
New Peptide-Based Pharmacophore Activates 20S Proteasome

 , , , and
, , , and 
Abstract
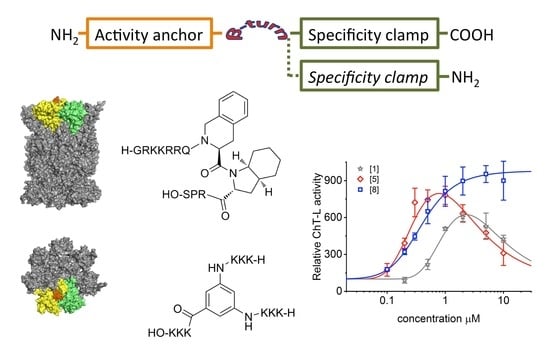
1. Introduction
2. Results and Discussion
2.1. Design of TAT Peptides
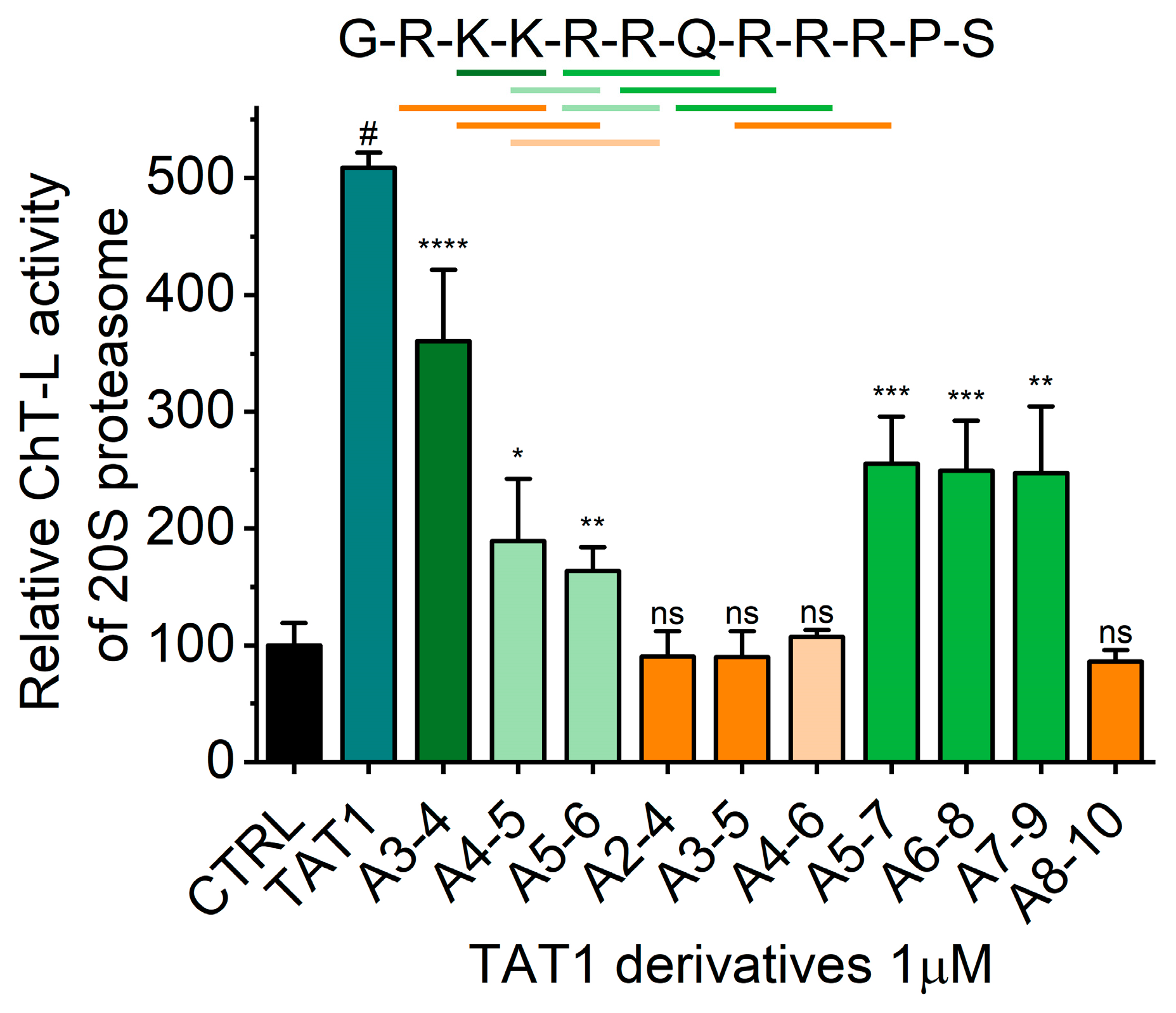
2.2. TAT Peptides Activate the ChT-L Peptidase of the Human Proteasome in vitro and in Cellulo
2.3. The α1/α2 Inter-Subunit Pocket on the α Face of Core Proteasome is the Primary Binding Site of TAT Peptides. The Binding Changes Conformational Equilibrium of the Proteasome’s Gate
3. Materials and Methods
3.1. Synthesis of Selected Peptides
3.2. Synthesis of TAT1-Den Peptide (8)
3.2.1. Peptide Cleavage from the Solid Support
3.2.2. Purification
3.2.3. Characterization of the Product with HPLC and Mass Spectrometry
3.3. Determination of Proteasome Activity
3.4. Cell Culture
3.5. Molecular Docking
3.6. Atomic Force Microscopy (AFM) Imaging
3.7. Statistical Analysis
4. Patents
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jankowska, E.; Stoj, J.; Karpowicz, P.; Osmulski, P.A.; Gaczynska, M. The proteasome in health and disease. Curr Pharm Des. 2013, 19, 1010–1028. [Google Scholar] [PubMed]
- Adams, J. The proteasome: A suitable antineoplastic target. Nat. Rev. Cancer. 2004, 4, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Besse, A.; Besse, L.; Kraus, M.; Mendez-Lopez, M.; Bader, J.; Xin, B.-T. Proteasome Inhibition in Multiple Myeloma: Head-to-Head Comparison of Currently Available Proteasome Inhibitors. Cell Chem Biol. 2019, 26, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, K.A.; Gaczynska, M.; Osmulski, P.A. Molecular mechanisms of proteasome plasticity in aging. Mech Ageing Dev. 2010, 131, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Chondrogianni, N.; Gonos, E.S. Proteasome dysfunction in mammalian aging: Steps and factors involved. Exp. Gerontol. 2005, 40, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.N.; Hanni, K.B.; Markesbery, W.R. Impaired proteasome function in Alzheimer’s disease. J. Neurochem. 2000, 75, 436–439. [Google Scholar] [CrossRef]
- McNaught, K.S.; Belizaire, R.; Jenner, P.; Olanow, C.W.; Isacson, O. Selective loss of 20S proteasome α-subunits in the substantia nigra pars compacta in Parkinson’s disease. Neurosci Lett. 2002, 326, 155–158. [Google Scholar] [CrossRef]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006, 443, 780–786. [Google Scholar] [CrossRef]
- Bedford, L.; Hay, D.; Devoy, A.; Paine, S.; Powe, D.G.; Seth, R. Depletion of 26S Proteasomes in Mouse Brain Neurons Causes Neurodegeneration and Lewy-Like Inclusions Resembling Human Pale Bodies. J. Neurosci. 2008, 28, 8189–8198. [Google Scholar] [CrossRef]
- Jones, C.L.; Tepe, J.J. Proteasome Activation to Combat Proteotoxicity. Molecules. 2019, 24, 2841. [Google Scholar] [CrossRef]
- Coleman, R.A.; Trader, D.J. Methods to Discover and Evaluate Proteasome Small Molecule Stimulators. Molecules. 2019, 24, 2341. [Google Scholar] [CrossRef] [PubMed]
- Njomen, E.; Tepe, J.J. Proteasome Activation as a New Therapeutic Approach To Target Proteotoxic Disorders. J. Med. Chem. 2019, 62, 6469–6481. [Google Scholar] [CrossRef] [PubMed]
- Chondrogianni, N.; Georgila, K.; Kourtis, N.; Tavernarakis, N.; Gonos, E.S. 20S proteasome activation promotes life span extension and resistance to proteotoxicity in Caenorhabditis elegans. FASEB J. 2015, 29, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Munkácsy, E.; Chocron, E.S.; Quintanilla, L.; Gendron, C.M.; Pletcher, S.D.; Pickering, A.M. Neuronal-specific proteasome augmentation via Prosβ5 overexpression extends lifespan and reduces age-related cognitive decline. Aging Cell. 2019, 18, 5. [Google Scholar]
- Chocron, S.E.; Munkacsy, E.; Harper, S.; Jiang, N.; Van Skike, C.E.; DeRosa, N. Genetic and pharmacologic proteasome augmentation ameliorates Alzheimer’s disease-like symptoms through increased turnover of β-amyloid machinery. Nat. Commun. 2020. under revision. [Google Scholar]
- Huang, L.; Ho, P.; Chen, C.-H. Activation and inhibition of the proteasome by betulinic acid and its derivatives. FEBS Lett. 2007, 581, 4955–4959. [Google Scholar] [CrossRef]
- Coleman, R.A.; Muli, C.S.; Zhao, Y.; Bhardwaj, A.; Newhouse, T.R.; Trader, D.J. Analysis of chain length, substitution patterns, and unsaturation of AM-404 derivatives as 20S proteasome stimulators. Bioorg Med. Chem Lett. 2019, 29, 420–423. [Google Scholar] [CrossRef]
- Jones, C.L.; Njomen, E.; Sjögren, B.; Dexheimer, T.S.; Tepe, J.J. Small Molecule Enhancement of 20S Proteasome Activity Targets Intrinsically Disordered Proteins. ACS Chem Biol. 2017, 12, 2240–2247. [Google Scholar] [CrossRef]
- Njomen, E.; Osmulski, P.A.; Jones, C.L.; Gaczynska, M.; Tepe, J.J. Small Molecule Modulation of Proteasome Assembly. Biochemistry. 2018, 57, 4214–4224. [Google Scholar] [CrossRef]
- Giżyńska, M.; Witkowska, J.; Karpowicz, P.; Rostankowski, R.; Chocron, E.S.; Pickering, A.M. Proline- and Arginine-Rich Peptides as Flexible Allosteric Modulators of Human Proteasome Activity. J. Med. Chem. 2019, 62, 359–370. [Google Scholar] [CrossRef]
- Huang, X.; Seifert, U.; Salzmann, U.; Henklein, P.; Preissner, R.; Henke, W. The RTP Site Shared by the HIV-1 Tat Protein and the 11S Regulator Subunit α is Crucial for their Effects on Proteasome Function Including Antigen Processing. J. Mol. Biol. 2002, 323, 771–782. [Google Scholar] [CrossRef]
- Jankowska, E.; Gaczynska, M.; Osmulski, P.; Sikorska, E.; Rostankowski, R.; Madabhushi, S. Potential allosteric modulators of the proteasome activity. Biopolymers. 2010, 93, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Witkowska, J.; Karpowicz, P.; Gaczynska, M.; Osmulski, P.A.; Jankowska, E. Dissecting a role of a charge and conformation of Tat2 peptide in allosteric regulation of 20S proteasome. J. Pept Sci. 2014, 20, 649–656. [Google Scholar] [CrossRef]
- Karpowicz, P.; Osmulski, P.A.; Witkowska, J.; Sikorska, E.; Giżyńska, M.; Belczyk-Ciesielska, A. Interplay between Structure and Charge as a Key to Allosteric Modulation of Human 20S Proteasome by the Basic Fragment of HIV-1 Tat Protein. PLoS ONE 2015, 10, E0143038. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wu, J.; Lu, Y.; Ma, Y.B.; Lee, B.H.; Yu, Z. Structural basis for dynamic regulation of the human 26S proteasome. Proc. Natl. Acad. Sci. USA 2016, 113, 12991–12996. [Google Scholar] [CrossRef] [PubMed]
- Jeang, K.-T.; Xiao, H.; Rich, E.A. Multifaceted Activities of the HIV-1 Transactivator of Transcription. Tat. J. Biol Chem. 1999, 274, 28837–28840. [Google Scholar] [CrossRef]
- Rana, T.M.; Jeang, K.-T. Biochemical and Functional Interactions between HIV-1 Tat Protein and TAR RNA. Arch. Biochem Biophys. 1999, 365, 175–185. [Google Scholar] [CrossRef]
- Gaczynska, M.; Osmulski, P.A. Targeting Protein–Protein Interactions in the Ubiquitin–Proteasome Pathway. In Advances in Protein Chemistry and Structural Biology. Protein-Protein Interactions in Human Disease, Part A; Elsevier Inc.: Amsterdam, The Netherlands, 2018; pp. 123–165. [Google Scholar]
- Schwarze, S.R.; Hruska, K.A.; Dowdy, S.F. Protein transduction: Unrestricted delivery into all cells? Trends Cell Biol. 2000, 10, 290–295. [Google Scholar] [CrossRef]
- Lata, S.; Mishra, R.; Banerjea, A.C. Proteasomal Degradation Machinery: Favorite Target of HIV-1 Proteins. Front. Microbiol. 2018, 9, 2738. [Google Scholar] [CrossRef]
- Kisselev, A.F.; Kaganovich, D.; Goldberg, A.L. Binding of hydrophobic peptides to several non-catalytic sites promotes peptide hydrolysis by all active sites of 20 S proteasomes-Evidence for peptide-induced channel opening in the alpha-rings. J. Biol Chem. 2002, 277, 22260–22270. [Google Scholar] [CrossRef]
- Tanaka, K.; Mizushima, T.; Saeki, Y. The proteasome: Molecular machinery and pathophysiological roles. Biol Chem. 2012, 393, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Vakser, I.A. Long-distance potentials: An approach to the multiple-minima problem in ligand-receptor interaction. “Protein Eng. Des. Sel. 1996, 9, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Vakser, I.A. Protein-Protein Docking: From Interaction to Interactome. Biophys, J. 2014, 107, 1785–1793. [Google Scholar] [CrossRef] [PubMed]
- Schrader, J.; Henneberg, F.; Mata, R.A.; Tittmann, K.; Schneider, T.R.; Stark, H. The inhibition mechanism of human 20S proteasomes enables next-generation inhibitor design. Science 2016, 353, 594–598. [Google Scholar] [CrossRef]
- Whitby, F.G.; Masters, E.I.; Kramer, L.; Knowlton, J.R.; Yao, Y.; Wang, C.C. Structural basis for the activation of 20S proteasomes by 11S regulators. Nature 2000, 408, 115–120. [Google Scholar] [CrossRef]
- Ortega, J.; Heymann, J.B.; Kajava, A.V.; Ustrell, V.; Rechsteiner, M.; Steven, A.C. The axial channel of the 20S proteasome opens upon binding of the PA200 activator. J. Mol. Biol. 2005, 346, 1221–1227. [Google Scholar] [CrossRef]
- Smith, D.M.; Chang, S.-C.; Park, S.; Finley, D.; Cheng, Y.; Goldberg, A.L. Docking of the proteasomal ATPases’ carboxyl termini in the 20S proteasome’s alpha ring opens the gate for substrate entry. Mol. Cell. 2007, 27, 731–744. [Google Scholar] [CrossRef]
- Giletto, M.B.; Osmulski, P.A.; Jones, C.L.; Gaczynska, M.E.; Tepe, J.J. Pipecolic esters as minimized templates for proteasome inhibition. Org. Biomol Chem. 2019, 17, 2734–2746. [Google Scholar] [CrossRef]
- Toste Rêgo, A.; da Fonseca, P.C.A. Characterization of Fully Recombinant Human 20S and 20S-PA200 Proteasome Complexes. Mol. Cell 2019, 76, 138–147. [Google Scholar] [CrossRef]
- Witkowska, J.; Giżyńska, M.; Grudnik, P.; Golik, P.; Karpowicz, P.; Giełdoń, A. Crystal structure of a low molecular weight activator Blm-pep with yeast 20S proteasome-insights into the enzyme activation mechanism. Sci Rep. 2017, 7, 6177. [Google Scholar] [CrossRef]
- Forster, A.; Whitby, F.G.; Hill, C.P. The pore of activated 20S proteasomes has an ordered 7-fold symmetric conformation. EMBO J. 2003, 22, 4356–4364. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, A.; Aufderheide, A.; Rudack, T.; Beck, F.; Pfeifer, G.; Plitzko, J.M. Structure of the human 26S proteasome at a resolution of 3.9 Å. Proc. Natl. Acad. Sci. 2016, 113, 7816–7821. [Google Scholar] [CrossRef] [PubMed]
- Osmulski, P.A.; Gaczynska, M. Nanoenzymology of the 20S proteasome: Proteasomal actions are controlled by the allosteric transition. Biochemistry. 2002, 41, 7047–7053. [Google Scholar] [CrossRef] [PubMed]
- Osmulski, P.A.; Hochstrasser, M.; Gaczynska, M. A Tetrahedral Transition State at the Active Sites of the 20S Proteasome Is Coupled to Opening of the α-Ring Channel. Structure. 2009, 17, 1137–1147. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, S.; Wu, Z.; Li, X.; Wang, W.L.; Zhu, Y. Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature 2019, 565, 49–55. [Google Scholar] [CrossRef]
- Gaczynska, M.; Osmulski, P.A. Harnessing proteasome dynamics and allostery in drug design. Antioxidants Redox Signal. 2014, 21, 2286–2301. [Google Scholar] [CrossRef]
- Wakata, A.; Lee, H.-M.; Rommel, P.; Toutchkine, A.; Schmidt, M.; Lawrence, D.S. Simultaneous Fluorescent Monitoring of Proteasomal Subunit Catalysis. J. Am. Chem Soc. 2010, 132, 1578–1582. [Google Scholar] [CrossRef]
- Asano, S.; Fukuda, Y.; Beck, F.; Aufderheide, A.; Forster, F.; Danev, R. A molecular census of 26S proteasomes in intact neurons. Science 2015, 347, 439–442. [Google Scholar] [CrossRef]
- Yoshimura, T.; Kameyama, K.; Takagi, T.; Ikai, A.; Tokunaga, F.; Koide, T. Molecular Characterization of the “26S” Proteasome Complex from Rat Liver. J. Struct Biol. 1993, 111, 200–211. [Google Scholar] [CrossRef]
- Tanahashi, N.; Murakami, Y.; Minami, Y.; Shimbara, N.; Hendil, K.; Tanaka, K. Hybrid Proteasomes. J. Biol Chem. 2000, 275, 14336–14345. [Google Scholar] [CrossRef]
- Nunan, J.; Williamson, N.A.; Hill, A.F.; Sernee, M.F.; Masters, C.L.; Small, D.H. Proteasome-mediated degradation of the C-terminus of the Alzheimer’s disease ?-amyloid protein precursor: Effect of C-terminal truncation on production of ?-amyloid protein. J. Neurosci Res. 2003, 74, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Qing, H.; Zhou, W.; Christensen, M.A.; Sun, X.; Tong, Y.; Song, W. Degradation of BACE by the ubiquitin-proteasome pathway. FASEB J. 2004, 18, 1571–1573. [Google Scholar] [CrossRef] [PubMed]
- Osmulski, P.A.; Gaczynska, M. Rapamycin allosterically inhibits the proteasome. Mol. Pharmacol. 2013, 84, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger, L.L.C. The AxPyMOL Molecular Graphics Plugin for Microsoft PowerPoint; Version 1.5; 2015. [Google Scholar]
- Schrodinger, L.L.C. The JyMOL Molecular Graphics Development Component; Version 1.5; 2015. [Google Scholar]
- Schrodinger, L.L.C. The PyMOL Molecular Graphics System; Version 1.5; 2015. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | R1 GRKKRRQ R2 RQRRRPS R3 GRK R4 RQ R5 RPS R6 KKK R7 AAA | |
| (1) | TAT1 | GRKKRRQRRRPS | |
| (2) | TAT1-4,5 TO 8,9-TOD | R3-Tic-Oic-R4-Tic-D-Oic-R5 | |
| (3) | TAT1-4,5 TO | R3-Tic-Oic-R2 | |
| (4) | TAT1-8,9 TO | R1-Tic-Oic-R5 | |
| (5) | TAT1-8,9 TOD | R1-Tic-D-Oic-R5 | |
| (6) | 3K-TO-3K | R6-TicOic-R6 | |
| (7) | 3A-TO-3A | R7-TicOic-R7 | |
| (8) | TAT1-Den | R6 R6-DABA< R6 | |
| (9) | TAT1 A8-10 | GRKKRRQ-R7-PS |
| Compound | AC50 [nM] ± SE | Max Activation Fold ± SE | |
|---|---|---|---|
| (1) | Tat1 | 710 ± 12 | 6.2 ± 0.20 |
| (2) | Tat1-4,5 TO 8,9-TOD | 1528 ± 208 | 1.5 ± 0.26 |
| (3) | Tat1-4,5 TO | 643 ± 74 | 3.3 ± 0.23 |
| (4) | Tat1-8,9 TO | 499 ± 62 | 5.2 ± 0.76 |
| (5) | Tat1-8,9 TOD | 220 ± 89 | 7.9 ± 0.74 |
| (6) | 3K-TO-3K | 1199 ± 118 | 8.0 ± 0.12 |
| (7) | 3A-TO-3A | 1744 ± 414 | 1.2 ± 0.04 |
| (8) | Tat1-Den | 374 ± 13 | 9.8 ± 0.18 |
| (9) | TAT1 A8-10 | ND | 0.9 ± 0.12 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osmulski, P.A.; Karpowicz, P.; Jankowska, E.; Bohmann, J.; Pickering, A.M.; Gaczyńska, M. New Peptide-Based Pharmacophore Activates 20S Proteasome. Molecules 2020, 25, 1439. https://doi.org/10.3390/molecules25061439
Osmulski PA, Karpowicz P, Jankowska E, Bohmann J, Pickering AM, Gaczyńska M. New Peptide-Based Pharmacophore Activates 20S Proteasome. Molecules. 2020; 25(6):1439. https://doi.org/10.3390/molecules25061439
Chicago/Turabian StyleOsmulski, Paweł A., Przemysław Karpowicz, Elżbieta Jankowska, Jonathan Bohmann, Andrew M. Pickering, and Maria Gaczyńska. 2020. "New Peptide-Based Pharmacophore Activates 20S Proteasome" Molecules 25, no. 6: 1439. https://doi.org/10.3390/molecules25061439
APA StyleOsmulski, P. A., Karpowicz, P., Jankowska, E., Bohmann, J., Pickering, A. M., & Gaczyńska, M. (2020). New Peptide-Based Pharmacophore Activates 20S Proteasome. Molecules, 25(6), 1439. https://doi.org/10.3390/molecules25061439