
A Reliable Enantioselective Route to Mono-Protected N1-Cbz Piperazic Acid Building Block


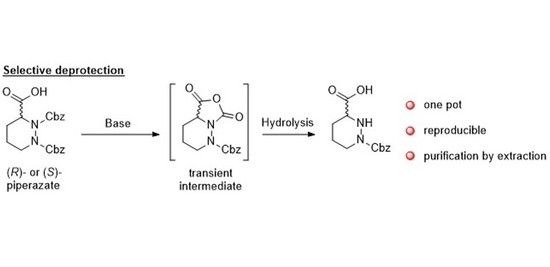
Abstract
:
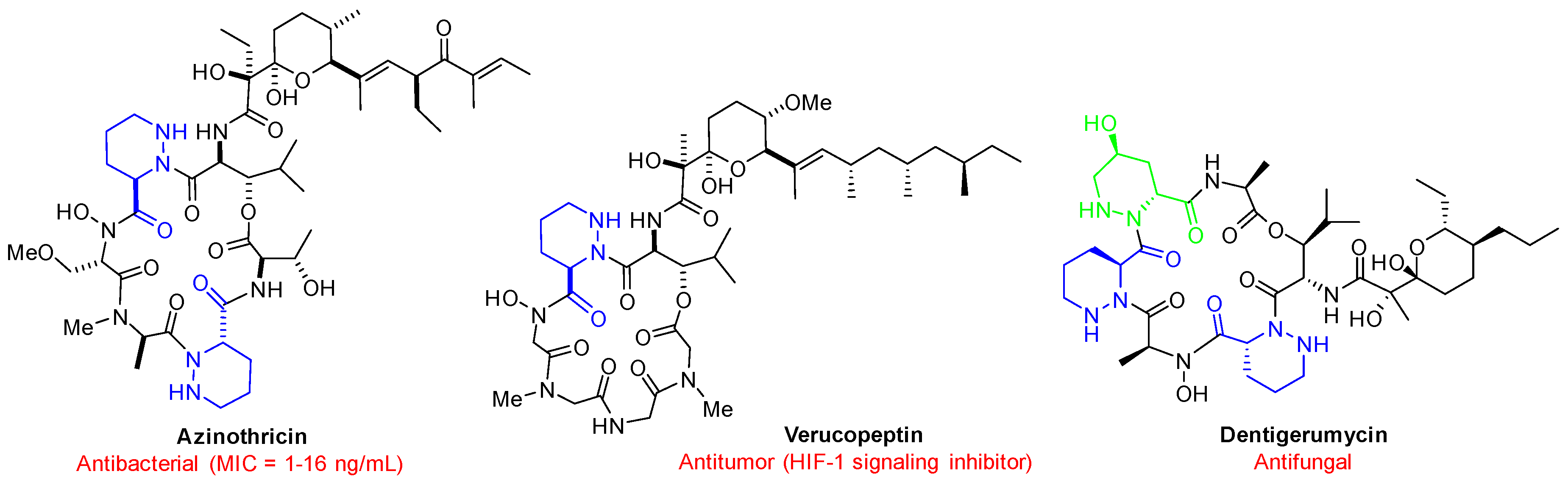
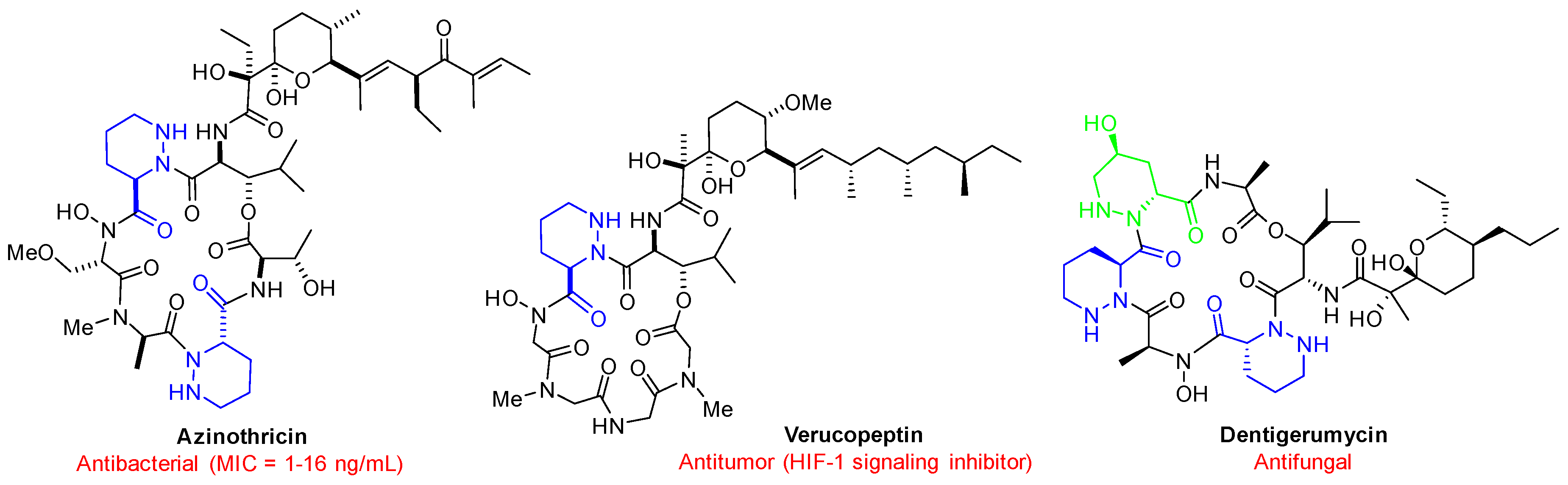
1. Introduction
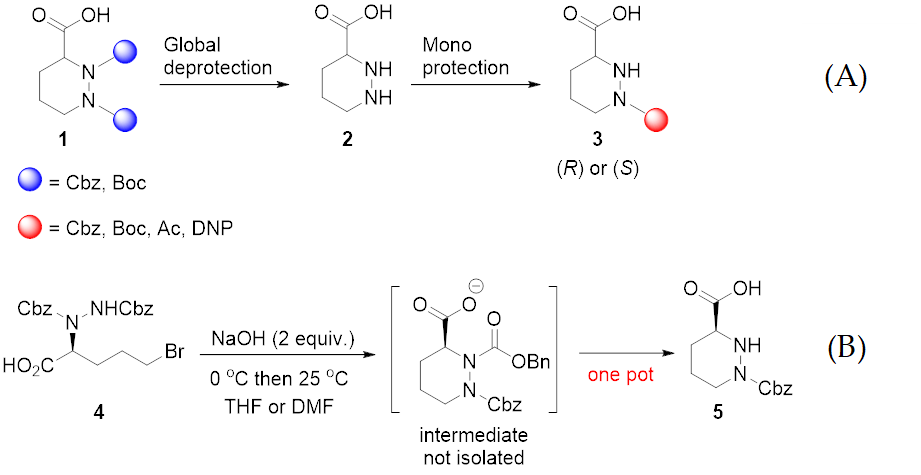
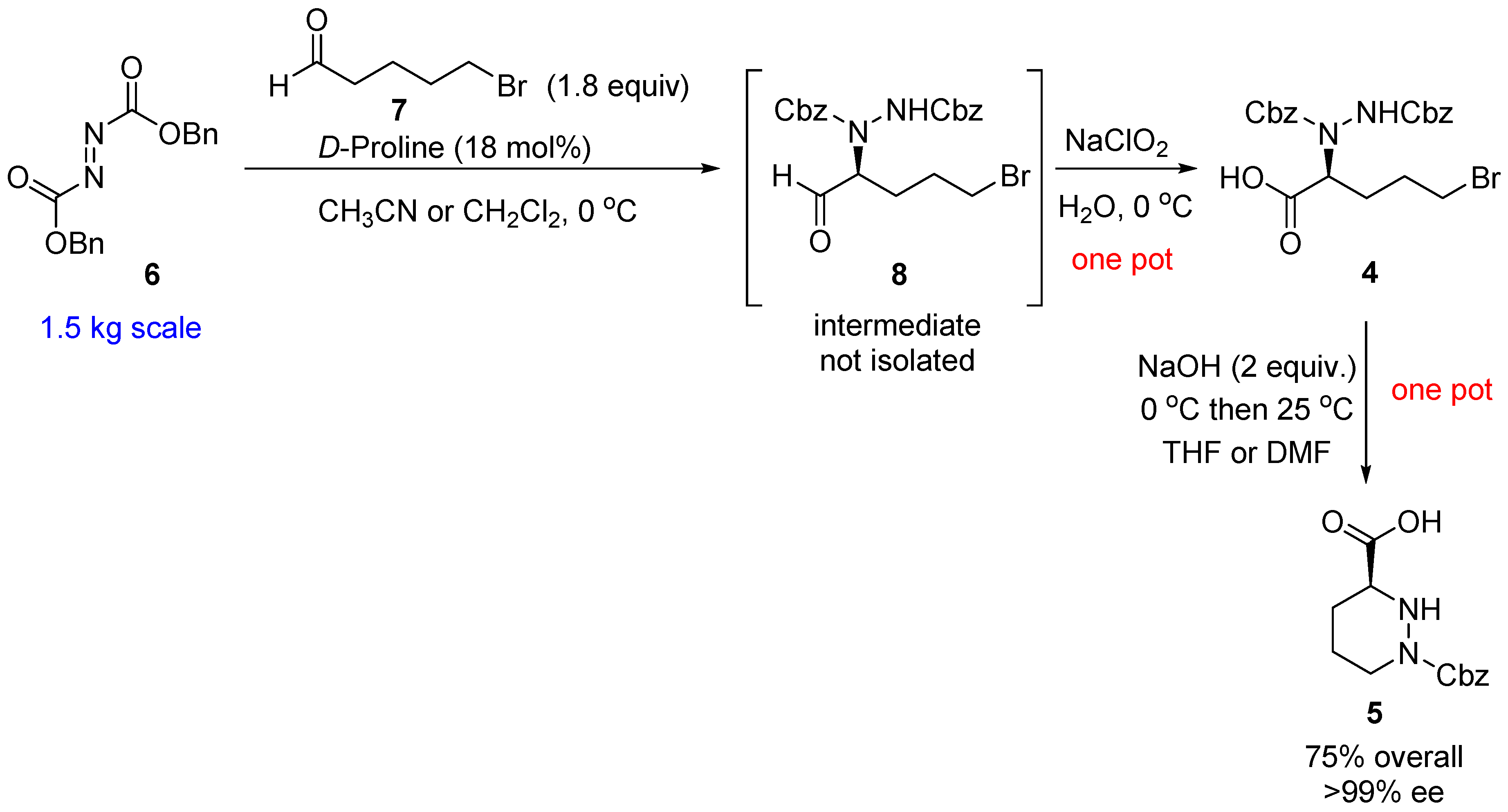
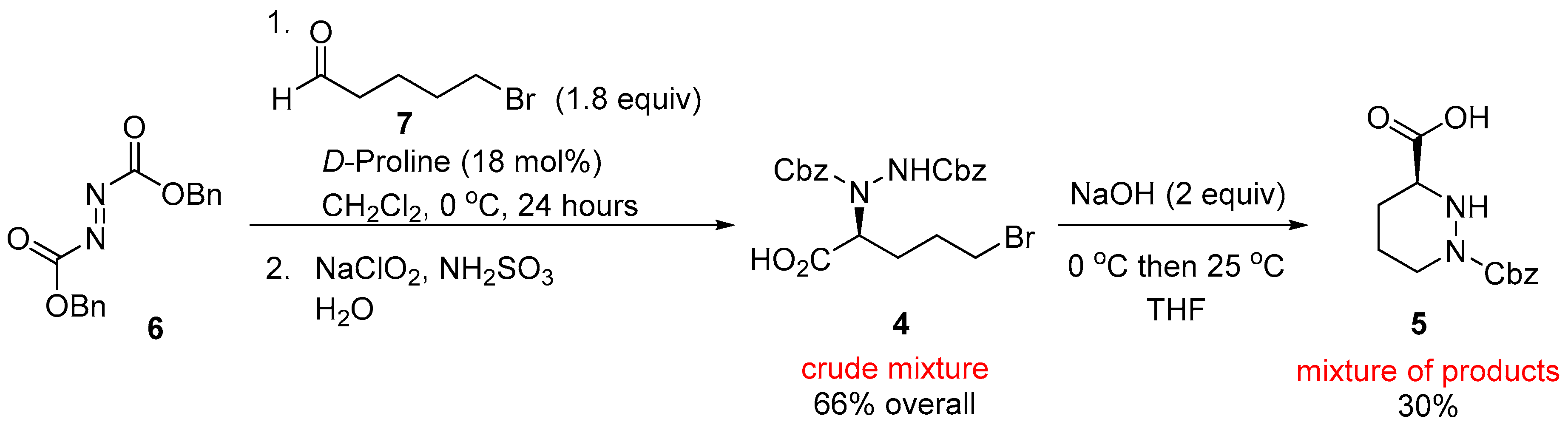
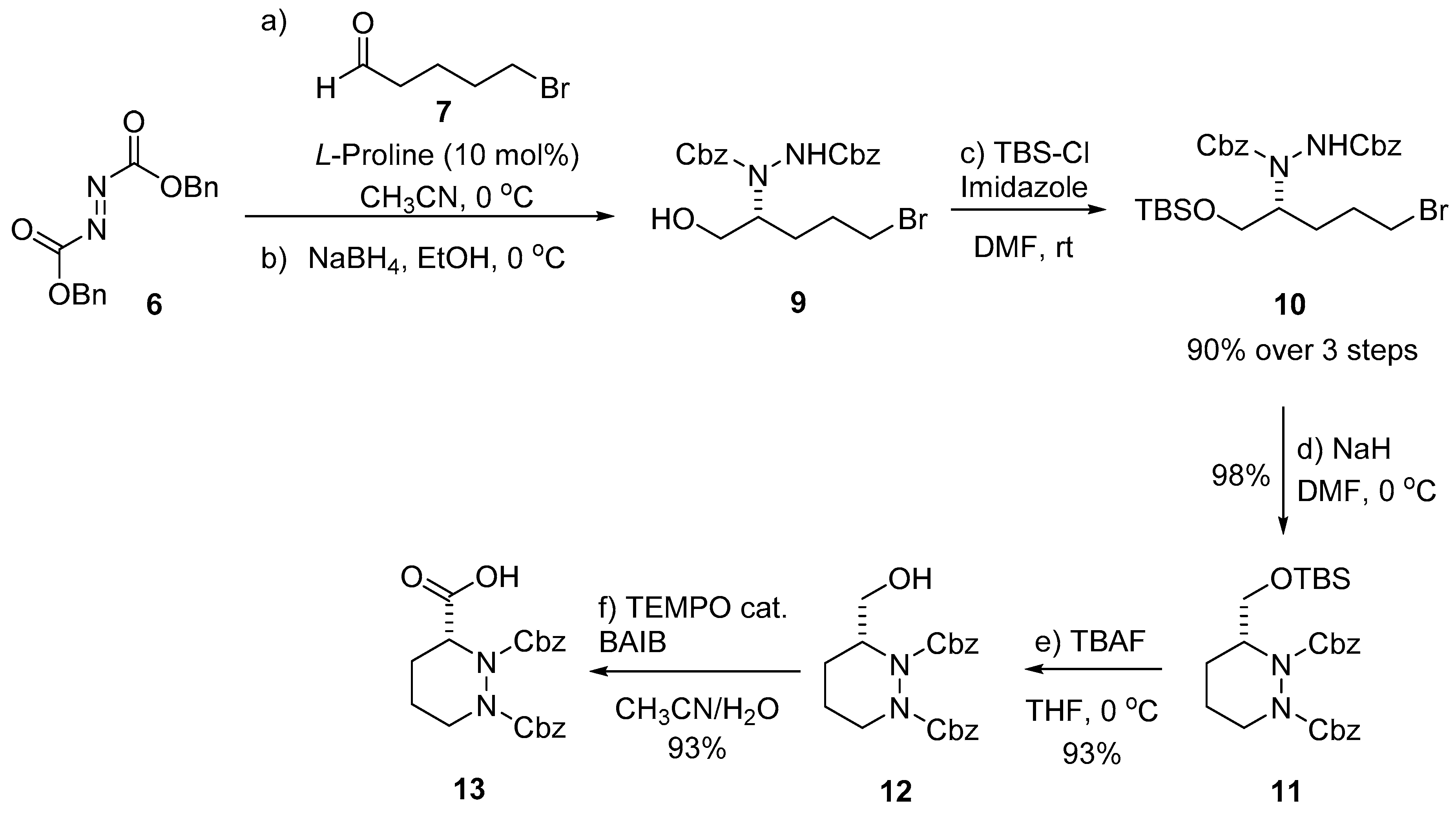
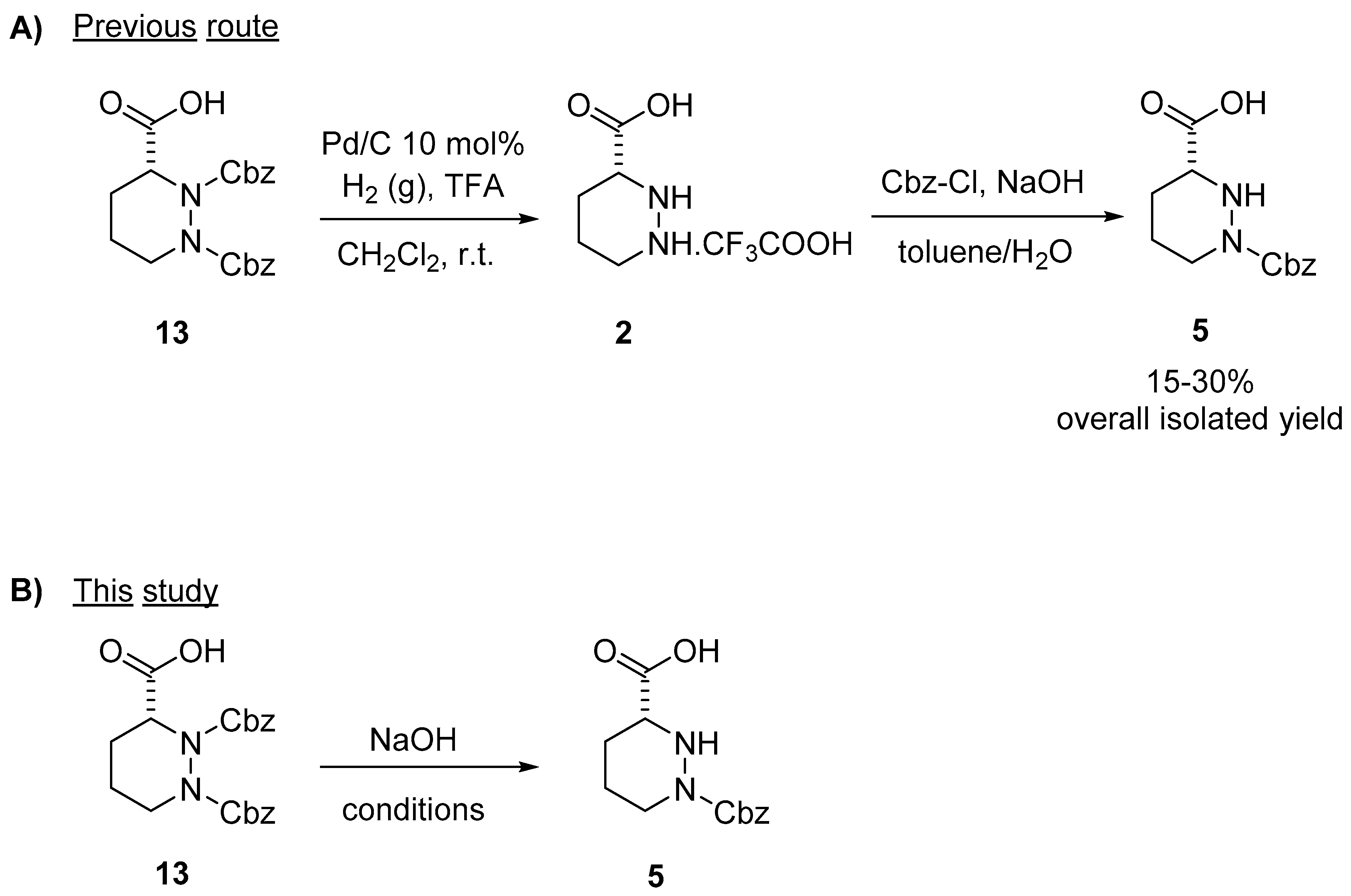
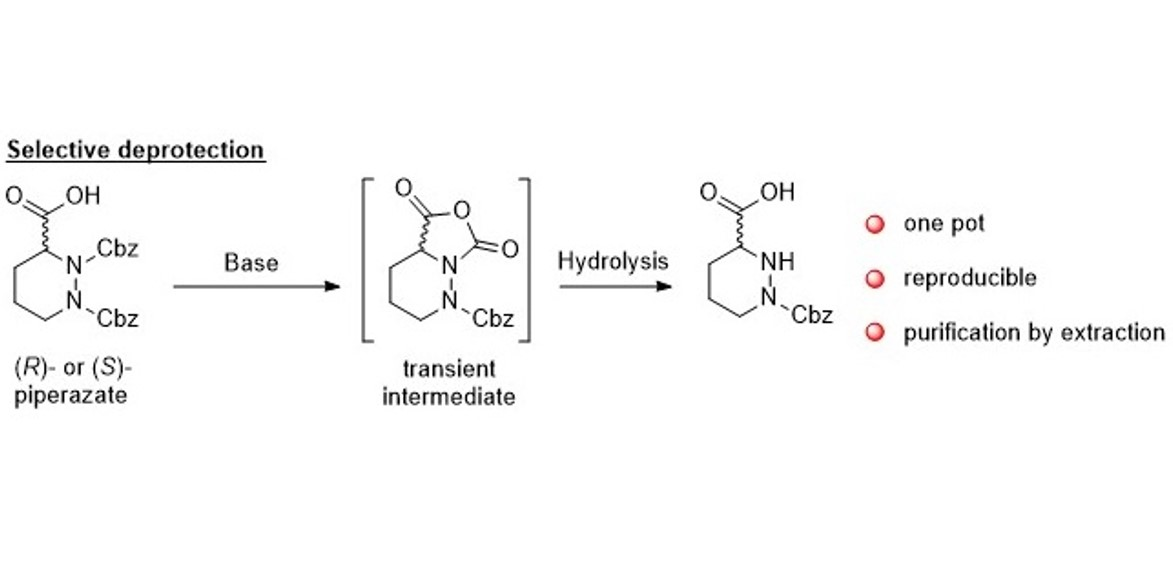
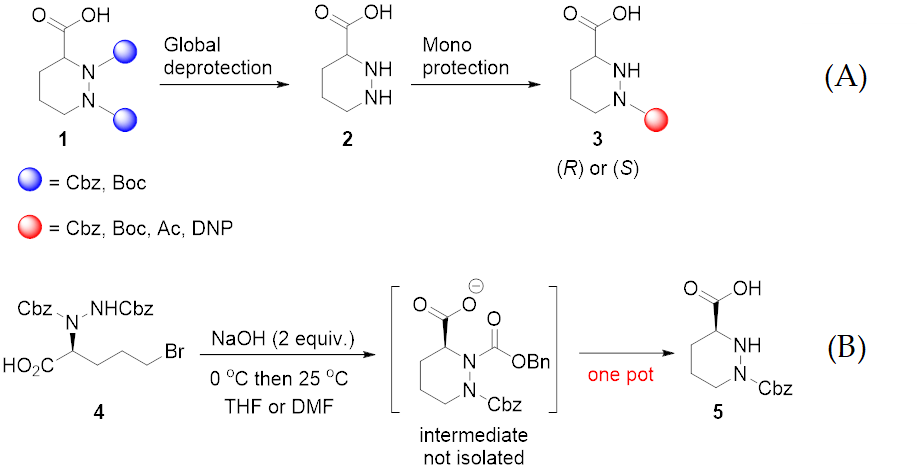
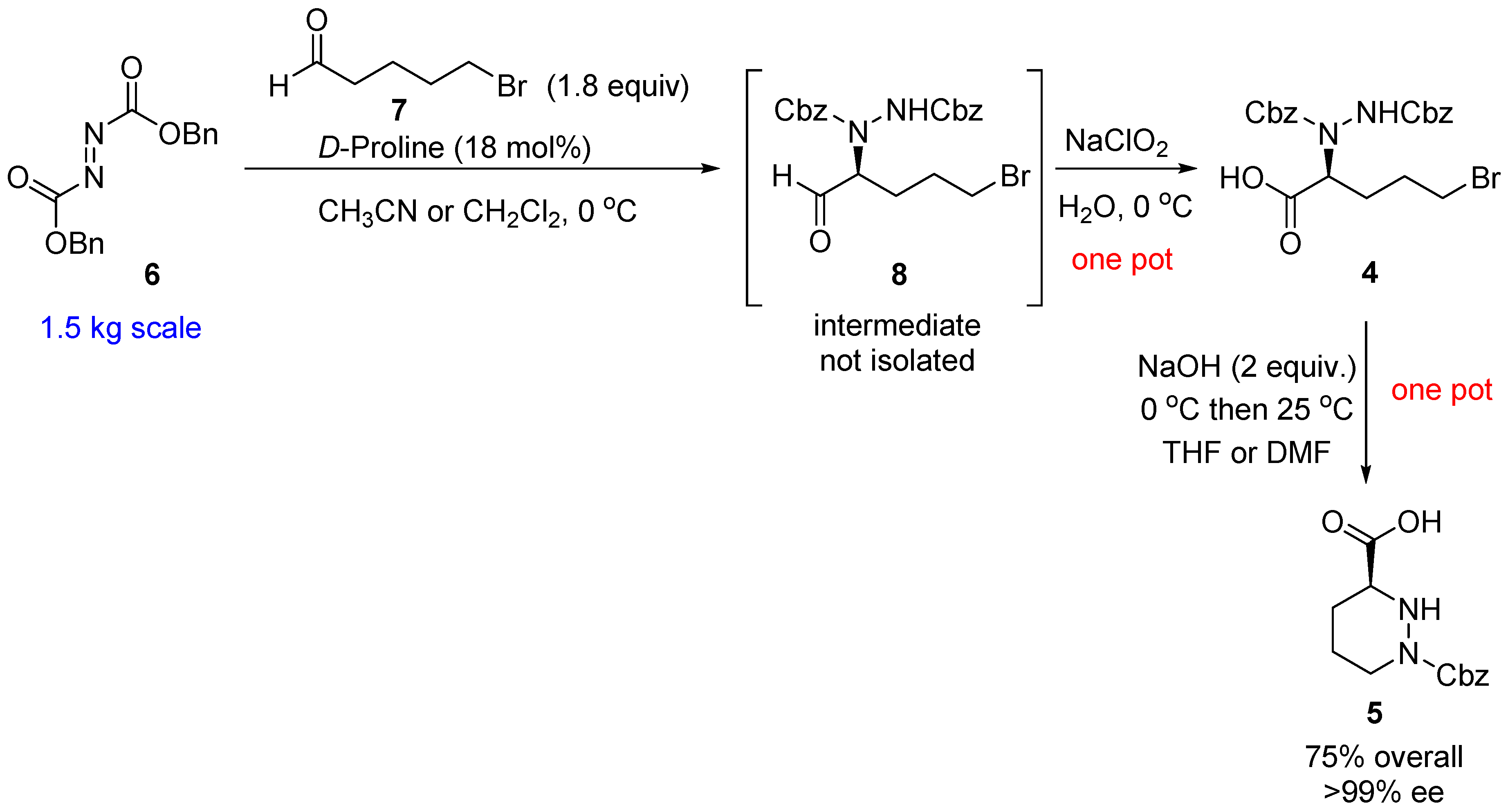
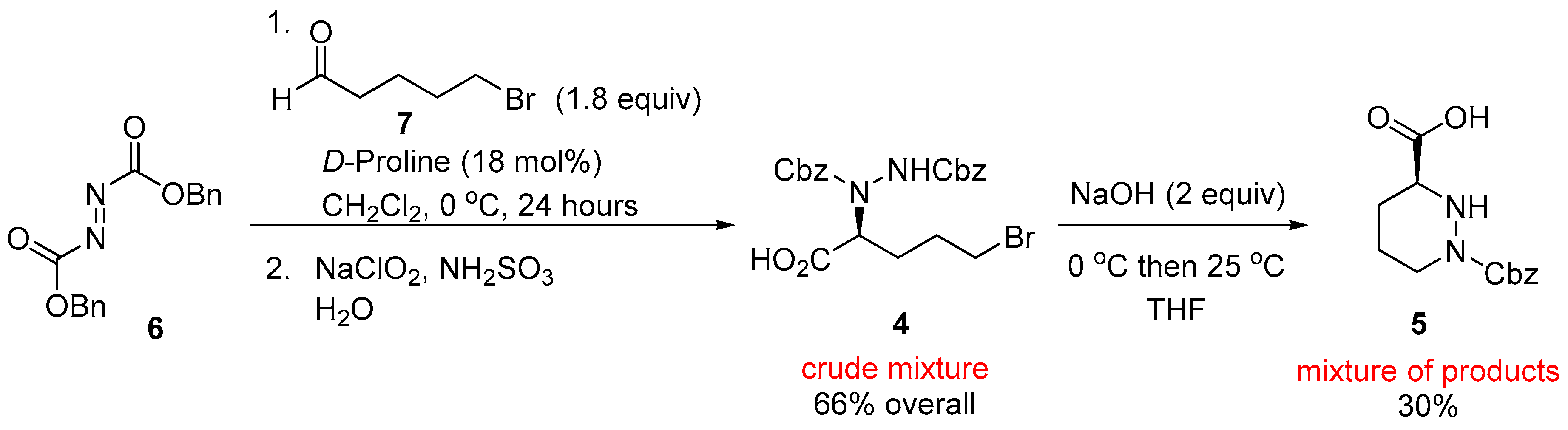
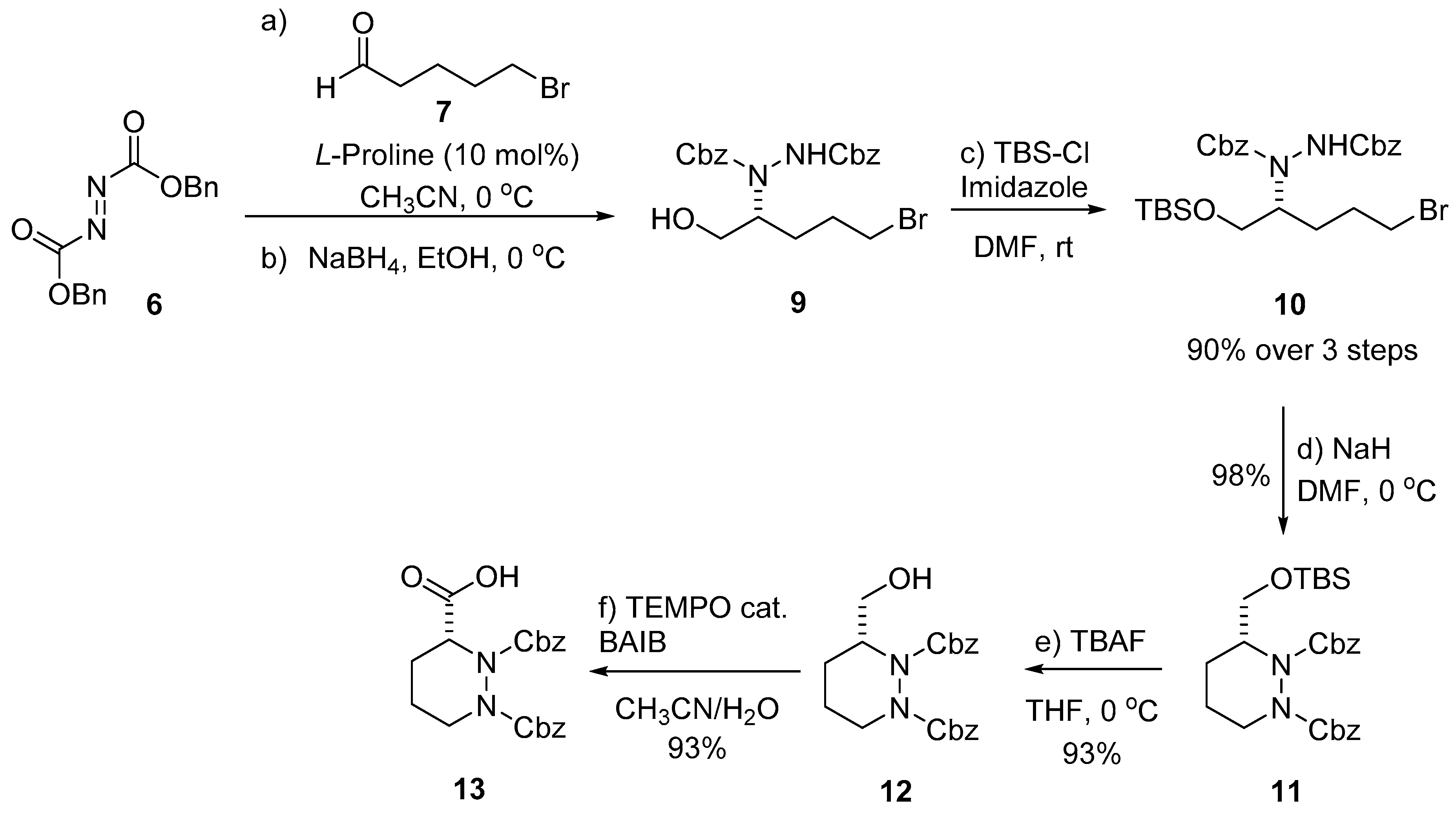
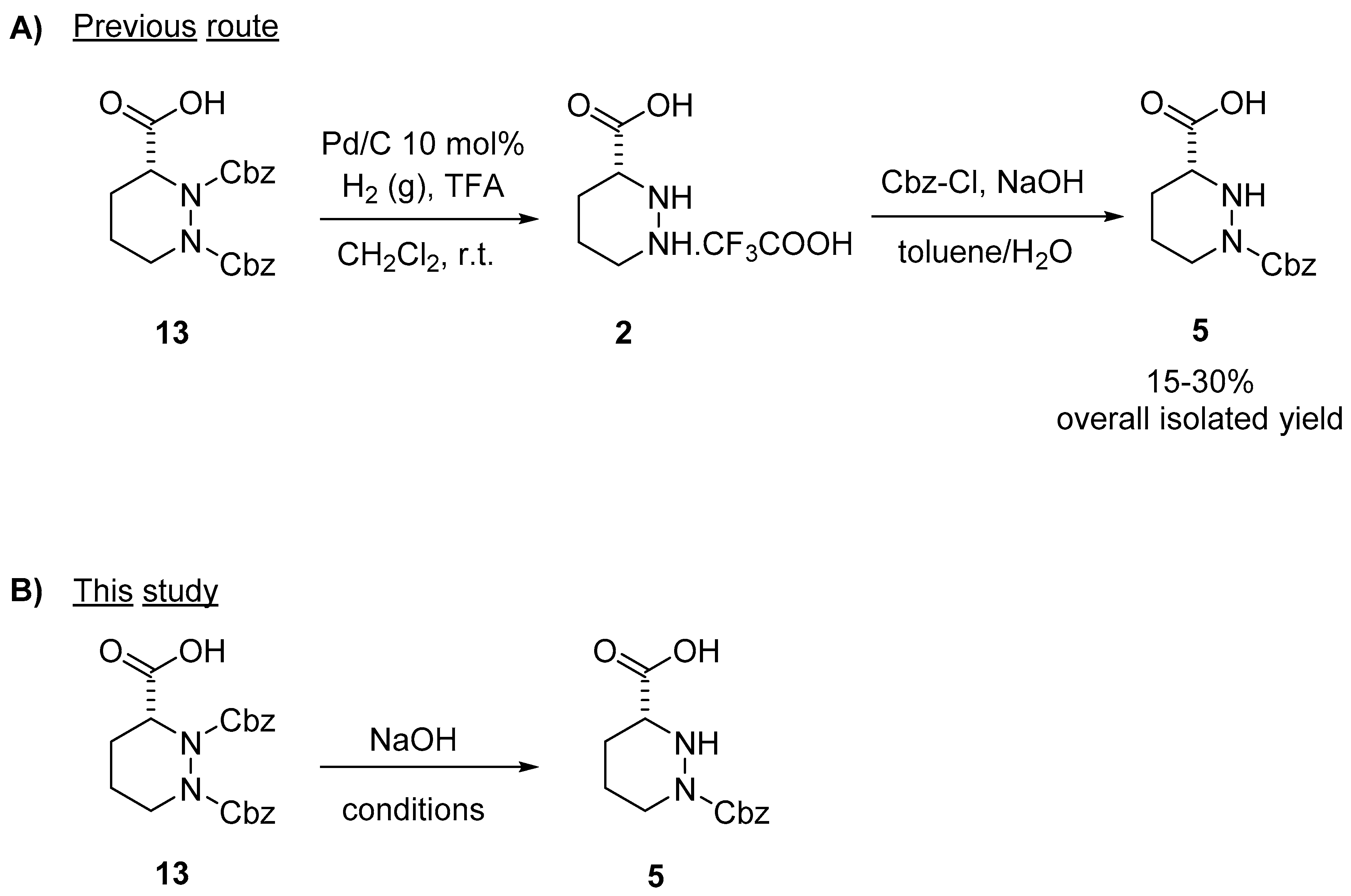
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bevan, K.; Davies, J.S.; Hassall, C.H.; Morton, R.B.; Phillips, D.A.S. Amino-acids and peptides. Part X. Characterisation of the Monamycins, Members of a New Family of Cyclodepsipeptide Antibiotics. J. Chem. Soc. C. 1971, 514–522. [Google Scholar] [CrossRef]
- Ciufolini, M.A.; Xi, N. Synthesis, chemistry and conformational properties of piperazic acids. Chem. Soc. Rev. 1998, 27, 437–445. [Google Scholar] [CrossRef]
- Oelke, A.J.; France, D.J.; Hofmann, T.; Wuitschik, G.; Ley, S.V. Piperazic acid-containing natural products: Isolation, biological relevance and total synthesis. Nat. Prod. Rep. 2011, 28, 1445–1471. [Google Scholar] [CrossRef] [PubMed]
- Morgan, K.D.; Andersen, R.J.; Ryan, K.S. Piperazic acid-containing natural products: Structures and biosynthesis. Nat. Prod. Rep. 2019, 36, 1628–1653. [Google Scholar] [CrossRef] [PubMed]
- Shibahara, S.; Matsubara, T.; Takahashi, K.; Ishihara, J.; Hatakeyama, S. Total Synthesis of NW-G01, a Cyclic Hexapeptide Antibiotic, and 34-epi-NW-G01. Org. Lett. 2011, 13, 4700–4703. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Sekioka, N.; Izumikawa, M.; Kozone, I.; Takagi, M.; Shin-ya, K.; Doi, T. Total Synthesis and Structure Elucidation of JBIR-39: A Linear Hexapeptide Possessing Piperazic Acid and γ-Hydroxypiperazic Acid Residues. Chem. Eur. J. 2015, 21, 3031–3041. [Google Scholar] [CrossRef] [PubMed]
- Elbatrawi, Y.M.; Kang, C.W.; Del Valle, J.R. Total Synthesis of L-156,373 and an oxoPiz Analogue via a Submonomer Approach. Org. Lett. 2018, 20, 2707–2710. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Gan, J.; Ma, D. Total Synthesis of Piperazimycin A: A Cytotoxic Cyclic Hexadepsipeptide. Angew. Chem. Int. Ed. 2009, 48, 8891–8895. [Google Scholar] [CrossRef] [PubMed]
- Ciufolini, M.A. Chapter 1 The total synthesis of luzopeptins. Strateg. Tactics Org. Synth. 2005, 6, 1–37. [Google Scholar]
- Kuchenthal, C.H.; Maison, W. Synthesis of Cyclic Hydrazino α-Carboxylic Acids. Synthesis 2010, 5, 719–740. [Google Scholar]
- Hale, K.J.; Delisser, V.M.; Manaviazar, S. Azinothricin synthetic studies. 1. Efficient asymmetric syntheses of (3R)- and (3S)-piperazic acids. Tetrahedron Lett. 1992, 33, 7613–7616. [Google Scholar] [CrossRef]
- Hale, K.J.; Cai, J.; Delisser, V.; Manaviazar, S.; Peak, S.A.; Bhatia, G.S.; Collins, T.C.; Jogiya, N. Enantioselective Synthesis of (3R)- and (3S)-Piperazic Acids. The Comparative Unimportance of DMPU Mediated Retro-Hydrazination. Tetrahedron 1996, 52, 1047–1068. [Google Scholar] [CrossRef]
- Hale, K.J.; Manaviazar, S.; George, J.H.; Walters, M.A.; Dalby, S.M. Total synthesis of (+)-Azinothricin and (+)-Kettapeptin. Org. Lett. 2009, 11, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lu, Y.; Zou, Q.; Chen, H.; Ma, D. A Scalable Process to the Key Intermediate of Cilazapril: (S)-1-Benzyloxycarbonylhexahydropyridazine-3-carboxylic acid, Through a Novel Cascade Course. Org. Process. Res. Dev. 2013, 17, 1209–1213. [Google Scholar] [CrossRef]
- List, B. Direct Catalytic Asymmetric α-Amination of Aldehydes. J. Am. Chem. Soc. 2002, 124, 5656–5657. [Google Scholar] [CrossRef] [PubMed]
- Henmi, Y.; Makino, K.; Yoshitomi, Y.; Hara, O.; Hamada, Y. Highly efficient synthesis of (R)- and (S)-piperazic acids using proline-catalyzed asymmetric α-hydrazination. Tetrahedron Asymmetry 2004, 15, 3477–3481. [Google Scholar] [CrossRef]
- Chong, J.M.; Heuft, M.A.; Rabbat, P. Solvent effects on the monobromination of α,ω-diols: A convenient preparation of ω-bromoalkanols. J. Org. Chem. 2000, 65, 5837–5838. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.E.; Aguilar, D.; Hertel, S.; Knight, W.H.; Paterson, J. Preparation of 1-(Benzyloxycarbonyl) Hexahydro-3-Pyridazine Carboxylic Acid, A Protected Piperazic Acid. Synth. Commun. 1988, 18, 2225–2231. [Google Scholar] [CrossRef]
- Coats, R.A.; Lee, S.-L.; Davis, K.A.; Patel, K.M.; Rhoads, E.K.; Howard, M.H. Stereochemical Definition and Chirospecific Synthesis of the Peptide Deformylase Inhibitor Sch 382583. J. Org. Chem. 2004, 69, 1734–1737. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, U.; Braun, C.; Sutoris, H. Enantioselective Syntheses of (R)- and (S)-Hexahydropyridazine-3-carboxylic Acid Derivatives. Synthesis 1996, 223–229. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
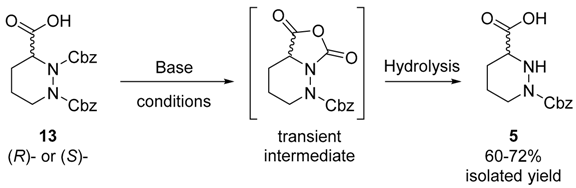
| Entry a | Base (equiv.) | Solvent | Temperature (°C) | Full Consumption of 13 b |
|---|---|---|---|---|
| 1 | NaOH (1.2) | THF | 23 |  |
| 2 | NaOH (2.0) | THF | 23 | |
| 3 | KOH (2.0) | THF | 23 | |
| 4 | LiOH (2.0) | THF | 23 | |
| 5 | NaH (1.1) | THF | 23 | |
| 6 | NaOH (2.0) | MeOH | 23 | |
| 7 | NaOH (2.0) | THF | 45 |  |
| 8 c | NaOH (2.0) | THF | 45 | |
Sample Availability: Samples of the compounds are available from the authors. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papadaki, E.; Georgiadis, D.; Tsakos, M. A Reliable Enantioselective Route to Mono-Protected N1-Cbz Piperazic Acid Building Block. Molecules 2020, 25, 5939. https://doi.org/10.3390/molecules25245939
Papadaki E, Georgiadis D, Tsakos M. A Reliable Enantioselective Route to Mono-Protected N1-Cbz Piperazic Acid Building Block. Molecules. 2020; 25(24):5939. https://doi.org/10.3390/molecules25245939
Chicago/Turabian StylePapadaki, Evanthia, Dimitris Georgiadis, and Michail Tsakos. 2020. "A Reliable Enantioselective Route to Mono-Protected N1-Cbz Piperazic Acid Building Block" Molecules 25, no. 24: 5939. https://doi.org/10.3390/molecules25245939
APA StylePapadaki, E., Georgiadis, D., & Tsakos, M. (2020). A Reliable Enantioselective Route to Mono-Protected N1-Cbz Piperazic Acid Building Block. Molecules, 25(24), 5939. https://doi.org/10.3390/molecules25245939