
- Di, K.; Lloyd, G.K.; Abraham, V.; MacLaren, A.; Burrows, F.J.; Desjardins, A.; Trikha, M.; Bota, D.A. Marizomib activity as a single agent in malignant gliomas: Ability to cross the blood-brain barrier. Neuro-Oncol. 2016, 18, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Manton, C.A.; Johnson, B.; Singh, M.; Bailey, C.P.; Bouchier-Hayes, L.; Chandra, J. Induction of cell death by the novel proteasome inhibitor marizomib in glioblastoma in vitro and in vivo. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Le Rhun, E.; Preusser, M.; Tonn, J.-C.; Roth, P. How we treat glioblastoma. Esmo Open 2019. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Singh, A.V.; Aujay, M.; Kirk, C.J.; Bandi, M.; Ciccarelli, B.; Raje, N.; Richardson, P.; Anderson, K.C. A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood 2010, 116, 4906–4915. [Google Scholar] [CrossRef]
- Ghobrial, I.M.; Vij, R.; Siegel, D.; Badros, A.; Kaufman, J.; Raje, N.; Jakubowiak, A.; Savona, M.R.; Obreja, M.; Berdeja, J.G. A Phase Ib/II Study of Oprozomib in Patients with Advanced Multiple Myeloma and Waldenström Macroglobulinemia. Clin. Cancer Res. 2019, 25, 4907–4916. [Google Scholar] [CrossRef]
- Hari, P.; Matous, J.V.; Voorhees, P.M.; Shain, K.H.; Obreja, M.; Frye, J.; Fujii, H.; Jakubowiak, A.J.; Rossi, D.; Sonneveld, P. Oprozomib in patients with newly diagnosed multiple myeloma. Blood Cancer J. 2019. [Google Scholar] [CrossRef]
- Hari, P.; Paba-Prada, C.E.; Voorhees, P.M.; Frye, J.; Chang, Y.-L.; Moreau, P.; Zonder, J.; Boccia, R.; Shain, K.H. Efficacy and safety results from a phase 1b/2, multicenter, open-label study of oprozomib and dexamethasone in patients with relapsed and/or refractory multiple myeloma. Leuk. Res. 2019. [Google Scholar] [CrossRef]
- Shah, J.; Usmani, S.; Stadtmauer, E.A.; Rifkin, R.M.; Berenson, J.R.; Berdeja, J.G.; Lyons, R.M.; Klippel, Z.; Chang, Y.-L.; Niesvizky, R. Oprozomib, pomalidomide, and Dexamethasone in Patients with Relapsed and/or Refractory Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2019, 19, 570–578. [Google Scholar] [CrossRef]
- Infante, J.R.; Mendelson, D.S.; Burris, H.A.; Bendell, J.C.; Tolcher, A.W.; Gordon, M.S.; Gillenwater, H.H.; Arastu-Kapur, S.; Wong, H.L.; Papadopoulos, K.P. A first-in-human dose-escalation study of the oral proteasome inhibitor oprozomib in patients with advanced solid tumors. Investig. New Drugs 2016, 34, 216–224. [Google Scholar] [CrossRef]
- Johnson, H.W.B.; Lowe, E.; Anderl, J.L.; Fan, A.; Muchamuel, T.; Bowers, S.; Moebius, D.C.; Kirk, C.; McMinn, D.L. Required Immunoproteasome Subunit Inhibition Profile for Anti-Inflammatory Efficacy and Clinical Candidate KZR-616 ((2 S,3 R)- N-((S)-3-(Cyclopent-1-en-1-yl)-1-((R)-2-methyloxiran-2-yl)-1-oxopropan-2-yl)-3-hydroxy-3-(4-methoxyphenyl)-2-((S)-2-(2-morpholinoacetamido)propanamido)propenamide). J. Med. Chem. 2018, 61, 11127–11143. [Google Scholar]
- Muchamuel, T.; Basler, M.; Aujay, M.A.; Suzuki, E.; Kalim, K.W.; Lauer, C.; Sylvain, C.; Ring, E.R.; Shields, J.; Jiang, J.; et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med. 2009, 15, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wan, C.; Ding, Y.; Han, R.; He, Y.; Xiao, J.; Hao, J. PR-957, a selective inhibitor of immunoproteasome subunit low-MW polypeptide 7, attenuates experimental autoimmune neuritis by suppressing Th17-cell differentiation and regulating cytokine production. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 1756–1766. [Google Scholar] [CrossRef] [PubMed]
- von Brzezinski, L.; Säring, P.; Landgraf, P.; Cammann, C.; Seifert, U.; Dieterich, D.C. Low Neurotoxicity of ONX-0914 Supports the Idea of Specific Immunoproteasome Inhibition as a Side-Effect-Limiting, Therapeutic Strategy. Eur. J. Microbiol. Immunol. 2017, 7, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, M.; Busch, M.; Esdar, C.; Friese-Hamim, M.; Krier, M.; Ma, J.; Musil, D.; Rohdich, F.; Sloot, W.; Walter, G.; et al. Abstract DDT02-01: First-time disclosure of M3258: A selective inhibitor of the immunoproteasome subunit LMP7 with potential for improved therapeutic utility in multiple myeloma compared to pan-proteasome inhibitors. In Proceedings of the Molecular and Cellular Biology/Genetics; American Association for Cancer Research: Atlanta, GA, USA, 2019; p. DDT02-01. [Google Scholar]
- Yao, T.; Cohen, R.E. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature 2002, 419, 403–407. [Google Scholar] [CrossRef]
- Altmann, E.; Erbel, P.; Renatus, M.; Schaefer, M.; Schlierf, A.; Druet, A.; Kieffer, L.; Sorge, M.; Pfister, K.; Hassiepen, U.; et al. Azaindoles as Zinc-Binding Small-Molecule Inhibitors of the JAMM Protease CSN5. Angew. Chem. Int. Ed. Engl. 2017, 56, 1294–1297. [Google Scholar] [CrossRef]
- Tran, H.J.T.T.; Allen, M.D.; Löwe, J.; Bycroft, M. Structure of the Jab1/MPN domain and its implications for proteasome function. Biochemistry 2003, 42, 11460–11465. [Google Scholar] [CrossRef]
- Gallery, M.; Blank, J.L.; Lin, Y.; Gutierrez, J.A.; Pulido, J.C.; Rappoli, D.; Badola, S.; Rolfe, M.; Macbeth, K.J. The JAMM motif of human deubiquitinase Poh1 is essential for cell viability. Mol. Cancer Ther. 2007, 6, 262–268. [Google Scholar] [CrossRef]
- Perez, C.; Li, J.; Parlati, F.; Rouffet, M.; Ma, Y.; Mackinnon, A.L.; Chou, T.-F.; Deshaies, R.J.; Cohen, S.M. Discovery of an Inhibitor of the Proteasome Subunit Rpn11. J. Med. Chem. 2017, 60, 1343–1361. [Google Scholar] [CrossRef]
- Verma, R.; Peters, N.R.; D’Onofrio, M.; Tochtrop, G.P.; Sakamoto, K.M.; Varadan, R.; Zhang, M.; Coffino, P.; Fushman, D.; Deshaies, R.J.; et al. Ubistatins inhibit proteasome-dependent degradation by binding the ubiquitin chain. Science 2004, 306, 117–120. [Google Scholar] [CrossRef]
- Nakasone, M.A.; Lewis, T.A.; Walker, O.; Thakur, A.; Mansour, W.; Castañeda, C.A.; Goeckeler-Fried, J.L.; Parlati, F.; Chou, T.-F.; Hayat, O.; et al. Structural Basis for the Inhibitory Effects of Ubistatins in the Ubiquitin-Proteasome Pathway. Structure 2017, 25, 1839–1855. [Google Scholar] [CrossRef]
- Randles, L.; Anchoori, R.K.; Roden, R.B.S.; Walters, K.J. The Proteasome Ubiquitin Receptor hRpn13 and Its Interacting Deubiquitinating Enzyme Uch37 Are Required for Proper Cell Cycle Progression. J. Biol. Chem. 2016, 291, 8773–8783. [Google Scholar] [CrossRef]
- Chen, W.; Hu, X.-T.; Shi, Q.-L.; Zhang, F.-B.; He, C. Knockdown of the novel proteasome subunit Adrm1 located on the 20q13 amplicon inhibits colorectal cancer cell migration, survival and tumorigenicity. Oncol. Rep. 2009, 21, 531–537. [Google Scholar]
- Fejzo, M.S.; Dering, J.; Ginther, C.; Anderson, L.; Ramos, L.; Walsh, C.; Karlan, B.; Slamon, D.J. Comprehensive analysis of 20q13 genes in ovarian cancer identifies ADRM1 as amplification target. Genes Chromosomes Cancer 2008, 47, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.H.; Park, J.W.; Kim, H.R.; Seong, J.K.; Kim, H.K. ADRM1 gene amplification is a candidate driver for metastatic gastric cancers. Clin. Exp. Metastasis 2014, 31, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Ray, A.; Li, S.; Das, D.S.; Tai, Y.T.; Carrasco, R.D.; Chauhan, D.; Anderson, K.C. Targeting proteasome ubiquitin receptor Rpn13 in multiple myeloma. Leukemia 2016, 30, 1877–1886. [Google Scholar] [CrossRef]
- Anchoori, R.K.; Karanam, B.; Peng, S.; Wang, J.W.; Jiang, R.; Tanno, T.; Orlowski, R.Z.; Matsui, W.; Zhao, M.; Rudek, M.A.; et al. A bis-benzylidine piperidone targeting proteasome ubiquitin receptor RPN13/ADRM1 as a therapy for cancer. Cancer Cell 2013, 24, 791–805. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Nowicka, U.; Sridharan, V.; Liu, F.; Randles, L.; Hymel, D.; Dyba, M.; Tarasov, S.G.; Tarasova, N.I.; Zhao, X.Z.; et al. Structure of the Rpn13-Rpn2 complex provides insights for Rpn13 and Uch37 as anticancer targets. Nat. Commun. 2017. [Google Scholar] [CrossRef] [PubMed]
- Anchoori, R.K.; Jiang, R.; Peng, S.; Soong, R.-S.; Algethami, A.; Rudek, M.A.; Anders, N.; Hung, C.-F.; Chen, X.; Lu, X.; et al. Covalent Rpn13-Binding Inhibitors for the Treatment of Ovarian Cancer. ACS Omega 2018, 3, 11917–11929. [Google Scholar] [CrossRef] [PubMed]
- Trader, D.J.; Simanski, S.; Kodadek, T. A reversible and highly selective inhibitor of the proteasomal ubiquitin receptor rpn13 is toxic to multiple myeloma cells. J. Am. Chem. Soc. 2015, 137, 6312–6319. [Google Scholar] [CrossRef]
- Baell, J.; Walters, M.A. Chemistry: Chemical con artists foil drug discovery. Nature 2014, 513, 481–483. [Google Scholar] [CrossRef]
- Song, Y.; Park, P.M.C.; Wu, L.; Ray, A.; Picaud, S.; Li, D.; Wimalasena, V.K.; Du, T.; Filippakopoulos, P.; Anderson, K.C.; et al. Development and preclinical validation of a novel covalent ubiquitin receptor Rpn13 degrader in multiple myeloma. Leukemia 2019, 33, 2685–2694. [Google Scholar] [CrossRef] [PubMed]
- Paiva, S.-L.; Crews, C.M. Targeted protein degradation: Elements of PROTAC design. Curr. Opin. Chem. Biol. 2019, 50, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, M.; Crews, C.M. PROteolysis TArgeting Chimeras (PROTACs) - Past, present and future. Drug Discov. Today Technol. 2019, 31, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Schapira, M.; Calabrese, M.F.; Bullock, A.N.; Crews, C.M. Targeted protein degradation: Expanding the toolbox. Nat. Rev. Drug Discov. 2019, 18, 949–963. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.-S.; Cai, D.; Archer, C.T.; Kodadek, T. Periodate-triggered cross-linking reveals Sug2/Rpt4 as the molecular target of a peptoid inhibitor of the 19S proteasome regulatory particle. J. Am. Chem. Soc. 2007, 129, 12936–12937. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.-S.; Archer, C.T.; Kodadek, T. Identification of a peptoid inhibitor of the proteasome 19S regulatory particle. J. Am. Chem. Soc. 2007, 129, 7750–7751. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Sokol, E.; Jin, D.; Brune, Z.; Thiru, P.; Ghandi, M.; Garraway, L.A.; Gupta, P.B.; Santagata, S.; Whitesell, L.; et al. Suppression of 19S proteasome subunits marks emergence of an altered cell state in diverse cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 382–387. [Google Scholar] [CrossRef]
- Acosta-Alvear, D.; Cho, M.Y.; Wild, T.; Buchholz, T.J.; Lerner, A.G.; Simakova, O.; Hahn, J.; Korde, N.; Landgren, O.; Maric, I.; et al. Paradoxical resistance of multiple myeloma to proteasome inhibitors by decreased levels of 19S proteasomal subunits. eLife 2015. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Mendillo, M.L.; Zhao, J.; Carette, J.E.; Merrill, P.H.; Cikes, D.; Varadarajan, M.; van Diemen, F.R.; Penninger, J.M.; Goldberg, A.L.; et al. Compromising the 19S proteasome complex protects cells from reduced flux through the proteasome. eLife 2015. [Google Scholar] [CrossRef]
- Wallington-Beddoe, C.T.; Sobieraj-Teague, M.; Kuss, B.J.; Pitson, S.M. Resistance to proteasome inhibitors and other targeted therapies in myeloma. Br. J. Haematol. 2018, 182, 11–28. [Google Scholar] [CrossRef]
- Pritchard, J.R.; Lauffenburger, D.A.; Hemann, M.T. Understanding resistance to combination chemotherapy. Drug Resist. Updat. Rev. Comment. Antimicrob. Anticancer Chemother. 2012, 15, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; André, T.; Atreya, C.E.; Schellens, J.H.M.; Yoshino, T.; Bendell, J.C.; Hollebecque, A.; McRee, A.J.; Siena, S.; Middleton, G.; et al. Combined BRAF, EGFR, and MEK Inhibition in Patients with BRAFV600E-Mutant Colorectal Cancer. Cancer Discov. 2018, 8, 428–443. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef]
- Steffen, J.; Seeger, M.; Koch, A.; Krüger, E. Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD-dependent feedback loop. Mol. Cell 2010, 40, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, S.K.; Lee, C.S.; Young, P.; Beskow, A.; Chan, J.Y.; Deshaies, R.J. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell 2010, 38, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, S.K.; den Besten, W.; Deshaies, R.J. p97-dependent retrotranslocation and proteolytic processing govern formation of active Nrf1 upon proteasome inhibition. eLife 2014. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, S.; Irie, T.; Hirayama, S.; Sakurai, Y.; Yashiroda, H.; Naguro, I.; Ichijo, H.; Hamazaki, J.; Murata, S. The aspartyl protease DDI2 activates Nrf1 to compensate for proteasome dysfunction. eLife 2016. [Google Scholar] [CrossRef]
- Lehrbach, N.J.; Ruvkun, G. Proteasome dysfunction triggers activation of SKN-1A/Nrf1 by the aspartic protease DDI-1. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Tomlin, F.M.; Gerling-Driessen, U.I.M.; Liu, Y.-C.; Flynn, R.A.; Vangala, J.R.; Lentz, C.S.; Clauder-Muenster, S.; Jakob, P.; Mueller, W.F.; Ordoñez-Rueda, D.; et al. Inhibition of NGLY1 Inactivates the Transcription Factor Nrf1 and Potentiates Proteasome Inhibitor Cytotoxicity. ACS Cent. Sci. 2017, 3, 1143–1155. [Google Scholar] [CrossRef]
- Lehrbach, N.J.; Breen, P.C.; Ruvkun, G. Protein Sequence Editing of SKN-1A/Nrf1 by Peptide:N-Glycanase Controls Proteasome Gene Expression. Cell 2019, 177, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Huang, C.; Fujihira, H. The cytoplasmic peptide:N-glycanase (NGLY1) - Structure, expression and cellular functions. Gene 2016, 577, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Detappe, A.; Cai, K.; Keys, H.R.; Brune, Z.; Ying, W.; Thiru, P.; Reidy, M.; Kugener, G.; Rossen, J.; et al. Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nat. Chem. Biol. 2019, 15, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.; Tang, W.K.; Ye, Y. Structure and function of the AAA+ ATPase p97/Cdc48p. Gene 2016, 583, 64–77. [Google Scholar] [CrossRef]
- Ye, Y.; Tang, W.K.; Zhang, T.; Xia, D. A Mighty “Protein Extractor” of the Cell: Structure and Function of the p97/CDC48 ATPase. Front. Mol. Biosci. 2017. [Google Scholar] [CrossRef]
- Van den Boom, J.; Meyer, H. VCP/p97-Mediated Unfolding as a Principle in Protein Homeostasis and Signaling. Mol. Cell 2018, 69, 182–194. [Google Scholar] [CrossRef]
- Ye, Y.; Meyer, H.H.; Rapoport, T.A. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 2001, 414, 652–656. [Google Scholar] [CrossRef]
- Meyer, H.; Bug, M.; Bremer, S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 2012, 14, 117–123. [Google Scholar] [CrossRef]
- Bodnar, N.O.; Kim, K.H.; Ji, Z.; Wales, T.E.; Svetlov, V.; Nudler, E.; Engen, J.R.; Walz, T.; Rapoport, T.A. Structure of the Cdc48 ATPase with its ubiquitin-binding cofactor Ufd1-Npl4. Nat. Struct. Mol. Biol. 2018, 25, 616–622. [Google Scholar] [CrossRef]
- Bodnar, N.; Rapoport, T. Toward an understanding of the Cdc48/p97 ATPase. F1000Research 2017. [Google Scholar] [CrossRef]
- Banerjee, S.; Bartesaghi, A.; Merk, A.; Rao, P.; Bulfer, S.L.; Yan, Y.; Green, N.; Mroczkowski, B.; Neitz, R.J.; Wipf, P.; et al. 2.3 Å resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science 2016, 351, 871–875. [Google Scholar] [CrossRef] [PubMed]
- Bruderer, R.M.; Brasseur, C.; Meyer, H.H. The AAA ATPase p97/VCP interacts with its alternative co-factors, Ufd1-Npl4 and p47, through a common bipartite binding mechanism. J. Biol. Chem. 2004, 279, 49609–49616. [Google Scholar] [CrossRef] [PubMed]
- Le Moigne, R.; Aftab, B.T.; Djakovic, S.; Dhimolea, E.; Valle, E.; Murnane, M.; King, E.M.; Soriano, F.; Menon, M.-K.; Wu, Z.Y.; et al. The p97 Inhibitor CB-5083 Is a Unique Disrupter of Protein Homeostasis in Models of Multiple Myeloma. Mol. Cancer Ther. 2017, 16, 2375–2386. [Google Scholar] [CrossRef] [PubMed]
- Watts, G.D.J.; Wymer, J.; Kovach, M.J.; Mehta, S.G.; Mumm, S.; Darvish, D.; Pestronk, A.; Whyte, M.P.; Kimonis, V.E. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 2004, 36, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.K.; Xia, D. Mutations in the Human AAA+ Chaperone p97 and Related Diseases. Front. Mol. Biosci. 2016. [Google Scholar] [CrossRef]
- Blythe, E.E.; Olson, K.C.; Chau, V.; Deshaies, R.J. Ubiquitin- and ATP-dependent unfoldase activity of P97/VCP•NPLOC4•UFD1L is enhanced by a mutation that causes multisystem proteinopathy. Proc. Natl. Acad. Sci. USA 2017, 114, 4380–4388. [Google Scholar] [CrossRef]
- Blythe, E.E.; Gates, S.N.; Deshaies, R.J.; Martin, A. Multisystem Proteinopathy Mutations in VCP/p97 Increase NPLOC4·UFD1L Binding and Substrate Processing. Structure 2019, 27, 1820–1829. [Google Scholar] [CrossRef]
- Yang, Y.; Kitagaki, J.; Dai, R.-M.; Tsai, Y.C.; Lorick, K.L.; Ludwig, R.L.; Pierre, S.A.; Jensen, J.P.; Davydov, I.V.; Oberoi, P.; et al. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res. 2007, 67, 9472–9481. [Google Scholar] [CrossRef]
- Hyer, M.L.; Milhollen, M.A.; Ciavarri, J.; Fleming, P.; Traore, T.; Sappal, D.; Huck, J.; Shi, J.; Gavin, J.; Brownell, J.; et al. A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment. Nat. Med. 2018, 24, 186–193. [Google Scholar] [CrossRef]
- Landré, V.; Rotblat, B.; Melino, S.; Bernassola, F.; Melino, G. Screening for E3-ubiquitin ligase inhibitors: Challenges and opportunities. Oncotarget 2014, 5, 7988–8013. [Google Scholar] [CrossRef]
- O’Donnell, M.A.; Legarda-Addison, D.; Skountzos, P.; Yeh, W.C.; Ting, A.T. Ubiquitination of RIP1 regulates an NF-kappaB-independent cell-death switch in TNF signaling. Curr. Biol. CB 2007, 17, 418–424. [Google Scholar] [CrossRef]
- LaCasse, E.C.; Mahoney, D.J.; Cheung, H.H.; Plenchette, S.; Baird, S.; Korneluk, R.G. IAP-targeted therapies for cancer. Oncogene 2008, 27, 6252–6275. [Google Scholar] [CrossRef]
- Wang, L.; Du, F.; Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef]
- Swords, R.T.; Coutre, S.; Maris, M.B.; Zeidner, J.F.; Foran, J.M.; Cruz, J.; Erba, H.P.; Berdeja, J.G.; Tam, W.; Vardhanabhuti, S.; et al. Pevonedistat, a first-in-class NEDD8-activating enzyme inhibitor, combined with azacitidine in patients with AML. Blood 2018, 131, 1415–1424. [Google Scholar] [CrossRef]
- Sarantopoulos, J.; Shapiro, G.I.; Cohen, R.B.; Clark, J.W.; Kauh, J.S.; Weiss, G.J.; Cleary, J.M.; Mahalingam, D.; Pickard, M.D.; Faessel, H.M.; et al. Phase I Study of the Investigational NEDD8-Activating Enzyme Inhibitor Pevonedistat (TAK-924/MLN4924) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 847–857. [Google Scholar] [CrossRef]
- Kategaya, L.; Di Lello, P.; Rougé, L.; Pastor, R.; Clark, K.R.; Drummond, J.; Kleinheinz, T.; Lin, E.; Upton, J.-P.; Prakash, S.; et al. USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature 2017, 550, 534–538. [Google Scholar] [CrossRef]
- Turnbull, A.P.; Ioannidis, S.; Krajewski, W.W.; Pinto-Fernandez, A.; Heride, C.; Martin, A.C.L.; Tonkin, L.M.; Townsend, E.C.; Buker, S.M.; Lancia, D.R.; et al. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature 2017, 550, 481–486. [Google Scholar] [CrossRef]
- Lamberto, I.; Liu, X.; Seo, H.-S.; Schauer, N.J.; Iacob, R.E.; Hu, W.; Das, D.; Mikhailova, T.; Weisberg, E.L.; Engen, J.R.; et al. Structure-Guided Development of a Potent and Selective Non-covalent Active-Site Inhibitor of USP7. Cell Chem. Biol. 2017, 24, 1490–1500. [Google Scholar] [CrossRef]
- Gavory, G.; O’Dowd, C.R.; Helm, M.D.; Flasz, J.; Arkoudis, E.; Dossang, A.; Hughes, C.; Cassidy, E.; McClelland, K.; Odrzywol, E.; et al. Discovery and characterization of highly potent and selective allosteric USP7 inhibitors. Nat. Chem. Biol. 2018, 14, 118–125. [Google Scholar] [CrossRef]
- Wang, S.; Song, Y.; Yan, F.; Liu, D. Mechanisms of resistance to third-generation EGFR tyrosine kinase inhibitors. Front. Med. 2016, 10, 383–388. [Google Scholar] [CrossRef]
- Cross, D.A.E.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.V.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef]
- Hallenbeck, K.K.; Turner, D.M.; Renslo, A.R.; Arkin, M.R. Targeting Non-Catalytic Cysteine Residues Through Structure-Guided Drug Discovery. Curr. Top. Med. Chem. 2017, 17, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Bond, D.A.; Woyach, J.A. Targeting BTK in CLL: Beyond Ibrutinib. Curr. Hematol. Malig. Rep. 2019, 14, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, M.; Liu, D. Acalabrutinib (ACP-196): A selective second-generation BTK inhibitor. J. Hematol. Oncol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Lee, B.-H.; Hanna, J.; King, R.W.; Finley, D. Trimming of ubiquitin chains by proteasome-associated deubiquitinating enzymes. Mol. Cell Proteom. MCP 2011. [Google Scholar] [CrossRef]
- Lee, B.-H.; Lee, M.J.; Park, S.; Oh, D.-C.; Elsasser, S.; Chen, P.-C.; Gartner, C.; Dimova, N.; Hanna, J.; Gygi, S.P.; et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 2010, 467, 179–184. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, Y.; Ding, S.; Li, J.; Song, N.; Ren, Y.; Hong, D.; Wu, C.; Li, B.; Wang, F.; et al. Small molecule inhibitors reveal allosteric regulation of USP14 via steric blockade. Cell Res. 2018, 28, 1186–1194. [Google Scholar] [CrossRef]
- D’Arcy, P.; Brnjic, S.; Olofsson, M.H.; Fryknäs, M.; Lindsten, K.; De Cesare, M.; Perego, P.; Sadeghi, B.; Hassan, M.; Larsson, R.; et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat. Med. 2011, 17, 1636–1640. [Google Scholar] [CrossRef]
- Woodle, E.S.; Alloway, R.R.; Girnita, A. Proteasome inhibitor treatment of antibody-mediated allograft rejection. Curr. Opin. Organ. Transpl. 2011, 16, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Ensor, C.R.; Yousem, S.A.; Marrari, M.; Morrell, M.R.; Mangiola, M.; Pilewski, J.M.; D’Cunha, J.; Wisniewski, S.R.; Venkataramanan, R.; Zeevi, A.; et al. Proteasome Inhibitor Carfilzomib-Based Therapy for Antibody-Mediated Rejection of the Pulmonary Allograft: Use and Short-Term Findings. Am. J. Transpl. Off. J. Am. Soc. Transpl. Am. Soc. Transpl. Surg. 2017, 17, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, N.M.; Reed, E.F. Antibody-mediated rejection across solid organ transplants: Manifestations, mechanisms, and therapies. J. Clin. Investig. 2017, 127, 2492–2504. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, H.L.; Terasaki, P.I.; Feroz, A.; Everly, M.J.; Vanikar, A.V.; Shankar, V.; Trivedi, V.B.; Kaneku, H.; Idica, A.K.; Modi, P.R.; et al. Abrogation of anti-HLA antibodies via proteasome inhibition. Transplantation 2009, 87, 1555–1561. [Google Scholar] [CrossRef] [PubMed]
- Ejaz, N.S.; Alloway, R.R.; Halleck, F.; Dürr, M.; Budde, K.; Woodle, E.S. Review of bortezomib treatment of antibody-mediated rejection in renal transplantation. Antioxid. Redox Signal. 2014, 21, 2401–2418. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Francis, J.; Gautam, A.; Pelletier, L.; Sanchorawala, V.; Quillen, K. Durable renal response after combination of bortezomib, corticosteroids, rituximab, and plasmapheresis for late antibody-mediated renal transplant rejection. Clin. Nephrol. 2018, 89, 252–259. [Google Scholar] [CrossRef]
- Morrow, W.R.; Frazier, E.A.; Mahle, W.T.; Harville, T.O.; Pye, S.E.; Knecht, K.R.; Howard, E.L.; Smith, R.N.; Saylors, R.L.; Garcia, X.; et al. Rapid reduction in donor-specific anti-human leukocyte antigen antibodies and reversal of antibody-mediated rejection with bortezomib in pediatric heart transplant patients. Transplantation 2012, 93, 319–324. [Google Scholar] [CrossRef]
- Govil, A.; Walsh, R.C.; Tevar, A.; Alloway, R.; Roy-Chaudhury, P.; Mogilishetty, G.; Wall, G.E.; Brailey, P.; Girnita, A.; Woodle, E.S. Bortezomib-based treatment of antibody mediated rejection in pancreas allograft recipients. Clin. Transpl. 2009, 45, 443–453. [Google Scholar]
- Baum, C.; Reichenspurner, H.; Deuse, T. Bortezomib rescue therapy in a patient with recurrent antibody-mediated rejection after lung transplantation. J. Heart Lung Transpl. Off. Publ. Int. Soc. Heart Transpl. 2013, 32, 1270–1271. [Google Scholar] [CrossRef]
- Krishnan, K.M.; Williamson, K.C. The proteasome as a target to combat malaria: Hits and misses. Transl. Res. J. Lab. Clin. Med. 2018, 198, 40–47. [Google Scholar] [CrossRef]
- Li, H.; O’Donoghue, A.J.; van der Linden, W.A.; Xie, S.C.; Yoo, E.; Foe, I.T.; Tilley, L.; Craik, C.S.; da Fonseca, P.C.A.; Bogyo, M. Structure- and function-based design of Plasmodium-selective proteasome inhibitors. Nature 2016, 530, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Le Chapelain, C.; Groll, M. Rational Design of Proteasome Inhibitors as Antimalarial Drugs. Angew. Chem. Int. Ed. Engl. 2016, 55, 6370–6372. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; van der Linden, W.A.; Verdoes, M.; Florea, B.I.; McAllister, F.E.; Govindaswamy, K.; Elias, J.E.; Bhanot, P.; Overkleeft, H.S.; Bogyo, M. Assessing subunit dependency of the Plasmodium proteasome using small molecule inhibitors and active site probes. ACS Chem. Biol. 2014, 9, 1869–1876. [Google Scholar] [CrossRef] [PubMed]
- Aminake, M.N.; Arndt, H.-D.; Pradel, G. The proteasome of malaria parasites: A multi-stage drug target for chemotherapeutic intervention? Int. J. Parasitol. Drugs Drug Resist. 2012, 2, 1–10. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Czesny, B.; Goshu, S.; Cook, J.L.; Williamson, K.C. The proteasome inhibitor epoxomicin has potent Plasmodium falciparum gametocytocidal activity. Antimicrob. Agents Chemother. 2009, 53, 4080–4085. [Google Scholar] [CrossRef]
- Li, H.; Ponder, E.L.; Verdoes, M.; Asbjornsdottir, K.H.; Deu, E.; Edgington, L.E.; Lee, J.T.; Kirk, C.J.; Demo, S.D.; Williamson, K.C.; et al. Validation of the proteasome as a therapeutic target in Plasmodium using an epoxyketone inhibitor with parasite-specific toxicity. Chem. Biol. 2012, 19, 1535–1545. [Google Scholar] [CrossRef]
- Becker, S.H.; Darwin, K.H. Bacterial Proteasomes: Mechanistic and Functional Insights. Microbiol. Mol. Biol. Rev. MMBR 2017. [Google Scholar] [CrossRef]
- Lin, G.; Hu, G.; Tsu, C.; Kunes, Y.Z.; Li, H.; Dick, L.; Parsons, T.; Li, P.; Chen, Z.; Zwickl, P.; et al. Mycobacterium tuberculosis prcBA genes encode a gated proteasome with broad oligopeptide specificity. Mol. Microbiol. 2006, 59, 1405–1416. [Google Scholar] [CrossRef]
- Hu, G.; Lin, G.; Wang, M.; Dick, L.; Xu, R.-M.; Nathan, C.; Li, H. Structure of the Mycobacterium tuberculosis proteasome and mechanism of inhibition by a peptidyl boronate. Mol. Microbiol. 2006, 59, 1417–1428. [Google Scholar] [CrossRef]
- Darwin, K.H.; Lin, G.; Chen, Z.; Li, H.; Nathan, C.F. Characterization of a Mycobacterium tuberculosis proteasomal ATPase homologue. Mol. Microbiol. 2005, 55, 561–571. [Google Scholar] [CrossRef]
- Darwin, K.H.; Ehrt, S.; Gutierrez-Ramos, J.-C.; Weich, N.; Nathan, C.F. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science 2003, 302, 1963–1966. [Google Scholar] [CrossRef] [PubMed]
- Agins, B.D.; Ikeda, D.J.; Reid, M.J.A.; Goosby, E.; Pai, M.; Cattamanchi, A. Improving the cascade of global tuberculosis care: Moving from the “what” to the “how” of quality improvement. Lancet Infect. Dis. 2019, 19, 437–443. [Google Scholar] [CrossRef]
- Zhan, W.; Hsu, H.-C.; Morgan, T.; Ouellette, T.; Burns-Huang, K.; Hara, R.; Wright, A.G.; Imaeda, T.; Okamoto, R.; Sato, K.; et al. Selective Phenylimidazole-Based Inhibitors of the Mycobacterium tuberculosis Proteasome. J. Med. Chem. 2019, 62, 9246–9253. [Google Scholar] [CrossRef] [PubMed]
- Totaro, K.A.; Barthelme, D.; Simpson, P.T.; Jiang, X.; Lin, G.; Nathan, C.F.; Sauer, R.T.; Sello, J.K. Rational Design of Selective and Bioactive Inhibitors of the Mycobacterium tuberculosis Proteasome. ACS Infect. Dis. 2017, 3, 176–181. [Google Scholar] [CrossRef] [PubMed]




| Name | Type | IC50 (nM) | β5 Dissoc. T1/2 (min) | Administration | ||
|---|---|---|---|---|---|---|
| β5 Chymotrypsin-Like | β1 Caspase- Like | β2 Trypsin- Like | ||||
| Bortezomib (PS-341) | Boronate | 2.4–7 *,# | 24–74 *,# | 1200–4200 *,# | 110 # | Intravenous |
| Carfilzomib (PR-171) | Epoxyketone | 6 * | 2400 * | 3600 * | Irreversible | Intravenous |
| Ixazomib (MLN9708) | Boronate | 3.4# | 31 # | 3500 # | 18 # | Oral |
| Marizomib (Salinosporamide A) | β-lactone | 3.5 * | 430 * | 28 * | Irreversible | Intravenous |
| Oprozomib (ONX0912) | Epoxyketone | 36 ^ | NA | NA | Irreversible | Oral |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sherman, D.J.; Li, J. Proteasome Inhibitors: Harnessing Proteostasis to Combat Disease. Molecules 2020, 25, 671. https://doi.org/10.3390/molecules25030671
Sherman DJ, Li J. Proteasome Inhibitors: Harnessing Proteostasis to Combat Disease. Molecules. 2020; 25(3):671. https://doi.org/10.3390/molecules25030671
Chicago/Turabian StyleSherman, David J., and Jing Li. 2020. "Proteasome Inhibitors: Harnessing Proteostasis to Combat Disease" Molecules 25, no. 3: 671. https://doi.org/10.3390/molecules25030671
APA StyleSherman, D. J., & Li, J. (2020). Proteasome Inhibitors: Harnessing Proteostasis to Combat Disease. Molecules, 25(3), 671. https://doi.org/10.3390/molecules25030671